
Abstract
Background
Hyperthermia can play a synergistic role with chemotherapy in combination therapy. Although the association between caspase activation, apoptosis, and pyroptosis have been published for both cisplatin (CDDP) and hyperthermia therapies independently, the interactions between these molecular pathways in combination therapy are unknown. The present study aimed to investigate the possible interactions between caspase 8 activation, apoptosis, and pyroptosis in combination therapy.
Methods
Cells were treated with CDDP (15 µg/ml), followed by hyperthermia at optimized temperature (42.5 °C) in water-bath. After combination therapy, cell viability was analyzed by CCK-8, and cell death was analyzed by Annexin-V-FITC/PI and caspases activation. Immuno-staining and co-immuno-precipitation were used to examine the interaction between p62 and caspase-8. Pyroptosis was investigated by western blotting and transmission electron microscopy. E3 ligase Cullin 3 was knockdown by siRNA. In addition, caspase-8 activation was modulated by CRISPR-Cas9 gene-editing or pharmacological inhibition.
Results
Combination therapy promoted K63-linked polyubiquitination of caspase-8 and cellular accumulation of caspase-8. In turn, polyubiquitinated caspase-8 interacted with p62 and led to the activation of caspase-3. Knockdown of the E3 ligase Cullin 3 by siRNA reduced caspase-8 polyubiquitination and activation. In addition, combination therapy induced release of the pore-forming N-terminus from gasdermins and promoted pyroptosis along with caspase-8 accumulation and activation. Knockdown of caspase-8 by CRISPR/Cas9 based gene editing reduced the sensitivity of tumor cells to apoptosis and pyroptosis.
Conclusions
Our study presented a novel mechanism in which hyperthermia synergized with chemotherapy in promoting apoptosis and pyroptosis in a caspase-8 dependent manner.
1. Introduction
Hyperthermia is a form of physical therapy, which is usually combined with radiotherapy, chemotherapy or other programs to change the physiological state of tumors and improve the efficacy of radiotherapy and chemotherapy [Citation1]. The term hyperthermia refers to the utilization of various physical factors (such as radio frequency, microwave, ultrasound, and laser) to elevate the temperature of tumor tissue and/or the entire body. This approach employs high temperature for tumor eradication along with its associated secondary effects in cancer treatment. Hyperthermia, combined with radiotherapy, has been successfully applied in the treatment of tumors, such as breast cancer [Citation2] and cervical cancer [Citation3]. In addition, hyperthermia and chemotherapy can synergistically enhance the sensitivity of tumor to chemotherapy and improve the postoperative long-term survival rate of tumor patients [Citation4–7].
Apoptosis is a well characterized mechanism of programmed cell death (PCD) that is defined by the activation of a family of cysteine proteases known as caspases [Citation8]. Caspase activation occurs through extrinsic and intrinsic signaling pathways [Citation9–10]. In addition, apoptosis plays a key role in maintaining normal tissue homeostasis and preventing disease [Citation11–14].
Protein ubiquitination is sequentially mediated by three enzymes [Citation15–16]. E3 ubiquitin ligases are the most important because they determine the specificity and diversity of substrates [Citation17]. Cullin3 (CUL3) is a member of the cullin ubiquitin ligase family, and the CUL3 E3 ligase complex is involved in ubiquitination of many proteins [Citation18]. Ubiquitin is a small protein containing seven lysine sites that can be ubiquitinated. Among these ubiquitin linkages, the K48 and K63 linkages are the most abundant, accounting for approximately 80% of the total linkages in mammalian cells [Citation19–24]. It has been reported that CUL3 interacts with death-inducing signaling complex, which promotes the ubiquitination of caspase-8 with K63-linked polyubiquitination [Citation15]. Ubiquitinated casepase-8 can interact with p62 through its ubiquitin associated domain [Citation15].
Pyroptosis was defined as gasdermin-mediated PCD [Citation25]. Gasdermins are cleaved when cells are stimulated under certain conditions, releasing its N-terminal pore-forming domain. The released N-terminal domain binds to membrane lipids and perforates the cell membrane, resulting in loss of cell osmotic pressure and cell membrane rupture [Citation26–28]. It has been reported that apoptosis and pyroptosis are intricately interconnected [Citation29–36].
Multiple mechanisms of action for combination therapy have been identified. For example, the association between caspase activation, apoptosis, and pyroptosis with both CDDP and hyperthermia therapies have been published independently, while the links between these cell death pathways during combination therapy remain unknown. In this study, we found that hyperthermia in combination with CDDP chemotherapy promoted caspase-8 accumulation and activation, which in turn initiated both apoptosis and pyroptosis, independent of the intrinsic apoptosis pathway. Our results shed new insight into the synergizing effect of hyperthermia and chemotherapy combination.
2. Materials and methods
2.1. Cell culture
The HepG2 (Procell), RAW264.7 (ATCC) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Basal Media, L110KJ). Culture media were supplemented with 10% fetal bovine serum (FBS, ExCell Bio, FSP500), penicillin (100 U/ml) and streptomycin (100 µg/ml) (Sangon Biotech, E607011-0500). All the cells were cultured in a 37 °C incubator with a humidified 5% CO2 atmosphere.
2.2. Antibodies and reagents
The following antibodies and reagents used in the study were: CUL3 (rabbit; Cell Signaling Technology; 10450s), K48-linked ubiquitin (rabbit; Cell Signaling Technology; 8081s), Caspase-3 (rabbit; Cell Signaling Technology; 9662s), Gasdermin E (rabbit; Cell Signaling Technology; 19453s), Caspase-8 (rabbit; R&D Systems; AF1650), K63-linked ubiquitin (rabbit; Invitrogen; MA5-32573), SQSTM1/p62 (rabbit; Santa cruz, NBP1-48320), SQSTM1 (mouse; Abnova; H00008878-M01), Goat anti-mouse IgG(H + L)-HRP (Apexbio; K1221), Goat anti-rabbit IgG(H + L)-HRP (Apexbio; K1223), GAPDH (mouse; Proteintech; HRP-60004), Actin antibody (mouse; Proteintech; HRP-60008), Tubulin antibody (mouse; ORIGENE; TA503129), GSDMDC1(rabbit; Novus; NBP2-33422), LC3 antibody (rabbit; MBL; M186-3), MG132 (A2585), Z-VAD-FMK (A1902) were purchased from APExBio; CDDP(S1166), Rapamycin (S1039), BafA1(S8865) were purchased from Selleck.
2.3. Cell hyperthermia and chemotherapy
Cells (HepG2, RAW264.7) were treated with CDDP (15 µg/mL) overnight, then heated in a water bath at 42.5 °C for 1h in the presence of CDDP. The cells were harvested or fixed 4 h later for follow-up examination. CDDP chemotherapy or hyperthermia was performed as controls. CDDP concentration and hyperthermia temperature and duration were pre-optimized (Fresh purified water was replenished for each heat treatment in the water bath during this study).
2.4. Western blotting
Cells was collected and lysed in lysis buffer (1% triton X-100, 20 mM Hepes, 150 mM NaCl, 12.5 mM β-glycerol phosphate disodium salt, 1.5 mM MgCl2, 2 mM EGTA). The supernatant was collected and protein concentration was assayed by Bradford method. Twenty-five µg protein from each sample was separated by SDS-PAGE and transferred onto PVDF membranes (Millipore, IPVH00010). Membranes were visualized by ECL kit (BOSTER, AR1174).
2.5. Co-IP (ubiquitination)
Adherent cells were grown in 100 mm plates, lifted off the dish by scrapping. The cells were centrifuged and pellet was resuspended in 300 µL CO-IP buffer (pH7.4 lysis buffer, NaF Aladdin S111591 10 mM, Na3VO4 Aldrich 450243-50 G 1 mM, PMSF Aladdin P105539 1 mM, PI tablet Roche 05892970001 20 µM, NEM Aladdin E100552 10 mM) on ice for 30 min. To the cleared cell lysates, 1.0 µg mouse-anti-Caspase-8 or mouse-anti-p62 was added. Immunocomplexes were allowed to form by incubation at 4 °C for 2h. Thirty µL pre-cleared protein A/G magnetic Beads (Beyotime, P2108) were added to each immunocomplexes and incubated at 4 °C overnight. The protein A/G magnetic beads were collected using manual magnetic separator and washed. The captured immunocomplexes were boiled at 95 °C for 10 min and analyzed by Western blotting with 8% SDS-PAGE.
2.6. Immunofluorescence
Cells were grown on coverslips. After treatment, cells were fixed for 30 min in 4% paraformaldehyde at room temperature (RT), then permeabilized with digitonin (100 µg/ml) (Sigma, D141) for 20 min, and blocked with 1.5% BSA for 30 min at RT. Cells were incubated with diluted primary antibodies (1:1000 in blocking buffer) at 4 °C overnight. Afterward, cells were incubated with secondary antibodies (1:400 in PBS) for 3h at RT. Cell nucleus was counterstained with 1 µg/ml DAPI (Thermo, 62248) for 15 min at RT. The coverslips were mounted on glass slides and sealed with anti-fluorescence quenching reagent. The cells were examined using a Leica (TCS SP8) confocal microscopy with a 63× oil objective.
2.7. Cell apoptosis analyses
Cells were stained with the Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences, 556547) according to manufacturer’s instructions and the cells were analyzed with flow cytometry (BD FACSCalibur, USA) immediately.
2.8. Colony-formation assay
After treatment with various therapies (see 2.3. Cell hyperthermia and chemotherapy), the cells were seeded in 6-well plates (500 cells/well) and allowed to grow for 2 weeks. Then the cells were fixed with methanol for 30 min, add stained with 0.5% crystal violet at RT. After washing with PBS, the plates were photographed to observe and count the colonies.
2.9. Cell viability assay
Cell viability was measured according to the instructions of CCK-8 Kit (APExBio, K1018).
2.10. LDH release assay
LDH released into the cell culture supernatant was measure using LDH cytotoxicity assay Kit (Beyotime, C0016).
2.11. CRISPR/Cas9 knockout caspase-8 in HepG2 cells
Generation of knockout cell lines by the CRISPR/Cas9 technology was performed as described in another of manuscript [Citation37]. Briefly, dual gRNAs were cloned into pGL3 plasmid downstream of U6 promoter. The plasmids pGL3-gRNA and pST1374-Cas9 were co-transfected in a ratio of 1:3 into cells by Lipofactamine 3000. Cells were used for further studies 36 h after transfection.
2.12. RT-PCR
Total RNA was extracted from cultured cells using RNA-easy Isolation Reagent (Vazyme, R701-02). 1 µg of total RNA from each sample was subjected to reverse transcription using the HiScript® II Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme, R223) to synthesize cDNA. Real-time PCR was performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme, Q711) on a Fluorescent Quantitative PCR Detection system (BIOER, Quant Gene 9600) following the manufacturer’s instructions. All genes were assayed in triplicate to ensure reproducibility. The primers used in this study were as follows:
2.13. Caspases activity assay
Caspase-8 and caspase-3 activity were assayed with the Caspase-8 Fluorometric Assay Kit (APExBio, K2012) and Caspase-3 Fluorometric Assay Kit (APExBio, K2007) in cell lysates.
2.14. Scanning electron microscope (SEM)
Cells were seeded at 8.5 × 104 cells/mL in 12-well plates. After various treatments to the predetermined time, the cells were rinsed gently with PBS, fixed with 2-3 ml electron microscope fixative buffer (Servicebio, G1102) at RT in the dark. The morphological changes of cells undergoing pyroptosis were observed and imaged under SEM (SU8100) at 3.0 kV.
2.15. siRNAs
The siRNAs used in this study were as follows:
All siRNAs were synthesized by IBSBIO (Shanghai). SiHuman-CUL3-443(B) was used in the experiments if there is no specification [Citation15].
2.16. Statistical analyses
Densitometry of immunoblots was quantified with the imageJ software. The number of punctate foci in immunofluorescence images was counted manually using the photoshop 2020 software. Data are expressed as mean ± standard deviation (SD). Statistical analyses were performed using ANOVAs. Two-tailed Student’s t-test was employed to assess differences between two groups. p ≤ 0.05 was considered statistically significant.
3. Results
3.1. Hyperthermia synergized with chemotherapy in promoting caspase-8 accumulation in cancer cells
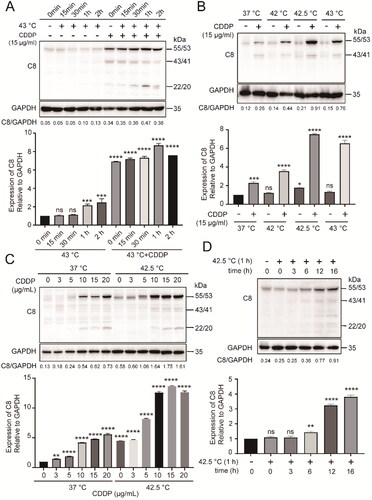
We found CDDP alone induced caspase-8 accumulation and activation, while combination therapy further enhanced this effect in cancer cells without extrinsic apoptosis stimulation (). Full-length caspase-8 is 55/53 kDa (the procaspase). The 43/41 kDa (the cleaved caspases) caspase-8 is the large subunit of processed caspase-8 and the 22/20 kDa caspase-8 is the small subunit. The optimized conditions for inducing caspase-8 accumulation in cancer cells was identified as heating at 42.5 °C for 1h () in the presence of 15 µg/mL CDDP (). Hyperthermia treatment alone induced caspase-8 accumulation but in a delayed manner (). These results indicate that hyperthermia synergized with chemotherapy in promoting caspase-8 accumulation without extrinsic apoptosis stimulation in cancer cells.
Figure 1. Hyperthermia synergized with chemotherapy in promoting caspase-8 accumulation in cancer cells. (A) HepG2 cells were heated at 43 °C in water-bath for indicated times in the absence or presence of CDDP. (B) HepG2 cells were heated at indicated temperatures for 1 h. (C) HepG2 cells were treated with CDDP at different concentrations in combination with hyperthermia at 42.5 °C. (D) HepG2 cells were heated at 42.5 °C for 1 h, then cultured for various times. The densitometry ratio of Western blots of caspase-8 to GAPDH was quantified (bottom panels). Data are expressed as mean ± standard deviation (SD), three independent experiments. * p < 0.05, **p < 0.01,***p < 0.001, ****p < 0.0001 by t-test. Ns: not significant, compared to control.
3.2. Combination therapy promoted apoptosis of cancer cells in correlation with caspase-8 activation
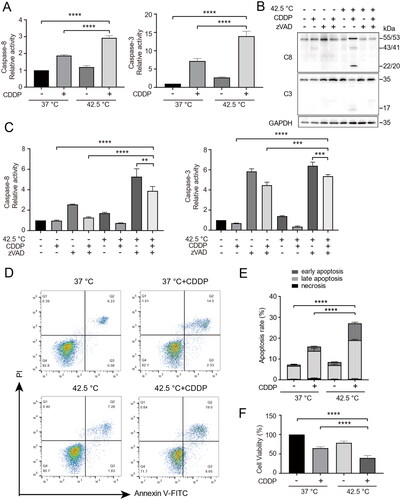
Next, caspase-8 and caspase-3 enzymatic activity in treated cancer cells were determined by substrate cleavage assay (Z-DEVD-AFC and Z-IETD-AFC respectively). In addition, caspase-8 and caspase-3 activation were determined by Western blotting. As shown in and B, combination therapy significantly increased the enzymatic activity and activation of caspase-8. Meanwhile, the enzymatic activity and activation of caspase-3 correlated with that of caspase-8 (). In the presence of the pan-caspase inhibitor zVAD-fmk, the enzymatic activities of both caspase-8 and caspase-3 (), as well as the activation of both casepase-8 and caspase-3 () were reduced. We found that combination therapy significantly increased early and late apoptosis (), as detected by Annexin V-FITC/PI. Consistently, combination therapy significantly reduced cell viability (). Together, these results indicate combination therapy promoted apoptosis of cancer cells in correlation with caspase-8 activation.
Figure 2. Combination therapy promoted apoptosis of cancer cells in correlation with caspase-8 activation. (A) HepG2 cells were heated at 42.5 °C for 1h in the presence of CDDP (15 µg/mL). Cells were harvested 16 h later and cell lysates were used for assay of enzymatic activities of caspase-8 and caspase-3. (B and C) Analysis of caspase-8 and caspase-3 activation (B) and enzymatic activities (C) in the presence of pan-caspase inhibitor. (D) Cells were treated as in (A), stained with Annexin V-FITC and PI, and analyzed by flow cytometry. (E) Quantification of apoptosis in (D). (F) Cells were treated as in (A), cell viability was measured by CCK-8 assay. Data are expressed as mean ± standard deviation (SD), three independent experiments. **p < 0.01, ***p < 0.001, ****p < 0.0001 by t-test.
3.3. Knockout of caspase-8 decreased the sensitivity of cancer cells to combination therapy
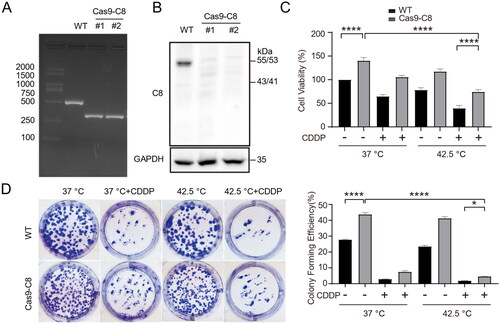
CRISPR/Cas9 mediated gene-editing was employed to knockout caspase-8 expression in HepG2 cells. Cas9 and gRNA encoding plasmid transfected cells were screened and single-cell cloned. PCR analysis revealed that precise genomic deletion of a fragment of caspase-8 gene occurred in cloned cells (). In addition, western blotting analysis of caspase-8 protein expression confirmed the successful gene-editing of caspase-8 (). Knockout of caspase-8 elevated cancer cell proliferation (). Moreover, knockout of caspase-8 rendered cancer cells resistance to combination therapy, as shown by CCK-8 assay () and clone formation assay (). These results indicate that caspase-8 is at least partially implicated in apoptosis activation in combination therapy treated cancer cells.
Figure 3. Knockout of caspase-8 decreased the sensitivity of cancer cells to combination therapy. (A) Caspase-8 gene was edited by CRISPR/Cas9 in HepG2 cells. Targeted PCR of genomic DNA of two cloned caspase-8 knockout cells (Cas9-C8) showed deletion of a segment of caspase-8 gene. (B) Western blotting analysis of caspase-8 protein expression in the two cloned Cas9-C8 cells in (A). (C) WT and Cas9-C8 cells were treated with CDDP (15 µg/mL) and heated at 42.5 °C for 1 h. Cell viability was measured by CCK-8 assay. (D) Cells were treated as in (C) and clone formation assay was performed. Cells were fixed and stained with crystal violet and clone formation rate was calculated (right panel). Data are expressed as mean ± standard deviation (SD), three independent experiments. * p < 0.05, ****p < 0.0001 by t-test.
3.4. Combination therapy promoted K63-linked ubiquitination of caspase-8 and enhanced the interaction between caspase-8 and p62
Caspase-8 has been implicated in apoptosis via interacting with p62 through its ubiquitin associated domain [Citation15]. To investigate how caspase-8 initiated apoptosis under combination therapy, we examined caspase-8 ubiquitination. Caspase-8 was immunoprecipitated from cell lysates and analyzed for K48 and K63-linked polyubiquitination by Western blotting. In cells treated with combination therapy, K63-linked polyubiquitination of caspase-8 increased while K48-linked polyubiquitination of caspase-8 decreased (). Inhibition of proteosome degradation with MG132 did not increase the levels of total caspase-8 or K63-linked polyubiquitination of caspase-8 (), indicating caspase-8 accumulation promoted by combination therapy was not due to inhibition of proteosome activity, but probably due to inhibition of an alternative protein degradation pathway: autophagy degradation (). Modification of autophagy in the presence of the lysosome inhibitor BafA1 or mTORC1 inhibitor rapamycin could regulate the degradation and activation of caspase-8 ().
Figure 4. Combination therapy increased K63-linked polyubiquitination of caspase-8 and enhanced its interaction with p62. (A) HepG2 cells were treated with combination therapy for 1 h and cultured for another 3 h. Cell lysates were used for immunoprecipitation with antibodies specific to caspase-8 and immunoblotting were performed to analyze ubiquitination. (B) Cells were treated as in (A) in the presence of 5 µM MG132 for 4 h. (C and D) Cells were transfected with siRNAs targeting CUL3 (50 nM) for 48 h. Cell lysates were subjected to immunoblotting (C) and immunoprecipitation (D). NTC, negative siRNA control. (E) The colocalization of p62 and caspase-8 proteins was examined by immunofluorescence staining and visualized under Confocal microscopy (DAPI, cell nuclei). Scale bar, 10 μm. (F) Cells were treated with combination therapy, and cell lysates were analyzed for CUL3 expression by western blotting. (G) Co-IP of caspase-8 by p62 in cells lysates. (H) Cells were treated as in (A) in the presence of 100 nM BafA1 for 10 h or 500 nM Rapamycin for 8 h, and then analyzed by immunoblotting to evaluated caspase-8 and autophagy proteins. Data are presentative of three independent experiments.
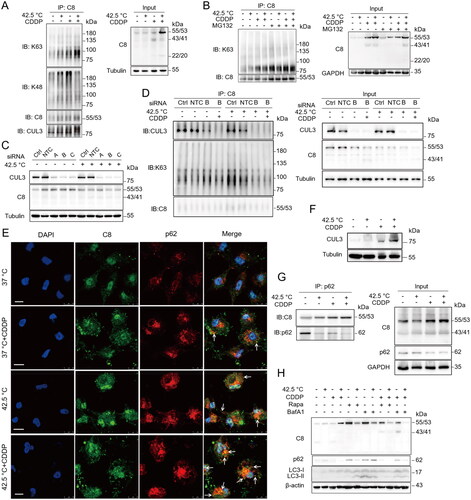
Cullin 3 (CUL3) is an E3 ligase of the Cullin-RING family involved in K63-linked polyubiquitination of caspase-8 [Citation15,Citation19,Citation38]. We found CUL3 expression increased in cancer cells treated with combination therapy (), in agreement with increased K63-linked polyubiquitination of caspase-8 (). siRNA gene silencing of CUL3 inhibited caspase-8 (55/53 kDa) accumulation/activation () and K63-linked polyubiquitination of caspase-8 induced by combination therapy ().
As caspase-8 might be degraded through autophagy as inferred from the above and p62 has a ubiquitin binding domain, we investigated the interaction between caspase-8 and p62. Confocal microscopy study indicated that combination therapy resulted in the highest colocalization of caspase-8 and p62 in cells (). We performed Co-immunoprecipitation (Co-IP) with p62 specific antibodies and found that combination therapy enhanced the interaction between p62 and caspase-8 (). Together, these results indicate combination therapy increased K63-linked polyubiquitination of caspase-8, which in turn enhances its binding to p62 and cell apoptosis.
3.5. Hyperthermia synergized with chemotherapy to promote pyroptosis of cancer cells
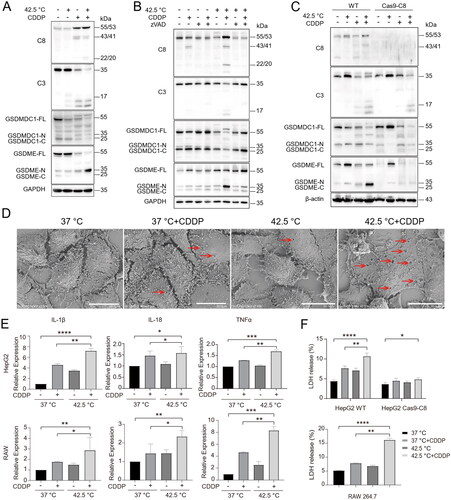
In addition to mediate the caspase-8/p62/caspase-3 death axis in cells treated with combination therapy, activated caspase-8 may also participate in the cleavage of gasdermins to induce pyroptosis [Citation26,Citation33]. We found that hyperthermia synergized with CDDP in inducing the cleavage of gasdermins, especially GSDME. The extent of gasdermin N-terminus releasing correlated with caspase-8 accumulation and activation (). Inhibition of caspase activity with zVAD-fmk reduced the cleavage of GSDME in cells treated with combination therapy, which correlated with reduced caspase-8 accumulation and activation (). In addition, knockout of casepase-8 expression by CRISPR/Cas9 reduced the cleavage of GSDME N-terminus (). Consistently, SEM examination revealed severe pyroptosis in cells treated with combination therapy, as shown by pores formed on cell membranes (). Pyroptosis has been reported to trigger the release of pro-cytokines such as IL-18 and IL-1β and cytoplasmic contents, such as lactate dehydrogenase (LDH). Indeed, we found combination therapy led to increased mRNA expression of inflammatory cytokines such as IL-1β, IL-18 and TNFα in both hepatocellular carcinoma cell line HepG2 cells and mouse monocyte cell line RAW 264.7 cells (). In addition, combination therapy led to increased release of LDH in wild type (WT) HepG2 cells and WT RAW264.7 cells but not in caspase-8 knockout HepG2 cells (). Together, these results indicate that combination therapy promotes pyroptois in a caspase-8 dependent manner.
Figure 5. Hyperthermia synergized with chemotherapy to promote caspase-8 dependent pyroptosis of cancer cells. (A) HepG2 cells were treated as indicated and cell lysates were analyzed for caspases and gasdermins by Western blotting with indicated antibodies. (B) HepG2 cells were pretreated with 40 µM pan-caspase inhibitor zVAD-fmk for 2 h and then treated with various therapies. Caspases and gasdermins were analyzed by Western blotting with antibodies as indicated. (C) WT and Caspase-8 knockout HepG2 cells were treated with various therapies and assessed as in (B). (D) HepG2 cells were treated with various therapies and pyroptosis was examined by scanning electron microscopy (SEM). Arrows indicate pores on cell membranes. Scale bar, 20 μm. (E) mRNA expression of inflammatory cytokines IL-1β, IL-18 and TNFα in HepG2 cells and RAW264.7 cells. (F) Measurement of LDH release in WT and Caspase-8 knockout HepG2 cells, and RAW264.7 cells. Data are expressed as mean ± standard deviation (SD), three independent experiments. * p < 0.05, **p < 0.01,***p < 0.001, ****p < 0.0001 compared to control; t-test.
4. Discussion
Apoptosis is important to sustain tissue homeostasis and protect against various diseases, including cancer. Caspases are a family of cysteine proteases, known as the central executioners of apoptosis [Citation39]. In this study, we found that hyperthermia and CDDP combination therapy led to accumulation of and activation of caspase-8. CDDP treatment was a precondition for caspase-8 accumulation and activation after hyperthermia in our experiment setting. We conjectured that CDDP treatment ‘primed’ cells to accumulate caspase-8 after hyperthermia, for example, translocating caspase-8 to proximity of the E3 ligase CUL3 so that it could be ubiquitinated in a fast fashion (in merely a few hours). Nevertheless, prolonged CDDP treatment slightly increased caspase-8 accumulation, and hyperthermia alone could lead to caspase-8 accumulation when cells were analyzed 16 h after the treatment (data not shown). These observations emphasize the synergistic effect of CDDP and hyperthermia from caspase-8 accumulation and activation aspect, but we did not exclude other mechanisms of action by combination therapy.
Further investigation found combination therapy resulted in elevated K63 linked polyubiquitination of caspase-8. K63-linked polyubiquitin regulates protein function, subcellular localization, protein-protein interactions, or autophagy degradation [Citation40–41]. Previously, it has been reported that CUL3 is a E3 ligase that conjugates K63-linked polyubiquitin to caspase-8 when death receptor ligation occurred. Although the upstream signal leading CUL3 to polyubiquitinate caspase-8 is unknown, we found that combination therapy resulted in elevated levels of CUL3 in cells (). CUL3 overexpression promotes ubiquitination and activation of caspase-8 [Citation15–16], thereby promoting the apoptosis of tumor cells.
Upon caspase-8 polyubiquitination, the ubiquitin-binding protein p62 could bind to caspase-8 and promote the aggregation of caspase-8 within p62-dependent foci, which might reduce the autophagic degradation of caspase-8 and lead to accumulation and activation of caspase-8, and activation of cell apoptosis [Citation15], i.e., the CUL3/caspase-8/p62/caspase-3 death axis. In agreement with a previous study, we not only found p62 interacted with caspase-8 by Co-IP (), but also found caspase-8 aggregated within p62 foci () in cancer cells underwent combination therapy. Interestingly, we found that the autophagy inhibitor BA1 reduced the levels of caspase-8 in cell lysates after hyperthermia/combination therapy (). One possible explanation is that hyperthermia/combination therapy promoted autophagy flux thus increased p62 foci formation, which provided a platform for caspase-8 aggregation and activation without extrinsic death signaling, and vice versa.
Pyroptosis is a form of cell death that is triggered by proinflammatory signals and associated with inflammation. Pyroptosis can be activated by the caspase-1 dependent canonical pathway [Citation42–46] and the caspase-4/-5/-11 dependent noncanonical pathway [Citation47]. More recently, caspase-8 has been reported to activate pyroptosis when apoptosis and necroptosis are inhibited [Citation48–49]. Here, we found caspase-8 could mediate both apoptosis and pyroptosis in cells underwent combination therapy. The difference may be reconciled by the two forms of caspase-8 found in cells treated with combination therapy: the inactive/pro-form of caspase-8 could be responsible for pyroptosis, while the active, p62 bounded caspase-8 could be responsible for apoptosis.
In the study of Shelmann et al. [Citation50], it was found that hyperthermia could not activate caspase-8, and only CDDP activated caspase-8. In this study, the results demonstrate that the combined application of hyperthermia and chemotherapy can significantly enhance the effectiveness of chemotherapy through accumulation and activation of caspase-8, probably due to we optimized the procedure of combination therapy, where prior CDDP treatment was followed by hyperthermia. This study is based on in vitro research at the cellular level, primarily focusing on in vitro heating and chemotherapy treatment, whether prior CDDP treatment followed by hyperthermia led to better outcome in the clinics warranted investigation. Furthermore, controlled delivery of CDDP to the site of tumor to effectively ‘prime’ cancer cells for hyperthermia is a challenge. We believe more in-depth research in this aspect in the future will benefit tumor treatment with hyperthermia and chemotherapy combination therapy.
In conclusion, our research uncovered a novel mechanism of inducing cell death by hyperthermia and chemotherapy combination therapy in which hyperthermia synergized with chemotherapy to promote caspase-8 accumulation and activation to accelerate tumor cell apoptosis and pyroptosis ( and ). Our finding provides further theoretical support for the implementation of hyperthermia and chemotherapy combination therapy for cancer treatment in clinical practice.
Authors’ contribution
B. P. designed the study. G. Z., J. C. and B. P. performed the experiments. Y. P. and Y. W. completed the experimental verification. B. P. and G. Z. analyzed the data, wrote the manuscript. All authors contributed in reviewing and editing the manuscript.
Supplemental Material
Download Zip (18.6 MB)Acknowledgements
We thank all members of the laboratory for their support during research implementation.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data are available upon request.
Additional information
Funding
References
- Dunne M, Regenold M, Allen C. Hyperthermia can alter tumor physiology and improve chemo- and radio-therapy efficacy. Adv Drug Deliv Rev. 2020;163–164:1–12. doi: 10.1016/j.addr.2020.07.007.
- Datta NR, Puric E, Klingbiel D, et al. Hyperthermia and radiation therapy in locoregional recurrent breast cancers: a systematic review and meta-analysis. Int J Radiat Oncol Biol Phys. 2016;94(5):1073–1087. doi: 10.1016/j.ijrobp.2015.12.361.
- Datta NR, Rogers S, Klingbiel D, et al. Hyperthermia and radiotherapy with or without chemotherapy in locally advanced cervical cancer: a systematic review with conventional and network meta-analyses. Int J Hyperthermia. 2016;32(7):809–821. doi: 10.1080/02656736.2016.1195924.
- Zheng N, Xu A, Lin X, et al. Whole-body hyperthermia combined with chemotherapy and intensity-modulated radiotherapy for treatment of advanced nasopharyngeal carcinoma: a retrospective study with propensity score matching. Int J Hyperthermia. 2021;38(1):1304–1312. doi: 10.1080/02656736.2021.1971778.
- Datta NR, Kok HP, Crezee H, et al. Integrating loco-regional hyperthermia into the current oncology practice: SWOT and TOWS analyses. Front Oncol. 2020;10:819. doi: 10.3389/fonc.2020.00819.
- Issels RD. Hyperthermia adds to chemotherapy. Eur J Cancer. 2008; 44(17):2546–2554. doi: 10.1016/j.ejca.2008.07.038.
- Shi Z, Lan B, Peng B, et al. Combination therapy with BH3 mimetic and hyperthermia tends to be more effective on anti-melanoma treatment. Biochem Biophys Res Commun. 2018; 503(1):249–256. doi: 10.1016/j.bbrc.2018.06.010.
- D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019;43(6):582–592. doi: 10.1002/cbin.11137.
- Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006; 25(34):4798–4811. doi: 10.1038/sj.onc.1209608.
- Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27(48):6194–6206. doi: 10.1038/onc.2008.297.
- Vaux DL, Korsmeyer SJ. Cell death in development. Cell CellPress. 1999;96(2):245–254. doi: 10.1016/S0092-8674(00)80564-4.
- Taylor R, Cullen S, Martin S. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–241. doi: 10.1038/nrm2312.
- Gonzalvez F, Lawrence D, Yang B, et al. TRAF2 sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol Cell. 2012;48(6):888–899. doi: 10.1016/j.molcel.2012.09.031.
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007; 35(4):495–516. doi: 10.1080/01926230701320337.
- Jin Z, Li Y, Pitti R, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137(4):721–735. doi: 10.1016/j.cell.2009.03.015.
- Li Y, Kong Y, Zhou Z, et al. The HECTD3 E3 ubiquitin ligase facilitates cancer cell survival by promoting K63-linked polyubiquitination of caspase-8. Cell Death Dis. 2013;4(11):e935–e935. doi: 10.1038/cddis.2013.464.
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67(1):425–479. doi: 10.1146/annurev.biochem.67.1.425.
- Roberts JZ, Holohan C, Sessler T, et al. The SCFSkp2 ubiquitin ligase complex modulates TRAIL-R2-induced apoptosis by regulating FLIP(L). Cell Death Differ. 2020;27(9):2726–2741. doi: 10.1038/s41418-020-0539-7.
- Ohtake F, Saeki Y, Ishido S, et al. The K48-K63 branched ubiquitin chain regulates NF-kB signaling. Mol Cell. 2016;64(2):251–266. doi: 10.1016/j.molcel.2016.09.014.
- Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26(4):399–422. doi: 10.1038/cr.2016.39.
- Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. 2016;18(6):579–586. doi: 10.1038/ncb3358.
- Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem. 2012;81(1):291–322. doi: 10.1146/annurev-biochem-051810-094654.
- Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81(1):203–229. doi: 10.1146/annurev-biochem-060310-170328.
- Broemer M, Meier P. Ubiquitin-mediated regulation of apoptosis. Trends Cell Biol. 2009;19(3):130–140. doi: 10.1016/j.tcb.2009.01.004.
- Chen KW, Demarco B, Heilig R, et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. Embo J. 2019; 38(10):e101638. doi: 10.15252/embj.2019101638.
- Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–158. doi: 10.1038/nature18629.
- Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–116. doi: 10.1038/nature18590.
- Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017; 42(4):245–254. doi: 10.1016/j.tibs.2016.10.004.
- Rogers C, Fernandes-Alnemri T, Mayes L, et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8(1):14128. doi: 10.1038/ncomms14128.
- Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103. doi: 10.1038/nature22393.
- Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017; Sep27(9):673–684. doi: 10.1016/j.tcb.2017.05.005.
- Zhang CC, Li CG, Wang YF, et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019;24(3–4):312–325. doi: 10.1007/s10495-019-01515-1.
- Zheng Z, Deng W, Bai Y, et al. The lysosomal rag-ragulator complex licenses RIPK1 and caspase-8-mediated pyroptosis by yersinia. Science. 2021; 372(6549):eabg0269. doi: 10.1126/science.abg0269.
- Chen KW, Demarco B, Broz P. Beyond inflammasomes: emerging function of gasdermins during apoptosis and NETosis. Embo J. 2020;39(2):e103397. doi: 10.15252/embj.2019103397.
- Laer L, Huizing E, Verstreken M, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet. 1998;20(2):194–197. doi: 10.1038/2503.
- Sarhan J, Liu BC, Muendlein HI, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during yersinia infection. Proc Natl Acad Sci U S A. 2018; 115(46):E10888–E10897. doi: 10.1073/pnas.1809548115.
- Munoz IM, Szyniarowski P, Toth R, et al. Improved genome editing in human cell lines using the CRISPR method. PLOS One. 2014; 9(10):e109752. doi: 10.1371/journal.pone.0109752.
- Camus S, Menéndez S, Cheok C, et al. Ubiquitin-independent degradation of p53 mediated by high-risk human papillomavirus protein E6. Oncogene. 2007;26(28):4059–4070. doi: 10.1038/sj.onc.1210188.
- Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid Redox Signal. 2013; 19(6):546–558. doi: 10.1089/ars.2012.4905.
- Chau V, Tobias JW, Bachmair A, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243(4898):1576–1583. doi: 10.1126/science.2538923.
- Li X, Yang KB, Chen W, et al. CUL3 (cullin 3)-mediated ubiquitination and degradation of BECN1 (beclin 1) inhibit autophagy and promote tumor progression. Autophagy. 2021; 17(12):4323–4340. doi: 10.1080/15548627.2021.1912270.
- Kayagaki N, Stowe IB, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–671. doi: 10.1038/nature15541.
- He W-T, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25(12):1285–1298. doi: 10.1038/cr.2015.139.
- Yu X, He S. GSDME as an executioner of chemotherapy-induced cell death. Sci China Life Sci. 2017;60(11):1291–1294. doi: 10.1007/s11427-017-9142-2.
- Jiang M, Qi L, Li L, et al. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020;6(1):112. doi: 10.1038/s41420-020-00349-0.
- Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi: 10.1038/nature15514.
- Gong W, Shi Y, Ren J. Research progresses of molecular mechanism of pyroptosis and its related diseases. Immunobiology. 2020; 225(2):151884. doi: 10.1016/j.imbio.2019.11.019.
- Schwarzer R, Laurien L, Pasparakis M. New insights into the regulation of apoptosis, necroptosis, and pyroptosis by receptor interacting protein kinase 1 and caspase-8. Curr Opin Cell Biol. 2020; 63:186–193. doi: 10.1016/j.ceb.2020.02.004.
- Wang L, Yan H, Chen X, et al. Caspase-8 is involved in pyroptosis, necroptosis and the maturation and release of IL-1β in Aspergillus fumigatus keratitis. Int Immunopharmacol. 2022; 113(Pt A):109275. doi: 10.1016/j.intimp.2022.109275.
- Shellman YG, Howe WR, Miller LA, et al. Hyperthermia induces endoplasmic reticulum-mediated apoptosis in melanoma and non-melanoma skin cancer cells. J Invest Dermatol. 2008; 128(4):949–956. doi: 10.1038/sj.jid.5701114.