
ABSTRACT
Aims
To isolate canine respiratory coronavirus (CRCoV) and canine pneumovirus (CnPnV) in cell culture and to compare partial genomic sequences of CRCoV and CnPnV from New Zealand with those from other countries.
Methods
Oropharyngeal swab samples from dogs affected by canine infectious respiratory disease syndrome that were positive for CnPnV (n = 15) or CRCoV (n = 1) by virus-specific reverse transcriptase quantitative PCR (RT-qPCR) in a previous study comprised the starting material. Virus isolation was performed in HRT-18 cells for CRCoV and RAW 264.7 and Vero cells for CnPnV. The entire sequence of CnPnV G protein (1,266 nucleotides) and most (8,063/9,707 nucleotides) of the 3’ region of CRCoV that codes for 10 structural and accessory proteins were amplified and sequenced. The sequences were analysed and compared with other sequences available in GenBank using standard molecular tools including phylogenetic analysis.
Results
Virus isolation was unsuccessful for both CRCoV and CnPnV. Pneumovirus G protein was amplified from 3/15 (20%) samples that were positive for CnPnV RNA by RT-qPCR. Two of these (NZ-048 and NZ-049) were 100% identical to each other, and 90.9% identical to the third one (NZ-007). Based on phylogenetic analysis of the G protein gene, CnPnV NZ-048 and NZ-049 clustered with sequences from the USA, Thailand and Italy in group A, and CnPnV NZ-007 clustered with sequences from the USA in group B. The characteristics of the predicted genes (length, position) and their putative protein products (size, predicted structure, presence of N- and O-glycosylation sites) of the New Zealand CRCoV sequence were consistent with those reported previously, except for the region located between open reading frame (ORF)3 (coding for S protein) and ORF6 (coding for E protein). The New Zealand virus was predicted to encode 5.9 kDa, 27 kDa and 12.7 kDa proteins, which differed from the putative coding capacity of this region reported for CRCoV from other countries.
Conclusions
This report represents the first characterisation of partial genomic sequences of CRCoV and CnPnV from New Zealand. Our results suggest that the population of CnPnV circulating in New Zealand is not homogeneous, and that the viruses from two clades described overseas are also present here. Limited conclusions can be made based on only one CRCoV sequence, but the putative differences in the coding capacity of New Zealand CRCoV support the previously reported variability of this region. The reasons for such variability and its biological implications need to be further elucidated.
Introduction
Canine pneumovirus (CnPnV) and canine respiratory coronavirus (CRCoV) are two of several pathogens that have been implicated in the development of canine infectious respiratory disease syndrome (CIRDS), also known as “kennel cough.” The disease most commonly affects dogs housed in groups (Schulz et al. Citation2014; Mitchell et al. Citation2017). Affected dogs present with acute onset of a hacking cough, accompanied in some cases by fever, sneezing, and nasal or ocular discharge. There is no surveillance for CIRDS in New Zealand and hence there are no reliable data on the incidence of CIRDS in this country. Anecdotally, however, outbreaks of kennel cough occur here, and there has been a perception among veterinarians that the numbers of CIRDS cases have increased within the past few years (Anonymous Citation2022).
Traditionally, CIRDS has been linked to infections with Bordetella bronchiseptica, canine parainfluenza virus, and canine adenovirus type 2 (McCandlish et al. Citation1978), but several other infectious agents have been detected from affected dogs in the past 20 years. These include CRCoV (Erles et al. Citation2003), CnPnV (Renshaw et al. Citation2010), canine influenza virus (Pecoraro et al. Citation2014), canine bocavirus (Kapoor et al. Citation2012) and canine hepacivirus (Kapoor et al. Citation2011), as well as bacteria (Streptococcus equi subsp. zooepidemicus (Chalker et al. Citation2003) and various Mycoplasma species (Chalker et al. Citation2004)). The exact role of these agents in the development of CIRDS needs to be elucidated as most have been detected both from clinically ill and healthy dogs, underscoring the impact of host-, environment- and pathogen-related factors on the expression of disease (Mitchell et al. Citation2017; Maboni et al. Citation2019). Of the CIRDS-associated pathogens, canine influenza virus, canine bocavirus, and canine hepacivirus have never been detected in New Zealand (Sowman et al. Citation2018; More et al. Citation2021).
Canine pneumovirus is an enveloped, negative sense, single-stranded RNA virus classified within the family Pneumoviridae, genus Orthopneumovirus (Rima et al. Citation2017). Pneumoviruses encode 9–11 proteins, including a heavily glycosylated attachment protein (G protein), which has a high degree of sequence variation within and between different species of pneumoviruses (Kumagai et al. Citation2021; Yu et al. Citation2021). CnPnV was first identified during a diagnostic investigation of acute infectious respiratory disease in dog breeding colonies in the USA (Renshaw et al. Citation2010, Citation2011). Since this initial report, the virus has been detected from dogs with respiratory disease in Thailand (Piewbang and Techangamsuwan Citation2019), China (Song et al. Citation2021), the UK, the Republic of Ireland (Mitchell et al. Citation2013a), and Italy (Decaro et al. Citation2014a). Based on serological evidence, infection with CnPnV is also common in many other European countries (Day et al. Citation2020). In a recently reported New Zealand-based study, CnPnV sequences were detected in 14/56 (25%) nasopharyngeal swabs collected from CIRDS-affected dogs and from 17/60 (28%) clinically normal dogs by reverse transcriptase quantitative PCR (RT-qPCR) (More et al. Citation2021).
Canine respiratory coronavirus is an enveloped positive sense, single-stranded RNA virus classified within the order Nidovirales, genus Betacoronavirus and species Betacoronavirus 1 (Erles et al. Citation2003). Typical for all nidoviruses, coronavirus genes are expressed from the 3′ co-terminal sets of mRNA generated through a process of discontinuous transcription () (Pasternak et al. Citation2006). Transcription regulatory sequences (TRS) are crucial for that process. The TRS are located at the 5’ end of the coronavirus genome, as well as upstream of structural and accessory genes located in the 3’ end of the genome, and commonly include a consensus sequence CYAAAC (Hofmann et al. Citation1993). The virus was first detected in samples collected from dogs with severe respiratory disease in a large re-homing centre in the UK in 2003 (Erles et al. Citation2003). Since then, CRCoV has been identified in other geographical areas including Italy (Lorusso et al. Citation2009), Korea (An et al. Citation2010), Japan (Soma et al. Citation2008), and China (Hong et al. Citation2015). Based on serological evidence, CRCoV infection is common among dogs in New Zealand, with more than half of the dogs sampled in one study testing positive for CRCoV antibodies (More et al. Citation2020). Despite this relatively high level of infection, the virus has never been isolated in New Zealand and only 1/116 dogs tested in a recent molecular survey was positive for CRCoV RNA by RT-qPCR (More et al. Citation2021).
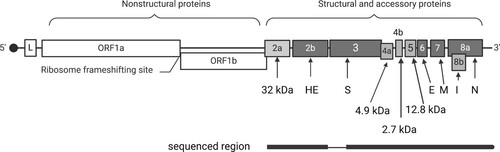
Figure 1. Schematic representation of the organisation of the genome of canine respiratory coronavirus (CRCoV; based on isolate BJ232; GenBank accession KX432213). The open reading frames (ORF) are numbered within or above the rectangles, and the protein products of the structural and accessory genes are specified below. The positive-sense, single-stranded, 30.8 kb RNA genome is capped at the 5’ end and expressed through a process of discontinuous transcription. A common leader sequence (L) encoded at the 5′ of the genome is present at the 5’ end of each mRNA. The region sequenced in the current study is shown below the genome, with a thin line indicating the missing sequence.
The aim of the current study was to culture CRCoV and CnPnV from samples collected from CIRDS-affected dogs that were positive for viral RNA in the previous study (More et al. Citation2021) and to compare partial genomic sequences of CRCoV and CnPnV form New Zealand with those from other countries.
Materials and methods
Source of samples
Oropharyngeal swab samples from dogs affected by CIRDS that were positive for CnPnV (n= 15) or CRCoV (n = 1) by virus-specific RT-qPCR in the previous study (More et al. Citation2021) constituted the starting material. The samples were stored at –80°C.
Virus isolation
Virus isolation was attempted in HRT-18 cells for CRCoV and in RAW 264.7 and Vero cells for CnPnV. The HRT-18 and Vero cell lines were commercially obtained from American Tissue Culture Collection (Manassas, VA, USA), while RAW 264.7 was donated by Dr. Patrick Reading (Melbourne, Australia). All three cell lines were grown in Advanced Dulbecco’s modified Eagle medium (Sigma-Aldrich, St Louis, MO, USA) supplemented with 2% v/v fetal bovine serum (FBS; Gibco Life Technologies, New York, NY, USA), 1% v/v antibiotic solution (5,000 U/mL penicillin and 5,000 µg/mL streptomycin; Thermo Fisher, Waltham, MA, USA), and 1% v/v Glutamax (Gibco Life Technologies) using standard laboratory procedures (Freshney Citation2005). Maintenance medium, where used, was the same as the growth medium, but with 1% v/v FBS. For isolation of CRCoV, the inoculum was supplemented with trypsin to 1 µg/mL, and the cells were maintained in maintenance medium containing 1 µg/mL trypsin. All cultures were incubated at 37°C in a humidified atmosphere with 5% CO2 for 5–6 days and observed daily for the presence of viral cytopathic effect. All cultures were passaged at least twice before considered negative.
The cell lysate from the third passage was used as a template for virus-specific RT-qPCR. Lysate from the fourth passage of CRCoV culture was subjected to hemagglutination (HA) test using 1% washed chicken red blood cells at 4°C until HA was observed in positive control wells containing bovine coronavirus (BCoV). Negative controls consisted of cell lysate from non-inoculated HRT-18 cells. Cultures were considered positive for CRCoV if both RT-qPCR and HA tests were positive, and positive for CnPnV when cell lysates from the third passage were positive for CnPnV RNA by RT-qPCR (performed as reported previously; More et al. Citation2021).
Nucleic acid extraction and cDNA synthesis
Total nucleic acids were extracted from the samples using a High Pure Viral Nucleic Acid Kit (Roche Diagnostics GmbH, Basel, Switzerland) as described previously (More et al. Citation2021). In order to obtain long cDNA fragments, RNA was reverse transcribed using the Transcriptor First Strand cDNA synthesis kit (Roche Diagnostics GmbH). The reaction consisted of 1 µg of RNA, 60 µM random hexamer primers, 4 µL of 1 X reaction buffer, 20 U of RNase inhibitor, 1 mM of deoxyribonucleotide mix and 10 U reverse transcriptase in a total reaction volume of 20 µL. The tubes were incubated at 25°C for 10 minutes, 55°C for 30 minutes and 85°C for 5 minutes.
Amplification and analysis of CnPnV G protein gene
The CnPnV G protein gene was amplified using overlapping primers previously described by Glineur et al. (Citation2013) (Supplementary Table 1). Each 25 µL PCR reaction consisted of 2.5 mM MgCl2, 0.3 mM of each dNTP, 0.5 units of KAPA HiFi HotStart DNA Polymerase (Kapa Biosystems, Wilmington, MA, USA) and 0.3 mM of each forward and reverse primer. The cycling conditions consisted of the initial denaturation at 95°C for 3 minutes followed by 30 cycles of denaturation at 98°C for 20 seconds, annealing at 67°C or 64°C for 15 seconds and extension at 72°C for 1 minute, with the final extension step at 72°C for 1 minute. PCR products of the expected size were excised from the gel following electrophoresis. DNA was recovered using a freeze-squeeze method and sent for sequencing to a commercial provider (Massey Genome Service, Massey University, Palmerston North, NZ). Sequencing was performed using a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) on an ABI PRISM 310 genetic analyser (Applied Biosystems). Obtained sequences were assembled and analysed using Geneious version 10.2.3 (Kearse et al. Citation2012). Nucleotide and amino acid sequence alignments were performed using either Clustal or Muscle algorithms within Geneious. A phylogenetic tree was constructed using the maximum likelihood method in MEGA 11 (Tamura et al. Citation2021) and visualised using Interactive Tree of Life (Letunic and Bork Citation2021).
Amplification and analysis of the 3’ end of the CRCoV genome
The 9,815 nucleotides from the 3’ genomic region of CRCoV comprising 10 open reading frames (ORF) coding for structural and accessory proteins () was targeted for amplification using previously published overlapping primers (Supplementary Table 2). Each PCR reaction included 1 X Platinum SuperFi Green PCR master mix (Invitrogen, Carlsbad, CA, USA), 0.5 µM of each primer and 2 µL of cDNA in a total reaction volume of 10 µL. The cycling conditions comprised the initial denaturation at 98°C for 30 seconds, followed by 30 cycles of denaturation (98°C for 10 seconds), annealing (various temperatures for 10 seconds) and extension (72°C for 2 minutes), followed by final extension (72°C for 5 minutes).
The PCR products were gel-purified and sequenced as described above for the CnPnV G gene. Obtained sequences were assembled and analysed using Geneious version 10.2.3 (Kearse et al. Citation2012). Putative ORF were identified based on the ORF predictor tool within Geneious combined with the position of the corresponding ORF in CRCoV-4182 from the UK (DQ682406); CRCoV-BJ232 from China (KX432213); CRCoV 240/05 from Italy (EU999954); and CRCoV K37 (EU983106), CRCoV K39 (EU983107), and CRCoV K9 (EU983105) from Korea, which was assessed based on multiple sequence alignments. The TRS for each gene of CRCoV was predicted based on locations previously described (Erles et al. Citation2007; Lorusso et al. Citation2009; An et al. Citation2010). The phylogenetic analysis was conducted using the nucleotide sequence of the partial S gene and the entire 3’ genomic sequence of the New Zealand CRCoV and overseas sequences available in GenBank. Nucleotide sequences were aligned using Clustal algorithms within Geneious. Maximum likelihood phylogenetic trees were generated using MEGA 11 (Tamura et al. Citation2021) and visualised using the Interactive Tree of Life program (Letunic and Bork Citation2021).
Putative N-glycosylation sites were predicted by using the NetNGlyc 1.0 server and O-glycosylation sites by using NetOGlyc 4.0 server (Julenius et al. Citation2005).
The partial sequence of CRCoV NZ-046/16 has been deposited in GenBank under accession number MH249786. The New Zealand sequence of CnPnV protein G was deposited in GenBank under accession numbers MK121747, MK121748 and MK121749.
Results
Virus isolation
All virus isolation attempts were unsuccessful. Neither CRCoV nor CnPnV were isolated from RT-qPCR-positive samples.
Sequence analysis of the CnPnV G gene
Out of the 15 samples positive for CnPnV by RT-qPCR, the G protein gene was amplified from only three samples: ITB-007, ITB-048 and ITB-049. These three samples had the highest calculated copy number of the CnPnV M gene per µL of template: 117 (ITB-048), 503 (ITB-049) and 933 (ITB-007), with the remaining M gene-positive samples containing low levels of target RNA (<10 copies per µL of template) based on previously reported data (More et al. Citation2021). The signalment and clinical history for these three dogs are presented in .
Table 1. Signalment of dogs that were the source of samples used for canine pneumovirus glycoprotein G sequencing.
Of the three CnPnV G gene sequences, NZ-048 and NZ-049 were identical to each other, and each was 90.9% identical to NZ-007. The G gene of CnPnV NZ-007 was 1,266 nucleotides long and encoded a predicted protein of 421 amino acids; while the G gene of CnPnV NZ-048 and 049 was 1,245 nucleotides long each and encoded a predicted protein of 414 amino acids, similar to other CnPnV sequences deposited in GenBank, except for CnPnV 126724-10-4DC (GenBank accession KC495967), which was 419 amino acids long. Differences in the predicted lengths in G proteins were due to the position of the stop codon, with the longer G proteins containing an additional 5–7 amino acids at the C-terminal.
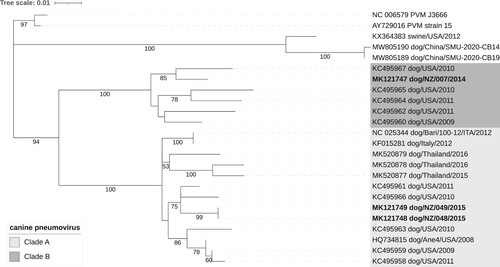
Based on phylogenetic analysis of the G protein, two New Zealand sequences (CnPnV NZ-048 and CnPnV NZ-049) clustered with sequences from the USA, Thailand and Italy in group A and one New Zealand sequence (CnPnV NZ-007) clustered with sequences from the USA in group B ().
Figure 2. Evolutionary relationships of canine pneumoviruses based on the analysis of glycoprotein G amino acid sequences (n = 24). The evolutionary history was inferred by using the maximum likelihood method and the JTT matrix-based model (Jones et al. Citation1992). The tree with the highest log likelihood ( –2,648.85) is shown. The percentage of trees in which the associated taxa clustered together in a bootstrap test (100 replicates) is shown below the branches (only values ≥ 50% are shown). Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (five categories (+G, parameter = 0.6156)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. All positions with 90% site coverage were eliminated (i.e. <10% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option)). There were a total of 414 positions in the final dataset. Evolutionary analyses were conducted in MEGA11 (Tamura et al. Citation2021). Tree was visualised using the Interactive Tree of Life online tool (Letunic and Bork Citation2021).
Sequence analysis of the partial CRCoV sequence
The target sequence was amplified with 9/10 primer pairs designed to amplify the entire 3’ end of the viral genome. The PCR with Sp-9 and Sp-10 primers, designed to generate a 4,200 bp region comprising the entire S gene, was unsuccessful. Hence, this region was attempted to be amplified using four separate PCR reactions to generate shorter (∼ 1 kb) overlapping fragments of the S gene (S1F/R, S2F/R, S3F/R and S4F/R; Supplementary Table 2). Of those, only primers S3F/R and S4F/R produced expected PCR products using the initial amplification conditions. Further optimisation of the PCR reactions with the remaining two primer pairs was not possible as there was no CRCoV-positive sample left. Subsequently, only a partial sequence of the region coding for the S protein was obtained (2,457 out of the predicted 4,092 nucleotides).
The characteristics of the predicted genes (length, position) and their putative protein products (size, predicted structure, presence of N- and O-glycosylation sites) of CRCoV NZ.046/16 () were consistent with those reported for CRCoV previously (Erles et al. Citation2007; An et al. Citation2010), with the exception of the region located between the ORF3 (coding for S protein) and ORF6 (coding for E protein). Sequence variation within this region has been reported previously, with predicted expression of two or three differently sized proteins (). A single base deletion (four instead of five uracil bases at nucleotide 6,647–6,650) in CRCoV NZ046/16 compared to CRCoV such as 240/05, BJ232, K9, K37 or K39 resulted in the absence of a stop codon and addition of eight amino acids to the accessory protein putatively encoded by ORF4a, with the predicted size of 5.9 kDa instead of 4.9 kDa in most other CRCoV. Several CRCoV sequences from the UK encode a putative 8.8 kDa protein in the same region, while CRCoV sequences from Thailand appear to lack the corresponding gene ().
Figure 3. Schematic representation of the genomic organisation of the region between the spike (S) protein gene and the envelope (E) protein gene of canine respiratory coronaviruses (CRCoV) and bovine coronavirus (BCoV). Representative viruses are listed on the right. The GenBank accession numbers for these viruses are: 240/05 (EU999954), K9 (GQ918141), K37 (JX860640), K39 (GQ918143), BJ232 (KX432213), 4182 (DQ682406), D187NS_THA2022 (OQ621724), D206NS_THA2022 (OQ621723), PP014_THA2013 (OQ621712), PP146_THA2015 (OQ621713), BCoV Mebus (BCU00735). Isolates T0715, T1030 and T1207 are included based on description in Erles et al. (Citation2007). Dark shading indicates non-expressed regions of the genome.
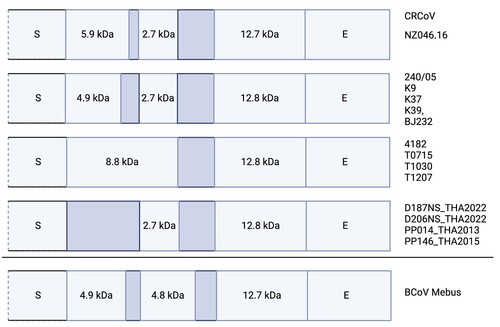
Table 2. Coding capacity and putative protein products expressed from the 3’ end of CRCoV 046/16.
The putative 2.7 kDa accessory protein gene (ORF4b) was 94.9% to 98.7% identical to the corresponding genes of other CRCoVs where it was present, and the gene for 12.5 kDa accessory protein was conserved in the genomes of all CRCoV examined, with 97.3% to 99.4% identity between NZ CRCoV and CRCoV from other countries.
Based on phylogenetic analysis of the partial sequence of the S protein, CRCoV NZ-046/16 clustered with CRCoV sequences from the USA (). When the phylogenetic analysis was repeated using the entire available 3’ sequence, the NZ-046/16 sequence clustered with sequences from the USA (USA 1 and USA 5) and Europe (240/05 from Italy and 4182 from the UK) (data not shown).
Figure 4. Evolutionary relationships of canine respiratory coronaviruses from various countries based on partial nucleotide sequences of the spike gene (n = 37). The New Zealand sequence is in bold. The evolutionary history was inferred by using the maximum likelihood method and Tamura-Nei model (Tamura and Nei Citation1993). The tree with the highest log likelihood (–5,109.77) is shown. The percentage of trees in which the associated taxa clustered together in the bootstrap test (100 replicates) is shown below the branches for values ≥ 50%. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura-Nei model, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (five categories (+G, parameter = 0.1139)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. All positions with <90% site coverage were eliminated (i.e. <10% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option)). There were a total of 2,457 positions in the final dataset. Evolutionary analyses were conducted in MEGA11 (Tamura et al. Citation2021). Tree was visualised using Interactive Tree of Life online tool (Letunic and Bork Citation2021).

Discussion
This report represents the first characterisation of partial genomic sequences of CRCoV and CnPnV from New Zealand. The small number of sequences analysed makes it impossible to make any inferences on the likely sources of these viruses in New Zealand or the timing of their introduction. The duration of quarantine for dogs imported into New Zealand from countries other than Australia ranges from 10 to 60 days (Anonymous Citation2023). Shedding of CRCoV from the oropharynx was detected up to 11 days following experimental infection (Mitchell et al. Citation2013b). There are no data on the duration of shedding of CnPnV, but the risk of transmission of respiratory syncytial virus (a human pneumovirus) between infected and uninfected household members was highest within the first week of infection (Otomaru et al. Citation2021). However, respiratory syncytial virus may establish persistent infections in immunocompromised people, with long-term shedding of up to 334 days reported in one study (Lehners et al. Citation2016). Whether or not persistent infections with CnPnV in immunocompromised dogs can also be established needs to be elucidated. Altogether, the quarantine requirements are likely to have substantially reduced, but not fully removed, the risk of introduction of CRCoV and CnPnV to the country through infected dogs. Another possibility is the introduction of those viruses on fomites. Both CRCoV and CnPnV are enveloped viruses and therefore unlikely to survive well outside of their hosts. However, the presence of viable virus on inanimate surfaces for as short as a couple of days would, in theory, allow for the inadvertent introduction of such a virus into New Zealand on fomites such as contaminated footwear (Geng and Wang Citation2023).
Canine pneumovirus sequence analysis
Glycoprotein G was amplified from only 3/15 (20%) samples that previously tested positive for CnPnV on qRT-PCR (More et al. Citation2021). The qRT-PCR used in the study targeted the M protein gene, which is relatively conserved compared to the G protein gene, which is more variable (Slovic et al. Citation2017; Kumagai et al. Citation2021; Yu et al. Citation2021). This may be one reason for the discrepancies between results of the two tests. The three protein G-positive samples had the highest calculated copy number of the CnPnV M gene per µL of template, and hence low levels of target RNA may be another reason for unsuccessful amplification of a G protein sequence from the remaining 12/15 samples used in the current study (More et al. Citation2021).
Pneumovirus G protein is responsible for viral attachment to the host cell receptor and is a major target for neutralising antibodies (Levine et al. Citation1987; Ling and Pringle Citation1989). Two distinct groups (group A and B) of CnPnV have been described based on sequence variation in the G protein gene of several CnPnV sequences from the USA (Glineur et al. Citation2013). Viruses from both groups were detected in the current study, suggesting that the variability of CnPnV circulating in New Zealand is similar to that described in the USA.
It has been suggested that the close similarity of CnPnV with the rodent viruses (MPV-15 and MPV-J3666 in ) may indicate cross-species transfer of a rodent pneumovirus to dogs (Decaro et al. Citation2014b). Interestingly, pneumoviruses recently detected from dogs in China were more similar to porcine pneumovirus than to murine pneumonia virus (MPV), raising a possibility that cross-species transmission may have also occurred from swine to dogs (Song et al. Citation2021). Alternatively, CnPnV and pneumoviruses of rodents, pigs and dogs may have co-evolved independently from a common ancestor. The limited number of CnPnV sequences available in GenBank precludes distinguishing between those possibilities.
Canine respiratory coronavirus
The sequence and genomic organisation of CRCoV is similar to that of BCoV (Erles et al. Citation2003, Citation2007). The most variable region is located between the spike (S) and envelope (E) genes. This region includes two or three small accessory proteins named based on their predicted molecular sizes. The UK CRCoV were predicted to encode two (8.8 or 4.9 kDa and 12.9 kDa) proteins in this region, while viruses from Italy and Korea were predicted to encode three proteins (4.9, 2.7 and 12.8 kDa) (Erles et al. Citation2007; Lorusso et al. Citation2009; An et al. Citation2010). Canine respiratory coronavirus sequences from Thailand were predicted to encode only 2.7 and 12.8 kDa accessory proteins (accessions OQ621707 to OQ621727, unpublished). The New Zealand CRCoV was predicted to encode three proteins of 5.9, 2.7 and 12.7 kDa in the same region, which is a combination that has not been previously described. This further highlights the variable nature of this genomic fragment. The expression and sizes of accessory proteins of different CRCoV need to be confirmed experimentally, as so far these are simply predictions based on the analysis of genomic sequences. The role of these putative proteins in the CRCoV life cycle and pathogenesis also remains to be elucidated.
Based on the phylogenetic analysis, CRCoV NZ-046/16 was more similar to CRCoV sequences from the USA than to sequences from Asia. However, it is difficult to draw any conclusions about evolutionary relationships between New Zealand CRCoV and viruses from other countries based on only one New Zealand sequence and a limited number of sequences from other countries. Such relationships will become clearer as more sequence data become available.
Virus isolation
Our attempts to isolate CRCoV and CnPnV from the oropharyngeal swab samples that were positive for these viruses by RT-qPCR were unsuccessful. This may reflect the absence of infectious viruses in the samples, with inactivation occurring during transportation or storage, or suboptimal culture conditions, particularly for CnPnV. Limited literature is available on in vitro growth requirements of CnPnV and so far, the virus has been successfully isolated only in canine fibroblast A-72 cells by some (Renshaw et al. Citation2010), but not other (Decaro et al. Citation2014b) researchers. Canine pneumovirus shares sequence similarity with MPV (Renshaw et al. Citation2011) and has been shown to replicate efficiently in the lung tissue of mice following experimental infection (Percopo et al. Citation2011; Glineur et al. Citation2013). Both Vero and RAW 264.7 cell lines were successfully used for the culture of MPV (Dyer et al. Citation2007), and hence we elected to use these cell lines to attempt CnPnV isolation. However, there are no data on susceptibility of these cell lines to CnPnV infection and hence, lack of successful isolation of the virus in the current study may reflect the choice of the cell lines used. The A-72 cells would have been preferred but were not available at the time of the study.
The HRT-18 cell line and its clone HRT-18G were shown to be supportive for the growth of CRCoV in previous studies, while several other cell lines including A-72, BHK-21, CHO, DH82, MDBK and MDCK were not (Erles et al. Citation2007; An et al. Citation2010). As CRCoV does not produce visible cytopathic effect in cell culture (Erles et al. Citation2007), RT-qPCR and HA were used to detect growth of the virus in the current study. Similar to our results, other researchers also failed to propagate CRCoV on HRT-18 cells from the samples that were positive for the virus using molecular methods (Yachi and Mochizuki Citation2006; Decaro et al. Citation2007; Lorusso et al. Citation2009; Lu et al. Citation2017), highlighting the difficulties in growing CRCoV in vitro.
Supplemental Material
Download PDF (93.1 KB)Acknowledgements
This work was supported by funds from the New Zealand Greyhound Association.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- An D-J, Jeong W, Yoon SH, Jeoung H-Y, Kim H-J, Park B-K. Genetic analysis of canine group 2 coronavirus in Korean dogs. Veterinary Microbiology 141, 46–52, 2010. https://doi.org/10.1016/j.vetmic.2009.09.002
- *Anonymous. Stay on Top of Canine Cough. https://vetcouncil.org.nz/Web/Web/News/Articles/2022/Stay_on_top_of_canine_cough.aspx (accessed 7 March 2024). Veterinary Council of New Zealand, Wellington, NZ, 2022
- *Anonymous. Bringing Cats and Dogs to New Zealand. https://www.mpi.govt.nz/bring-send-to-nz/pets-travelling-to-nz/bringing-cats-and-dogs-to-nz/ (accessed 27 February 2024). Ministry for Primary Industries, Wellington, NZ, 2023
- Chalker VJ, Owen WM, Paterson C, Barker E, Brooks H, Rycroft AN, Brownlie J. Mycoplasmas associated with canine infectious respiratory disease. Microbiology 150, 3491–7, 2004. https://doi.org/10.1099/mic.0.26848-0
- Chalker VJ, Brooks HW, Brownlie J. The association of Streptococcus equi subsp. zooepidemicus with canine infectious respiratory disease. Veterinary Microbiology 95, 149–56, 2003. https://doi.org/10.1016/S0378-1135(03)00155-X
- Day MJ, Carey S, Clercx C, Kohn B, MarsilIo F, Thiry E, Freyburger L, Schulz B, Walker DJ. Aetiology of canine infectious respiratory disease complex and prevalence of its pathogens in Europe. Journal of Comparative Pathology 176, 86–108, 2020. https://doi.org/10.1016/j.jcpa.2020.02.005
- Decaro N, Desario C, Elia G, Mari V, Lucente MS, Cordioli P, Colaianni ML, Martella V, Buonavoglia C. Serological and molecular evidence that canine respiratory coronavirus is circulating in Italy. Veterinary Microbiology 121, 225–30, 2007. https://doi.org/10.1016/j.vetmic.2006.12.001
- Decaro N, Pinto P, Mari V, Larocca V, Colaianni M, Losurdo M, Occhiogrosso L, Martella V, Buonavoglia C. Canine infectious respiratory disease associated to canine pneumovirus infection. Veterinaria (Cremona) 28, 7–12, 2014a
- Decaro N, Pinto P, Mari V, Elia G, Larocca V, Camero M, Terio V, Losurdo M, Martella V, Buonavoglia C. Full-genome analysis of a canine pneumovirus causing acute respiratory disease in dogs, Italy. PLoS One 9, e85220, 2014b. https://doi.org/10.1371/journal.pone.0085220
- Dyer KD, Schellens IM, Bonville CA, Martin BV, Domachowske JB, Rosenberg HF. Efficient replication of pneumonia virus of mice (PVM) in a mouse macrophage cell line. Virology Journal 4, 48, 2007. https://doi.org/10.1186/1743-422X-4-48
- Erles K, Toomey C, Brooks HW, Brownlie J. Detection of a group 2 coronavirus in dogs with canine infectious respiratory disease. Virology 310, 216–23, 2003. https://doi.org/10.1016/S0042-6822(03)00160-0
- Erles K, Shiu K-B, Brownlie J. Isolation and sequence analysis of canine respiratory coronavirus. Virus Research 124, 78–87, 2007. https://doi.org/10.1016/j.virusres.2006.10.004
- *Freshney RI. Culture of specific cell types. In: Freshney RI (ed). Culture of Animal Cells: a Manual of Basic Technique. 5th Edtn. John Wiley & Sons Inc. Hoboken, NJ, USA, 2005. https://doi.org/10.1002/0471747599.cac023
- Geng Y, Wang Y. Stability and transmissibility of SARS-CoV-2 in the environment. Journal of Medical Virology 95, e28103, 2023. https://doi.org/10.1002/jmv.28103
- Glineur SF, Renshaw RW, Percopo CM, Dyer KD, Dubovi EJ, Domachowske JB, Rosenberg HF. Novel pneumoviruses (PnVs): evolution and inflammatory pathology. Virology 443, 257–64, 2013. https://doi.org/10.1016/j.virol.2013.05.011
- Hofmann MA, Chang RY, Ku S, Brian DA. Leader-mRNA junction sequences are unique for each subgenomic mRNA species in the bovine coronavirus and remain so throughout persistent infection. Virology 196, 163–71, 1993. https://doi.org/10.1006/viro.1993.1464
- Hong S, Lee H-Y, Kim O. Detection of canine respiratory coronavirus from dogs with respiratory disease. Journal of Biomedical Research 16, 19–22, 2015. https://doi.org/10.12729/jbr.2015.16.1.019
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Bioinformatics (Oxford, England) 8, 275–82, 1992. https://doi.org/10.1093/bioinformatics/8.3.275
- Julenius K, Mølgaard A, Gupta R, Brunak S. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 15, 153–64, 2005. https://doi.org/10.1093/glycob/cwh151
- Kapoor A, Simmonds P, Gerold G, Qaisar N, Jain K, Henriquez JA, Firth C, Hirschberg DL, Rice CM, Shields S, et al. Characterization of a canine homolog of hepatitis C virus. Proceedings of the National Academy of Sciences of the United States of America 108, 11608–13, 2011. https://doi.org/10.1073/pnas.1101794108
- Kapoor A, Mehta N, Dubovi EJ, Simmonds P, Govindasamy L, Medina JL, Street C, Shields S, Lipkin WI. Characterization of novel canine bocaviruses and their association with respiratory disease. Journal of General Virology 93, 341–6, 2012. https://doi.org/10.1099/vir.0.036624-0
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics (Oxford, England) 28, 1647–9, 2012. https://doi.org/10.1093/bioinformatics/bts199
- Kumagai A, Kawauchi K, Andoh K, Hatama S. Sequence and unique phylogeny of G genes of bovine respiratory syncytial viruses circulating in Japan. Journal of Veterinary Diagnostic Investigation 33, 162–6, 2021. https://doi.org/10.1177/1040638720975364
- Lehners N, Tabatabai J, Prifert C, Wedde M, Puthenparambil J, Weissbrich B, Biere B, Schweiger B, Egerer G, Schnitzler P. Long-term shedding of influenza virus, parainfluenza virus, respiratory syncytial virus and nosocomial epidemiology in patients with hematological disorders. PLoS One 11, e0148258, 2016. https://doi.org/10.1371/journal.pone.0148258
- Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Research 49, W293–6, 2021. https://doi.org/10.1093/nar/gkab301
- Levine S, Klaiber-Franco R, Paradiso P. Demonstration that glycoprotein G is the attachment protein of respiratory syncytial virus. Journal of General Virology 68, 2521–4, 1987. https://doi.org/10.1099/0022-1317-68-9-2521
- Ling R, Pringle C. Polypeptides of pneumonia virus of mice. I. Immunological cross-reactions and post-translational modifications. Journal of General Virology 70, 1427–40, 1989. https://doi.org/10.1099/0022-1317-70-6-1427
- Lorusso A, Desario C, Mari V, Campolo M, Lorusso E, Elia G, Martella V, Buonavoglia C, Decaro N. Molecular characterization of a canine respiratory coronavirus strain detected in Italy. Virus Research 141, 96–100, 2009. https://doi.org/10.1016/j.virusres.2008.12.011
- Lu S, Wang Y, Chen Y, Wu B, Qin K, Zhao J, Lou Y, Tan W. Discovery of a novel canine respiratory coronavirus support genetic recombination among betacoronavirus 1. Virus Research 237, 7–13, 2017. https://doi.org/10.1016/j.virusres.2017.05.006
- Maboni G, Seguel M, Lorton A, Berghaus R, Sanchez S. Canine infectious respiratory disease: new insights into the etiology and epidemiology of associated pathogens. PLoS One 14, e0215817, 2019. https://doi.org/10.1371/journal.pone.0215817
- McCandlish IA, Thompson H, Cornwell HJ, Wright NG. A study of dogs with kennel cough. Veterinary Record 102, 293–301, 1978. https://doi.org/10.1136/vr.102.14.293
- Mitchell JA, Cardwell JM, Renshaw RW, Dubovi EJ, Brownlie J. Detection of canine pneumovirus in dogs with canine infectious respiratory disease. Journal of Clinical Microbiology 51, 4112–9, 2013a. https://doi.org/10.1128/JCM.02312-13
- Mitchell JA, Brooks HW, Szladovits B, Erles K, Gibbons R, Shields S, Brownlie J. Tropism and pathological findings associated with canine respiratory coronavirus (CRCoV). Veterinary Microbiology 162, 582–94, 2013b. https://doi.org/10.1016/j.vetmic.2012.11.025
- Mitchell JA, Cardwell JM, Leach H, Walker CA, Poder SL, Decaro N, Rusvai M, Egberink H, Rottier P, Fernandez M, et al. European surveillance of emerging pathogens associated with canine infectious respiratory disease. Veterinary Microbiology 212, 31–8, 2017. https://doi.org/10.1016/j.vetmic.2017.10.019
- More GD, Dunowska M, Acke E, Cave NJ. A serological survey of canine respiratory coronavirus in New Zealand. New Zealand Veterinary Journal 68, 54–9, 2020. https://doi.org/10.1080/00480169.2019.1667282
- More GD, Biggs PJ, Cave NJ, Acke E, Dunowska M. A molecular survey of canine respiratory viruses in New Zealand. New Zealand Veterinary Journal 69, 224–33, 2021. https://doi.org/10.1080/00480169.2021.1915211
- Otomaru H, Sornillo JBT, Kamigaki T, Bado SLP, Okamoto M, Saito-Obata M, Inobaya MT, Segubre-Mercado E, Alday PP, Saito M, et al. Risk of transmission and viral shedding from the time of infection for respiratory syncytial virus in households. American Journal of Epidemiology 190, 2536–43, 2021. https://doi.org/10.1093/aje/kwab181
- Pasternak AO, Spaan WJM, Snijder EJ. Nidovirus transcription: how to make sense … ? Journal of General Virology 87, 1403–21, 2006. https://doi.org/10.1099/vir.0.81611-0
- Pecoraro HL, Bennett S, Huyvaert KP, Spindel ME, Landolt GA. Epidemiology and ecology of H3N8 canine influenza viruses in US shelter dogs. Journal of Veterinary Internal Medicine 28, 311–8, 2014. https://doi.org/10.1111/jvim.12301
- Percopo CM, Dubovi EJ, Renshaw RW, Dyer KD, Domachowske JB, Rosenberg HF. Canine pneumovirus replicates in mouse lung tissue and elicits inflammatory pathology. Virology 416, 26–31, 2011. https://doi.org/10.1016/j.virol.2011.04.010
- Piewbang C, Techangamsuwan S. Phylogenetic evidence of a novel lineage of canine pneumovirus and a naturally recombinant strain isolated from dogs with respiratory illness in Thailand. BMC Veterinary Research 15, 300, 2019. https://doi.org/10.1186/s12917-019-2035-1
- Renshaw RW, Zylich NC, Laverack MA, Glaser AL, Dubovi EJ. Pneumovirus in dogs with acute respiratory disease. Emerging Infectious Diseases 16, 993–5, 2010. https://doi.org/10.3201/eid1606.091778
- Renshaw R, Laverack M, Zylich N, Glaser A, Dubovi E. Genomic analysis of a pneumovirus isolated from dogs with acute respiratory disease. Veterinary Microbiology 150, 88–95, 2011. https://doi.org/10.1016/j.vetmic.2011.01.013
- Rima B, Collins P, Easton A, Fouchier R, Kurath G, Lamb RA, Lee B, Maisner A, Rota P, Wang L, et al. ICTV virus taxonomy profile: Pneumoviridae. Journal of General Virology 98, 2912–3, 2017. https://doi.org/10.1099/jgv.0.000959
- Schulz BS, Kurz S, Weber K, Balzer HJ, Hartmann K. Detection of respiratory viruses and Bordetella bronchiseptica in dogs with acute respiratory tract infections. Veterinary Journal 201, 365–9, 2014. https://doi.org/10.1016/j.tvjl.2014.04.019
- Slovic A, Kosutic-Gulija T, Santak M, Ivancic-Jelecki J, Jagusic M, Ljubin-Sternak S, Mlinarić-Galinović G, Vilibić-Čavlek T, Tabain I, Forcic D. Genetic variability and sequence relatedness of matrix protein in viruses of the families Paramyxoviridae and Pneumoviridae. Intervirology 60, 181–9, 2017. https://doi.org/10.1159/000487049
- Soma T, Ishii H, Miyata K, Hara M. Prevalence of antibodies to canine respiratory coronavirus in some dog populations in Japan. Veterinary Record 163, 368–9, 2008. https://doi.org/10.1136/vr.163.12.368
- Song X, Li Y, Huang J, Cao H, Zhou Q, Sha X, Zhang B. An emerging orthopneumovirus detected from dogs with canine infectious respiratory disease in China. Transboundary and Emerging Diseases 68, 3217–21, 2021. https://doi.org/10.1111/tbed.14291
- Sowman HR, Cave NJ, Dunowska M. A survey of canine respiratory pathogens in New Zealand dogs. New Zealand Veterinary Journal 66, 236–42, 2018. https://doi.org/10.1080/00480169.2018.1490214
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution 10, 512–26, 1993. https://doi.org/10.1093/oxfordjournals.molbev.a040023
- Tamura K, Stecher G, Kumar S. MEGA 11: Molecular Evolutionary Genetics Analysis version 11. Molecular Biology and Evolution 38, 3022–7, 2021. https://doi.org/10.1093/molbev/msab120
- Yachi A, Mochizuki M. Survey of dogs in Japan for group 2 canine coronavirus infection. Journal of Clinical Microbiology 44, 2615–8, 2006. https://doi.org/10.1128/JCM.02397-05
- Yu JM, Fu YH, Peng XL, Zheng YP, He JS. Genetic diversity and molecular evolution of human respiratory syncytial virus A and B. Scientific Reports 11, 12941, 2021. https://doi.org/10.1038/s41598-021-92435-1
- *Non-peer-reviewed