
Abstract
Background
Hereditary angioedema with normal C1-inhibitor (HAE-nC1-INH) is a rare genetic disease. The symptoms can resemble other forms of hereditary angioedema (HAE), but the specific laboratory values are inconspicuous. The knowledge about treatment strategies in HAE-nC1-INH remains insufficient; most of the drugs are only licensed and approved for other types of HAE.
Methods
An analysis of all patients with HAE-nC1-INH was carried out in a certified angioedema treatment center in southern Germany. Only patients with a confirmed HAE-nC1-INH mutation were included. The impact of disease was monitored with validated questionnaires.
Results
Eighteen patients were included: two families with a factor XII mutation and seven families with a plasminogen mutation. All individuals received icatibant for on-demand therapy—efficient treatment response was reported. Three patients were severely affected, and prophylaxis was initiated with lanadelumab. According to the questionnaires, the clinical course and symptoms improved significantly under this prophylactic regime.
Conclusion
This is one of the first descriptions of the clinical outcomes as a response to prophylactic treatment with lanadelumab in HAE-nC1-INH patients with a known mutation. The therapeutic management of HAE-1 and HAE-2 should also be the basis of HAE-nC1-INH, including prophylaxis.
Background and objective
Hereditary angioedema (HAE) is a rare autosomal dominant inherited disease (Citation1). Patients suffer from recurrent, unpredictable attacks of angioedema, involving cutaneous and subcutaneous tissues, the gastrointestinal tract and (in life-threatening situations) the upper airway region. The swellings occur without wheals and without itching. The absence of itching and wheals however, is one of the most important clinical parameters to distinguish between bradykinin mediated HAE and mast cell-mediated forms of angioedema (Citation2,Citation3). Bradykinin, is a vascular circulating peptide which leads to the formation of angioedema due to vasodilation and increased vascular permeability (Citation4). HAE is a rare disease—it is estimated that 1:10,000 to 1:50,000 patients are affected (Citation5). The diagnosis ‘HAE due to C1-inhibitor (C1-INH) deficiency’ (HAE-1 and HAE-2) should be confirmed biologically (Citation4,Citation6). A third type of HAE is even rarer: Initially named HAE-3, it is now known as HAE with normal C1-INH levels (HAE-nC1-INH) (Citation7). The name describes one of the most important differences to HAE-1 and HAE-2: in HAE-nC1-INH, C1-INH level, C1-INH activity, and C4 level are within the normal range (Citation8). Nevertheless, patients with HAE-nC1-INH suffer from recurrent episodes of angioedema without wheals and without itching; in most of the cases other family members are also affected (Citation9). Facial and oropharyngeal swellings are more frequently reported in HAE-nC1-INH than in HAE-1 and HAE-2 patients (Citation10). It is discussed that HAE-nC1-INH follows an autosomal dominant inheritance pattern and was initially described in female patients (Citation11). There are publications about male patients with HAE-nC1-INH, but with a considerably lesser burden of disease (Citation12). Female patients with HAE-nC1-INH are often more severely affected by the symptoms when they have estrogen-containing medication or during pregnancy (Citation13). The courses of disease vary among patients with regard to the underlying mutation: estrogens appear to play a smaller role on triggering attacks in HAE-Plasminogen (PLG) compared with HAE-Factor XII (FXII) (Citation9). The mean age at onset of clinical symptoms is, in most of the cases, between the ages of 20 and 30 (sometimes triggered by the intake of estrogens or, in older patients, by ACE inhibitors), which is later than that of HAE-1 and HAE-2 patients (Citation14).
The clinically suspected diagnosis HAE-nC1-INH can only be confirmed by genetic testing (Citation6). During recent years, much progress has been made concerning the knowledge of mutations leading to HAE-nC1-INH.
As some mutations were published very recently, it can be assumed that in the future, more mutations will be identified to be responsible for HAE-nC1-INH. The influence of each mutation on the clinical course of disease also remains unclarified. Not all mutations do directly interact with the ‘bradykinin pathway’; in some mutations, the exact pathomechanism continues to be a matter of research and discussion (Citation15). There is evidence that bradykinin is the main regulator of some types of HAE-nC1-INH, depending on the mutation. In HAE-FXII, HAE-PLG and HAE-KNG bradykinin is assumed to play an important role in the pathomechanism of angioedema formation (Citation14–16). HAE-ANGPT1 and HAE-MYOF, on the other hand, result from mutations involving the vascular endothelium. According to the current gain of knowledge in this field, new classifications of angioedema have been initiated but not yet published.
The use of approved therapeutic agents for HAE-1 and HAE-2 in HAE-nC1-INH patients remains a further matter of discussion as the pivotal studies were often limited to patients diagnosed with HAE due to C1-INH deficiency (HAE-1 and HAE-2).
The management of patients with HAE-nC1-INH, according to the current guidelines, should be based on the principles of the HAE-1 and HAE-2 treatment strategies (Citation6, Citation12, Citation17). To date, there are no randomized, placebo-controlled clinical trials about specific therapies in patients with HAE-nC1-INH. Moreover, due to the general small number of patients with this rare disease, near-term studies with appropriate numbers of patients are unlikely. As it is assumed that the ‘antiallergic’ treatment with antihistamines and steroids is not effective in patients with HAE-nC1-INH like in HAE-1 and HAE-2 patients, it is crucial to improve the knowledge about treatment options.
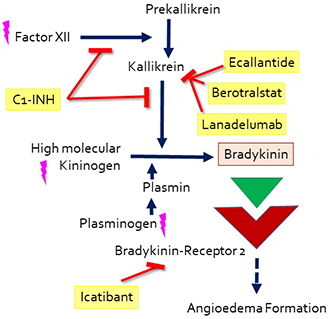
Concerning HAE therapy, one must distinguish between the (on-demand) treatment of acute attacks and a long-term prophylactic therapeutic regime. The latter is especially recommended in severely affected patients to prevent edema attacks. For the acute therapy of angioedema, C1-INH IV, icatibant SC, and ecallantide SC (the last-mentioned is only licensed in the US and a few Latin American countries) are approved. As first-line prophylactic options, C1-INH IV and SC, berotralstat PO and lanadelumab SC are approved (Citation6). A simplified overview of the presumed pathomechanism and mode of action of these drugs is summarized in .
Figure 1. A simplified overview of the presumed pathomechanism and mode of action of drugs approved for HAE-1 and HAE-2, acute and prophylactic treatment. The drugs are marked in yellow, pink lightnings mark known mutations leading to HAE-nC1-INH.
Methods
Design and patients
The clinical courses of disease of all patients with HAE-nC1-INH with a proven mutation were analyzed retrospectively. The monocentric analysis was performed in a certified angioedema center in southern Germany, University hospital, department of ORL. The study has been approved by the local institution’s ethics committee without the need to obtain written informed consent due to the retrospective setting and anonymous evaluation. There was no restriction regarding age and sex.
Data collection
The clinical records of all patients with the aforementioned inclusion criteria were analyzed retrospectively, collecting all available information regarding sex, date of birth, date of symptoms’ onset, family members, date of diagnosis of HAE-nC1-INH, relevant medical conditions, as well as laboratory values such as C1-INH-a, C1-INH-f, C4, the results of genetic testing, localization of the attacks, therapy and response. The burden of disease and treatment response were analyzed with the validated questionnaires AE-QoL (Angioedema Quality of Life Questionnaire) and AECT (Angioedema Control Test) for three months. Both patient-reported outcome measures are recommended tools, especially for HAE patients who take LTP, according to the current HAE guideline (Citation6). The AE-QoL addresses four domains (functioning, fatigue/mood, fears/shame, food). It can be used to record the quality of life and its change over a period of time or under therapy. The questionnaire comprises 17 questions. Each answer represents a score between 0 and 4 points. A maximum of 68 points is possible. The total score can then be converted into a quality of life scale with values from 0% to 100%. The total score (0–100 scale) shows, how the quality of life is impaired. The higher the score, the lower the quality of life (Citation18). The AECT addresses the question of how well the disease is currently controlled and is based on 4 questions. There are possible recall periods of four weeks and three months. Each answer represents a score of between 0 and 4 points. A maximum of 16 points is possible and means complete control, 0 points means no control at all. HAE patients with at least 10 points usually have sufficient disease control; fewer than 10 points implies the disease is poorly controlled, and a change of the therapeutic regime should be considered (Citation19).
In most of the cases, genetic testing (next generation sequencing, TWISTTM Exome Kit, sequencing: NextSeq High OutputKit v2.5; 300 cycles) was performed at the University of Ulm’s institute of human genetics. Two families received genetic testing elsewhere and were subsequently linked to the angioedema center of the authors due to a shorter driving distance.
The data presentations, calculations and statistical analyses were performed using Graph Pad Prism (version 5).
Results
Patient characteristics
Eighteen patients with HAE-nC1-INH with a known mutation were identified and analyzed. Fifteen of the patients were females, three patients were males.
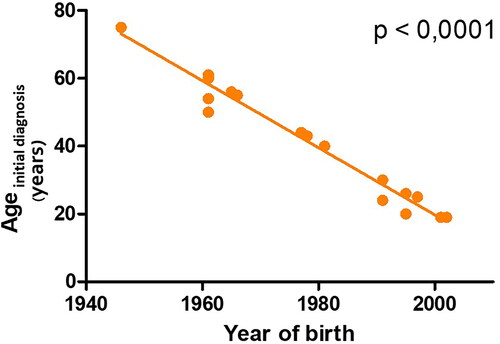
At the time of the data evaluation, the age of the patients ranged from 21 to 77 years with a mean age of 45 years. First symptoms of angioedema occurred between the ages of 16 and 53 with a mean age of 32 years. In four patients, the age of symptom onset could not be evaluated: two patients never had symptoms of angioedema and were only tested for and diagnosed with HAE-nC1-INH as family members had symptoms of angioedema; two patients initially suffered from very unspecific symptoms such as intense abdominal pain and could therefore not define an exact age of symptom onset. Regarding the date of initial diagnosis of HAE-nC1-INH, we took the point in time when HAE-nC1-INH was first diagnosed and treated, but in some cases the mutation was found some time later. HAE-nC1-INH was initially diagnosed between the ages of 19 and 75 with a mean age of 42 years. presents the highly significant negative correlation between the year of birth and the age of initial diagnosis (Spearman correlation; p = < .0001). Supplementary Figure 1 shows the significant negative correlation between the year of birth and the time between first symptoms of HAE to the initial diagnosis (Spearman correlation; p = .0024). Both figures demonstrate that younger HAE-nC1-INH patients (in the present case HAE-PLG and HAE-FXII) receive the first diagnosis of the rare disease significantly earlier compared to older patients. The number of years from symptom onset to the initial diagnosis of HAE-nC1-INH ranged from 1 to 38 years (mean: 14.5 years).
Figure 2. The highly significant negative correlation between the year of birth and the age of initial diagnosis (Spearman correlation; p = <.0001).
The 18 patients represented 9 families. In four cases, only one member of the family was treated in the department of the authors and thus included to this study, even though all of them had other family members diagnosed with HAE-nC1-INH. The most frequent reason, why family members were linked to different angioedema centers, was the distance from their homes to the departments.
Laboratory values and genetic testing
All symptomatic patients were primarily tested for HAE due to C1-INH deficiency. In these patients, at least one set of laboratory values was available for analysis and C1-INH activity and C1-INH concentration levels as well as C4 values were within the normal range. Then genetic testing was performed in an institute of human genetics. The mutations are summarized in . Six of the 18 patients had a factor XII mutation; 12 of the 18 patients had a plasminogen mutation ().
Table 1. All study patients (n = 18) with HAE-nC1-INH and the family members with a history of angioedema, the mutations and clinical information are listed.
Trigger factors and symptoms
The following trigger factors were reported by the patients:
Patients with factor XII mutation: estrogen-containing contraceptives, stress, mechanical and thermic stimuli. Two patients had no more episodes of angioedema after discontinuation of the estrogen-containing contraceptive.
Patients with plasminogen mutation: Hormonal factors (menstruation), stress, mechanical stimuli, infections. Three patients were treated with an ACE inhibitor, which triggered acute angioedema. Two of them were male and in one of them the ACE inhibitor induced edema was his only episode of HAE-nC1-INH ever.
The frequency of angioedema attacks in patients varied from no attacks at all to 10 attacks per month. The above-mentioned triggers were, in the majority of cases, an important influential factor regarding the number of attacks.
The localization of angioedema presents .
Therapy and treatment response
A minimum of one syringe of icatibant was prescribed for all 18 patients for on-demand treatment, as the further course of disease is not predictable. All the patients who treated the attacks with icatibant 30 mg SC reported efficient therapy response. The choice of icatibant as acute therapy was finally made by the patients: all drugs approved for acute treatment of HAE due to C1-INH deficiency were presented by the physician and discussed in accordance with the guidelines for other HAE patients. The main incentives to take icatibant as an acute therapy drug were the option of SC administration and the positive reports of family members. The assessment of the effectiveness of icatibant is based on the patients’ subjective reports. A structured descriptive or statistical evaluation of the treatment response was not possible due to the retrospective setting. The patients reported a complete resolution of edema within a few hours after icatibant application compared to several days without treatment. Icatibant was used for angioedema of all localizations: skin, extremities, gastrointestinal and the head and neck region (including the upper airway). Occasionally, the patients reported to not have treated mild swelling, contrary to medical advice, which was also observed in HAE-1 and HAE-2 patients. A variety of reasons were given for not treating edema, including ‘no heavy burden from peripheral swelling’ and ‘saving on the expensive medication.’
Three patients had severe courses of disease (measured by the questionnaires AE-QoL and AECT as well as the reported burden of disease in daily life), therefore prophylactic treatment was initiated:
Three female patients with plasminogen (n = 1) or factor XII (n = 2) mutations were treated with lanadelumab 300 mg SC in intervals ranging from two to four weeks. All drugs approved for LTP in HAE due to C1-INH deficiency were presented by the physician and discussed in accordance with the guidelines for other HAE patients. The main argument in favor of lanadelumab as LTP was the (already known) SC administration in two- to four-week intervals.
The patient (female, 61) with HAE-PLG was analyzed in two-week treatment intervals for a total of nine months. One patient (female, 44) with HAE-FXII was analyzed for a total of 12 months under lanadelumab prophylaxis; after 6 months, four-week intervals were introduced. Another patient (female, 45) with factor XII mutation was analyzed under lanadelumab prophylaxis in two-week intervals for a total of six months. Subsequently, four-week intervals were introduced as a result of the excellent treatment response. All three patients reported a clear improvement in the burden of their disease in everyday life. () None of them experienced negative side effects except occasional small local reactions at the site of injection.
Figure 3. (a) The course of all three patients with HAE-nC1-INH (factor XII: n = 2, PLG: n = 1) under long-term prophylaxis with lanadelumab 300 mg SC measured with AECT (Angioedema Control Test). Ten and more points in AECT reflect a sufficiently controlled disease situation. (b) The course of all three patients with HAE-nC1-INH (Factor XII: n = 2, PLG: n = 1) under long-term prophylaxis with lanadelumab 300 mg SC measured with AE-QoL (Angioedema Quality of Life Questionnaire). In AE-QoL (0–100), a high score reflects a low quality of life. (c) AECT results, before and after comparison, *= p < .05; one-tailed Kruskal–Wallis test.
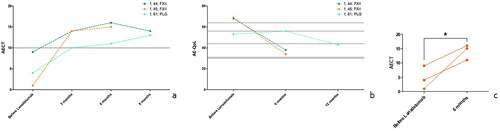
The AECT and AE-QoL results of the patients under prophylaxis with lanadelumab are demonstrated in .
Discussion
With its first description being only 23 years ago, HAE-nC1-INH is a comparatively young condition (Citation11, Citation20). The knowledge about mutations has notably advanced in recent times: during the last six years five of the six known mutations were discovered (Citation3). These facts, combined with an often unspecific course of symptoms, suggest that the estimated number of unrecorded cases may be considerably higher than the actual presumed incidence. Like in many rare and unspecific diseases, the time from symptom onset to the point of correct diagnosis can take up to decades. Time to diagnosis in HAE-nC1-INH often ranges from a short period to longer than 10 years (Citation21). In our analysis with 18 HAE-nC1-INH patients with known mutations, the clinical diagnosis of HAE-nC1-INH was, in some cases, done years before the mutation was known and found. Therefore, in our study, the timepoint of the first clinical diagnosis and treatment begin of HAE-nC1-INH was evaluated, even though in some cases, the mutation was found some time later. Similar to HAE-1 and HAE-2, we found the gratifying result, that younger patients are diagnosed earlier than older HAE participants are, possibly due to growing awareness and knowledge about the disease (Citation22).
The surge of knowledge in recent years indicates, that there are still more pending issues on the disease HAE-nC1-INH. As already mentioned in the introduction, the therapeutic strategy of HAE-nC1-INH is a ‘grey area.’ According to the current angioedema guidelines, the treatment regime should be based on the principles of the treatment strategies of HAE-1 and HAE-2, nevertheless the clinical trials are not designed for patients with HAE-nC1-INH (Citation12, Citation17). The small incidence of the disease and variety of mutations tend to complicate matters further. Even in our analysis with only 18 patients, we found hints that the different groups of HAE-nC1-INH have different courses of disease and perhaps differ regarding the treatment response. This makes further studies even more difficult, as the analyses of subgroups of each mutated gene (e.g., HAE-FXII, HAE-PLG, HAE-ANGPT1, …) are likely to be limited by insufficient statistical power due to a small number of known cases.
Another difficulty regarding the disease HAE-nC1-INH is the patients, who are diagnosed with HAE-nC1-INH but are without a detectable mutation. Riedl et al. remarked aptly: ‘both overdiagnosis and underdiagnosis of the condition (HAE-nC1-INH) are possible’ (Citation21). As it is likely that not all mutations and variants have been discovered yet, it is highly probable, that there are patients with the diagnosis HAE-nC1-INH who—with the current knowledge—can be diagnosed solely based on their (family) anamnesis and typical symptoms. On the other hand, the ambiguity of the clinical diagnosis criteria poses the risk of misdiagnosis: Patients with recurrent edema (of other etiology) could be misdiagnosed with HAE-nC1-INH.
Therefore, only patients with known mutations were analyzed in the present study in order to avoid misleading results (e.g., regarding the response to anti-bradykinin specific medication) from unreliably diagnosed patients. Riedl and colleagues recently presented an overview of how patients with HAE-nC1-INH are diagnosed and treated in the United States. One diagnostic ‘tool’ was using HAE-specific medications or alternatively antihistamines and corticosteroids to assess treatment response (Citation21). However, an observational study of patients with proven HAE-PLG found that treatment for acute attacks with corticosteroids had a certain benefit in some cases and C1-INH IV was not effective in all of the patients (Citation9).
Regarding the therapeutic regime and response in HAE-nC1-INH, the results of the literature vary, particularly in terms of long-term prophylaxis. Structured research concerning the known response to therapy for each subgroup of HAE-nC1-INH was performed by Bork and colleagues in 2020 (Citation9). Some aspects are highlighted in the following section. For acute therapy in many cases icatibant 30 mg SC is used, like in the current study (Citation21). The observational study performed by Bork and colleagues deemed C1-INH IV very effective for the treatment in most patients with HAE-PLG. Nevertheless, in some cases of HAE-nC1-INH C1-INH concentrate IV was less effective or even without response (Citation9). In patients with HAE-FXII, icatibant SC and C1-INH IV were found to be effective in most of the cases (Citation23, Citation24). For long-term prophylaxis in severely affected HAE-nC1-INH patients, the publications range from case reports to larger observational studies. The exact understanding of the pathomechanism of each of the mutations leading to edema may help to individually target the mechanism accordingly in the future. Hintze and colleagues recently analyzed the pathomechanism of HAE-PLG and summarized that a treatment approach for HAE-PLG should be aimed at plasminogen (Citation25). For long-term prophylactic treatment, C1-INH SC or IV are used with varying results (Citation17). Data about the use of lanadelumab and berotralstat in HAE-nC1-INH is scarce or not available at all. For example, a recent network meta-analysis for the indirect comparison of lanadelumab and berotralstat in the treatment of HAE summarizes studies and treatment responses to both agents; HAE-nC1-INH it not at all mentioned (Citation26). Available reports about treatment responses of HAE-nC1-INH patients to lanadelumab are also rare and incoherent—the largest study found, according to the best knowledge of the authors, was a retrospective study of Jones and colleagues including medical records from 10 US treatment centers. In total, 23 patients with HAE-nC1-INH were analyzed–with 7 of them treated with lanadelumab. In none of the patients was it mentioned, whether genetic testing was performed, no mutations were mentioned and in one of the seven patients with lanadelumab, an unusual and off-label dosing regimen (weekly treatment) was initiated (Citation17). In addition to that, the clinical courses of two HAE-FXII patients treated with lanadelumab were published by Bouillet and colleagues. From the onset of treatment with lanadelumab, one of the two patients experienced chronic urticaria but significantly fewer angioedema attacks (25 months follow up time), the other patient was attack-free during the follow up of 5 months after beginning lanadelumab without systemic side effects. AECT and AE-QoL were not assessed (Citation27).
Thus, the present results are one of the first to analyze the clinical course and treatment response to lanadelumab in patients with HAE-nC1-INH with a known mutation. In all three patients, a satisfying treatment response could be demonstrated with few side effects.
Unfortunately, the response to therapy is generally monitored differently in all analyses and ranges from subjective statements to the description of attack frequency to the results of validated questionnaires. In addition to the already mentioned drugs, the use and effect of tranexamic acid and progestins is described for long-term prophylaxis especially in patients with HAE-FXII (Citation9). Both were not evaluated in patients of the present study.
According to previous published results, it was found that patients with HAE-nC1-INH experience angioedema attacks involving different anatomic locations, such as extremities, face and tongue and the abdomen (Citation17). The involvement of the head and neck region is principally reported more frequently than in HAE-1 and HAE-2, which increases the need for efficient treatment due to life-threatening attacks. As shown in the analysis of the trigger factors, the intake of ACE inhibitors is often associated with severe angioedema attacks of the head and neck and is sometimes the first symptom of HAE-nC1-INH, especially in male patients. It may therefore be discussed whether ACE inhibitor induced angioedema is, in some cases, caused by an underlying HAE-nC1-INH, which goes unrecognized due to low disease activity.
Finally, the limitations of the current study should be mentioned: Like in most of the studies about rare diseases such as HAE, only a small number of patients were analyzed. Nevertheless, only patients with a known mutation were evaluated, which excluded bias due to uncertain or wrong diagnoses like chronic urticaria without wheals. A further limitation is the retrospective design, which however enabled a thorough analysis of the courses of disease of the 18 patients with the rare disease HAE-nC1-INH.
Conclusion
HAE-nC1-INH is a young disease, which is undergoing a current transformation in terms of knowledge and understanding of the disease. This study is one of the first descriptions of a treatment response in HAE-nC1-INH patients, with a known mutation to a long-term prophylaxis with lanadelumab. The detection of mutations and a better understanding of the pathomechanism of each of the subgroups leading to edema will help to develop more targeted therapeutic regimes in the future. As the head and neck region is more frequently affected than in HAE due to C1-INH deficiency, effective treatments are crucial.
Ethical approval
This study protocol was reviewed and approved by the local ethics committee (Ethikkommission der Universität Ulm), approval number: 299/22.
Supplemental Material
Download JPEG Image (50.4 KB){kind=link}
Acknowledgment
The authors thank cand. med. Hannah Ramanathan for editorial assistance with regard to proper English language.
Disclosure statement
R. Lochbaum has received funding to attend conferences/educational events from CSL Behring, BioCryst and Takeda. He has participated as an investigator in a clinical trial for Takeda and Pharvaris and received financial support for research projects from Takeda.
S. Trainotti has received grant research support and/or speaker/consultancy fees from CSL Behring and Takeda. She has also received funding to attend conferences/educational events from CSL Behring and Takeda. She has participated as an investigator in a clinical trial/registry for CSL Behring, BioCryst, IONIS Pharmaceuticals and Takeda.
J. Hahn has received speaker/consultancy fees from CSL Behring and Takeda. She has also received funding to attend conferences/educational events from CSL Behring and Takeda. She has participated as an investigator in a clinical trial/registry for CSL Behring, BioCryst, Pharvaris and Takeda.
J. Greve has received speaker/consultancy fees from CSL Behring, Kalvista Takeda and BioCryst. He has also received funding to attend conferences/educational events from CSL Behring and Takeda. He has participated as an investigator in a clinical trial/registry for CSL Behring, Pharvaris, BioCryst and Takeda.
T. K. Hoffmann has no conflict of interest to declare regarding this publication.
Additional information
Funding
References
- Sharma J, Jindal AK, Banday AZ, et al. Pathophysiology of hereditary angioedema (HAE) beyond the SERPING1 gene. Clin Rev Allergy Immunol. 2021;60(3):1–7. doi: 10.1007/s12016-021-08835-8.
- Grumach AS, Veronez CL, Csuka D, et al. Angioedema without wheals: challenges in laboratorial diagnosis. Front Immunol. 2021;12:785736. doi: 10.3389/fimmu.2021.785736.
- Santacroce R, D’Andrea G, Maffione AB, et al. The genetics of hereditary angioedema: a review. J Clin Med. 2021;10(9):2023. doi: 10.3390/jcm10092023.
- Lepelley M, Bernardeau C, Defendi F, et al. Update on bradykinin-mediated angioedema in 2020. Therapies. 2020;75(2):195–205. doi: 10.1016/j.therap.2020.02.011.
- Bernstein JA. HAE update: epidemiology and burden of disease. Allergy Asthma Proc. 2013;34(1):3–6. doi: 10.2500/aap.2013.34.3623.
- Maurer M, Magerl M, Betschel S, et al. The international WAO/EAACI guideline for the management of hereditary angioedema—the 2021 revision and update. Allergy. 2022;77(7):1961–1990. doi: 10.1111/all.15214.
- Riedl MA. Hereditary angioedema with normal C1-INH (HAE type III). J Allergy Clin Immunol Pract. 2013;1(5):427–432. doi: 10.1016/j.jaip.2013.06.004.
- Christiansen SC, Zuraw BL. Laboratory approaches for assessing contact system activation. Immunol Allergy Clin North Am. 2017;37(3):527–539. doi: 10.1016/j.iac.2017.04.008.
- Bork K, Machnig T, Wulff K, et al. Clinical features of genetically characterized types of hereditary angioedema with normal C1 inhibitor: a systematic review of qualitative evidence. Orphanet J Rare Dis. 2020;15(1):289. doi: 10.1186/s13023-020-01570-x.
- Bork K, Gül D, Hardt J, et al. Hereditary angioedema with normal C1 inhibitor: clinical symptoms and course. Am J Med. 2007;120(11):987–992. doi: 10.1016/j.amjmed.2007.08.021.
- Bork K, Barnstedt SE, Koch P, et al. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. 2000;356(9225):213–217. doi: 10.1016/S0140-6736(00)02483-1.
- Magerl M, Germenis AE, Maas C, et al. Hereditary angioedema with normal C1 inhibitor. Immunol Allergy Clin North Am. 2017;37(3):571–584. doi: 10.1016/j.iac.2017.04.004.
- Bork K. Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor; 2010 [cited 2023 June]. Available from: http://www.aacijournal.com/content/6/1/15 doi: 10.1186/1710-1492-6-15.
- Bork K, Wulff K, Witzke G, et al. Hereditary angioedema with normal C1-INH with versus without specific F12 gene mutations. Allergy. 2015;70(8):1004–1012. doi: 10.1111/all.12648.
- Shamanaev A, Dickeson SK, Ivanov I, et al. Mechanisms involved in hereditary angioedema with normal C1-inhibitor activity. Front Physiol. 2023;14:1146834. doi: 10.3389/fphys.2023.1146834.
- Bork K, Wulff K, Witzke G, et al. Treatment of patients with hereditary angioedema with the c.988A > G (p.Lys330Glu) variant in the plasminogen gene. Orphanet J Rare Dis. 2020;15(1):52. doi: 10.1186/s13023-020-1334-8.
- Jones DH, Bansal P, Bernstein JA, et al. Clinical profile and treatment outcomes in patients with hereditary angioedema with normal C1 esterase inhibitor. World Allergy Organ J. 2022;15(1):100621. doi: 10.1016/j.waojou.2021.100621.
- Weller K, Groffik A, Magerl M, et al. Development and construct validation of the angioedema quality of life questionnaire. Allergy. 2012;67(10):1289–1298. doi: 10.1111/all.12007.
- Weller K, Donoso T, Magerl M, et al. Development of the angioedema control test-a patient-reported outcome measure that assesses disease control in patients with recurrent angioedema. Allergy. 2020;75(5):1165–1177. doi: 10.1111/all.14144.
- Binkley KE, Davis A 3rd. Clinical, biochemical, and genetic characterization of a novel estrogen-dependent inherited form of angioedema. J Allergy Clin Immunol. 2000;106(3):546–550. doi: 10.1067/mai.2000.108106.
- Riedl MA, Danese M, Danese S, et al. Hereditary angioedema with normal C1 inhibitor: US survey of prevalence and provider practice patterns. J Allergy Clin Immunol Pract. 2023;11(8):2450.e6–2456.e6. doi: 10.1016/j.jaip.2023.01.023.
- Hahn J, Hoess A, Friedrich DT, et al. Unnecessary abdominal interventions in patients with hereditary angioedema. J Dtsch Dermatol Ges. 2018;16(12):1443–1449. doi: 10.1111/ddg.13698.
- Bouillet L, Boccon-Gibod I, Gompel A, et al. Hereditary angioedema with normal C1 inhibitor: clinical characteristics and treatment response with plasma-derived human C1 inhibitor concentrate (berinert®) in a french cohort. Eur J Dermatol. 2017;27(2):155–159. doi: 10.1684/ejd.2016.2948.
- Deroux A, Boccon-Gibod I, Fain O, et al. Hereditary angioedema with normal C1 inhibitor and factor XII mutation: a series of 57 patients from the french national center of reference for angioedema. Clin Exp Immunol. 2016;185(3):332–337. doi: 10.1111/cei.12820.
- Hintze S, Möhl BS, Beyerl J, et al. Mutant plasminogen in hereditary angioedema is bypassing FXII/kallikrein to generate bradykinin. Front Physiol. 2022;13:1090732. doi: 10.3389/fphys.2022.1090732.
- Watt M, Malmenäs M, Romanus D, et al. Network meta-analysis for indirect comparison of lanadelumab and berotralstat for the treatment of hereditary angioedema. J Comp Eff Res. 2023;12(6):e220188. doi: 10.57264/cer-2022-0188.
- Bouillet L, Bocquet A, Belbezier A, et al. Effectiveness of lanadelumab in patients with hereditary angioedema with normal C1 inhibitor and FXII mutation. Ann Allergy Asthma Immunol. 2021;127(3):391–392. doi: 10.1016/j.anai.2021.05.028.