
Abstract
Context
Podophyllotoxin (PPT) derivatives, used in cancer therapy, require development toward enhanced efficacy and reduced toxicity.
Objective
This study synthesizes PPT derivatives to assess their anticancer activities.
Materials and methods
Compounds E1-E16 antiproliferative activity was tested against four human cancer cell lines (H446, MCF-7, HeLa, A549) and two normal cell lines (L02, BEAS-2B) using the CCK-8 assay. The effects of compound E5 on A549 cell growth were evaluated through molecular docking, in vitro assays (flow cytometry, wound healing, Transwell, colony formation, Western blot), and in vivo tests in female BALB/c nude mice treated with E5 (2 and 4 mg/kg). E5 (4 mg/kg) significantly reduced xenograft tumor growth compared to the DMSO control group.
Results
Among the 16 PPT derivatives tested for cytotoxicity, E5 exhibited potent effects against A549 cells (IC50: 0.35 ± 0.13 µM) and exceeded the reference drugs PPT and etoposide to inhibit the growth of xenograft tumours. E5-induced cell cycle arrest in the S and G2/M phases accelerated tubulin depolymerization and triggered apoptosis and mitochondrial depolarization while regulating the expression of apoptosis-related proteins and effectively inhibited cell migration and invasion, suggesting a potential to limit metastasis. Molecular docking showed binding of E5 to tubulin at the colchicine site and to Akt, with a consequent down-regulation of PI3K/Akt pathway proteins.
Discussion and conclusions
This research lays the groundwork for advancing cancer treatment through developing and using PPT derivatives. The encouraging results associated with E5 call for extended research and clinical validation, leading to novel and more effective cancer therapies.
Introduction
Cancer remains one of the most formidable diseases and ranks second as the leading cause of death globally. It is characterized by uncontrolled proliferation of cells, disruption of cell cycle regulation, and resistance to programmed cell death, culminating in the formation of malignant tumours that can invade nearby tissues and ultimately lead to fatal outcomes (Siegel et al. Citation2022; Abd Emoniem et al. Citation2023). Throughout the disease, cancer patients often face significant physical and emotional challenges that profoundly affect their quality of life (Mouna et al. Citation2022). Chemotherapy and surgery are effective modalities for treating cancer (Mansoori et al. Citation2017; Panda and Biswal Citation2019). However, the efficacy of chemotherapy requires the continued development of new drugs to address the pressing issues of tumour resistance and adverse effects (Tshering Vogel et al. Citation2010; Bai et al. Citation2020). Natural products represent valuable resources due to their diverse structures, unique biological activities, and specific selectivity (Zi et al. Citation2019). Nature has consistently served as a crucial reservoir for medicinal compounds. However, numerous potential natural compounds lack tumour selectivity and frequently manifest noticeable toxic side effects, thus limiting their progress toward clinical applications (Zi et al. Citation2019).
Among natural compounds, lignans have attracted attention because of their distinctive characteristics. Podophyllotoxin (1) (PPT, ) is one of the most representative members of this family, primarily isolated from Podophyllum species (Berberidaceae). It inhibits the proliferation of multiple cancer cells by affecting mitotic microtubule assembly and cell division (Sk et al. Citation2013; Shah et al. Citation2021). Despite its high toxicity that prevents clinical use, PPT serves as a starting material for semi-synthetic anticancer drugs. First-generation PPT modifiers, such as etoposide (2, VP-16), teniposide (3, VM-26), or etopophos (4) (), are clinically used to combat various tumour types, including small-cell lung cancer, germ cell malignancies, lymphomas, and Kaposi′s sarcoma (Gordaliza et al. Citation2000; Mouna et al. Citation2022). These semisynthetic drugs differ from the parent compound PPT in their antitumor mechanism. VP-16 and its analogues act primarily on DNA topoisomerase II to form a stable reversible complex with drug-enzyme-DNA, preventing DNA repair (Pitts et al. Citation2011; Xiao et al. Citation2020). These drugs exhibit some adverse effects in clinical treatment, including alopecia, gastrointestinal irritation, tumour drug resistance, and toxic side effects on normal cells. Therefore, there is an urgent need to develop new drugs to meet demand.
Figure 1. Structures of podophyllotoxin, 4-phenylbutyrate, ABT-751, and their analogues.
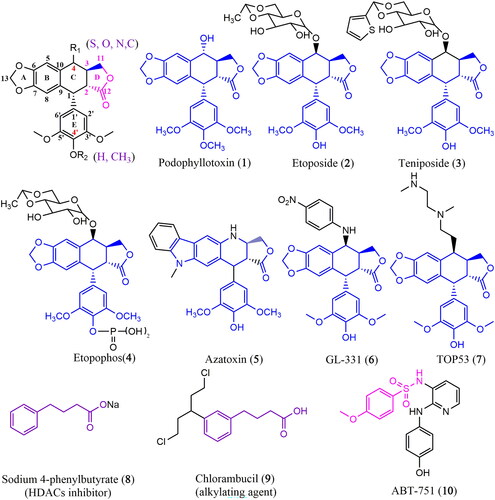
Recent evidence strongly suggests that combining PPT with other chemotherapeutic agents can overcome the limitations of PPT and VP-16. Novel molecules such as azatoxin (5), GL331 (6), and TOP53 (7) () have been synthesized. TOP53 is effective against metastatic lung cancer, while GL331 is effective against colorectal cancer (Xiao et al. Citation2020; Zhao et al. Citation2021). Consequently, PPT emerges as a leading compound for developing anticancer drugs.
We designed a series of PPT derivatives using the chemistry hybrid concept to enhance drug activity and targeting based on the above study findings. A comprehensive review of the existing literature identified sodium 4-phenylbutyrate (8, 4-PBA sodium) () as a histone deacetylase (HDAC) inhibitor capable of impeding the growth of non-small cell lung cancer (NSCLC) cell lines at 2 mM. Furthermore, combining 4-PBA sodium with ciglitizone has been found to intensify cancer cell growth arrest (Chang and Szabo Citation2002), indicating its potential sensitizing properties.
Chlorambucil (9, CB-1348) (), an orally active antineoplastic agent belonging to the nitrogen mustard group, is a bifunctional alkylating agent. Studies have shown its efficacy against lymphocytic leukaemia, ovarian and lung carcinomas, and Hodgkin’s disease (Salem et al. Citation2011; Guo et al. Citation2017). Furthermore, ABT-751 (10, E7010) () is a new bioavailable tubulin-binding and antimitotic sulphonamide agent, which exhibits an IC50 of approximately 1.5 and 3.4 μM in neuroblastoma and non-neuroblastoma cell lines, with the sulphonamide group playing a crucial role (Aggarwal et al. Citation2012).
Using a molecular hybridization strategy to test our hypothesis, we designed novel hybrids incorporating PPT, a benzene sulphonamide group, and 4-PBA in a single molecule (). Sixteen PPT derivatives were synthesized and assessed for their anticancer activities in vitro and in vivo.
Figure 2. Design of the target compounds.

Materials and methods
Reagents
All chemical reagents and solvents for synthesizing the compounds were of analytical grade.1H NMR and 13C NMR spectra were recorded using a Bruker AscendTM-400 and 600 model spectrometer (Bruker Company, USA) in CDCl3 with TMS as the internal standard. Melting points (uncorrected) were measured on a Beijing Taike XT4. Thin layer chromatography (TLC) was performed on silica gel plates (Silica Gel 60 GF254) and visualized under UV light (254 nm). PPT (1) was isolated from the Chinese medicinal herb Dysosma versipellis (Hance) M. Cheng ex Ying (Berberidaceae) and was the parent material for preparing all new PPT-like derivatives. Annexin V-FITC/PI cell apoptosis assay kit (Cat#40302ES60) and the cell cycle and apoptosis analysis kit (Cat#40301ES60) were purchased from YEASEN Biotech Co. Ltd. (Shanghai, China). JC-1 mitochondrial membrane potential (Cat#C2006) and LY294002 (Cat#S1737) were purchased from Beyotime Biotech (Shanghai, China).
All antibodies were diluted in blocking buffers: anti-PI3K (Rabbit, 1:1000, ABclonal, RRID: AB_2863407, Cat#A4992), anti-pPI3K (Rabbit, 1:1000, ABclonal, RRID: AB_2771417, Cat#AP0427), anti-GAPDH (Rabbit, 1:5000, Proteintech, RRID: AB_2263076, Cat#10494-1-AP), anti-AKT (Rabbit, 1:1000, Beyotime, Cat#AA326), anti-AKT (Rabbit, 1:1000, Beyotime, Cat#AA326), anti-pAKT (Rabbit, 1:1000, Beyotime, Cat#AA326), anti-AKT (Rabbit, 1:1000, Beyotime, Cat#AA326), anti-pAKT (Rabbit, 1:1000, Beyotime, Cat#AA329), anti-Tubulin (Mouse, 1:500, Beyotime, Cat#AT819), anti-Bax (Mouse, 1:800, Beyotime, Cat#AF0054), anti-Tubulin (Mouse, 1:1000, Abmart, Cat#M20005), anti-Bcl2 (Rabbit, 1:1000, Proteintech, RRID: AB_2818996, Cat#26593-1-AP), goat anti-mouse IgG-HRP (Mouse, 1:10000, Affinity, RRID: AB_2839430, Cat#S0002), and goat anti-rabbit IgG-HRP (Rabbit, 1:10000, Affinity, RRID: AB_2839429, Cat#S0001).
General procedure for the synthesis of compounds 3a-3p
The synthetic routes are detailed in Scheme 1. To synthesize the compounds, 4-(4-aminophenyl)butanoic acid (5.58 mmoL, 0.916 g) was dissolved in 20 mL of H2O, and saturated Na2CO3 was added dropwise at room temperature. The pH was regulated to 8-9, and the stirring continued until the acid completely dissolved in the water. Subsequently, the appropriate phenylsulfonyl chloride (6 mmoL, 2a-2p) was added and stirred overnight at room temperature. After the reaction, the pH was adjusted to 1-2 using a 1 M HCl solution. A significant amount of bright yellowish solid precipitated. After filtration, the solid was washed with water and dried to obtain a pale yellow solid.
Scheme 1. Synthesis of podophyllotoxin derivatives E1-E16. Reagents and reaction conditions: (a) room temperature; (b) (1) H2O, saturated Na2CO3, pH 8-9, room temperature, 2 h; (2) 1 M HCl, pH 1-2; (c) DCC, DMAP, CH2Cl2, 0 °C, 8 h.
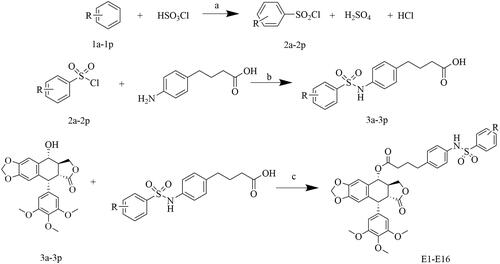
General procedure for the synthesis of compounds E1-E16
PPT (0.175 mmoL, 0.0725 g), 3a-3p (0.263 mmoL), 4-dimethyaminopyridine (DMAP), and N, N′-dicyclohexyl-carbodiimide (DCC) were dissolved in dichloromethane. The reaction mixture was stirred at 0 °C for 8 h and monitored by TLC. After the reaction was completed, the solvent was evaporated under reduced pressure. The crude compound was purified by silica gel chromatography using EtOAc-petroleum ether as a solvent to obtain the desired products E1-E16. Details are shown in .
Table 1. Eluent composition and proportion.
Cell culture
Human small-lung carcinoma cells (H446, Procell CL-0401, RRID: CVCL_1562), human breast cancer cell (MCF-7, Procell CL-0149, RRID: CVCL_0031), human cervical cancer (HeLa, Procell CL-0101, RRID: CVCL_0030), human lung adenocarcinoma epithelial cell (A549, Procell CL-0016, RRID: CVCL_ 0023), human normal hepatic (L02, Procell CL-0111, RRID: CVCL_6926), and human normal lung epithelial (BEAS-2B, Procell CL-0496, RRID: CVCL_0168) cell lines were purchased from Procell Life Science & Technology Co., Ltd. (Wuhan, China). Cells were kept in DMEM/F12 medium (A549) and RPMI 1640 medium (H446), while the rest were kept in Dulbecco′s modified Eagle medium (DMEM, Gibco; high glucose) with l-glutamine supplemented with 10% fetal bovine serum (FBS, BI), 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco), and incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Antiproliferation assay
Cell suspensions of A549, MCF-7, H446, HeLa, and non-tumorigenic (BEAS-2B, L02) were seeded in 96-well plates and incubated with E1-E16, PPT, and etoposide in a 37 °C incubator for 24 h. The mixture was further incubated after adding the CCK-8 solution for 2 h. Subsequently, the absorbance at 450 nm was measured using a microplate reader (ELx800, BioTek, USA).
Cell-cycle analysis
A549 cells were seeded in 6-well plates (5.0 × 104 cells/well) and incubated at 37 °C overnight to adhere. Exponentially growing cells were treated with 0.1% DMSO or E5 at 0.1, 0.5, 1.0 µM, and PPT at 1.0 µM for 24 h. Subsequently, cells were fixed with 70% ethanol at 4 °C for 24 h. Cells were centrifuged, harvested, resuspended with PBS and containing 10 µL of RNase A and 10 µL of propidium iodide (PI) at 4 °C for 0.5 h in the dark. For the time-dependent assay, cells were incubated with E5 at a concentration of 0.5 μM for 12, 24, and 36 h. Flow cytometric data analysis was conducted immediately after supravital stain.
The single-cell gating process established gates on forward scatter (FSC) and side scatter (SSC) plots. This approach is crucial to exclude debris, thus ensuring that the analysis is limited to single cells. In a separate but equally important procedure, DNA histogram gating, a histogram represents the fluorescence intensity of PI (denoted as FL2) versus cell counts. This histogram is instrumental in setting gates that accurately distinguish between the cell cycle′s G0/G1, S, and G2/M phases based on DNA content.
Cell apoptosis analysis
Approximately 5.0 × 104 cells/well of A549 were cultured in 6-well plates treated with compound E5 (0, 0.5, 1.0, and 2.0 µM) or PPT (2.0 µM) for 24 h and cells were treated with 1.0 µM E5 for 12, 24, and 36 h to induce cell apoptosis. The cells were then collected and washed twice with PBS. Subsequently, cells were resuspended in 500 µL 1 × binding buffer and then stained with 5 µL of Annexin V-FITC and 10 µL of PI 15 min at 0 °C in the dark. The stained cells were analyzed by flow cytometry (Beckman, USA). Statistical analysis was performed using the Flowjo 7.6.1 software.
In single-cell gating, gates were set on forward (FSC) and side scatter (SSC) plots. This procedure is essential to exclude debris, ensuring that the analysis is focused exclusively on single cells. Subsequently, in the process of apoptosis gating, a dot plot is created to show the relationship between Annexin V fluorescence (FL1) and PI fluorescence (FL2). Critical to this analysis is the establishment of gates that differentiate various cell populations: viable cells (Annexin V-/PI-), early apoptotic cells (Annexin V+/PI-), late apoptotic or necrotic cells (Annexin V+/PI+), and non-viable cells (Annexin V-/PI+). These techniques are essential for accurately discriminating cell states, particularly in cell viability and apoptosis studies.
Mitochondrial membrane potential analysis
Mitochondrial membrane potential (MMP, ΔΨm) was detected using the JC-1 assay kit. Briefly, A549 cells were seeded in 6-well plates (1.0 × 105 cells/well) and allowed to adhere. The cells were treated with compound E5 (0, 0.5, 1.0 and 2.0 µM) for 12 h. After incubation, cells were collected and incubated with JC-1 solution for 20 min, washed twice with JC-1 staining buffer, and resuspended in a medium. The stained cells were analyzed with a flow cytometer (Beckman, USA) and an Olympus confocal microscope.
Wound healing assay
The experiments were conducted using a previous method (Ma et al. Citation2021) with some modifications. A549 cells were seeded in a 6-well tissue culture plate and grown to 85% confluence in a 37 °C incubator for 24 h. The cells were then scratched using a micropipette tip, washed with PBS to remove non-adherent cells, and incubated with 0.1% DMSO or different concentrations of E5 at 37 °C for 24 h. Cell migration images were captured with an inverted fluorescent microscope at 0 and 24 h.
Colony formation assay
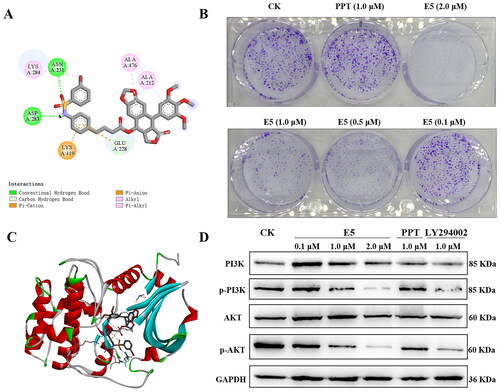
A549 cells were trypsinized and cultured in 6-well plates (1.0 × 103 cells/well), incubated for 24 h, and then treated with different concentrations of E5 for 24 h. Subsequently, cells were incubated for 2 weeks until the colony formed. Cells were washed twice with PBS, fixed with 4% paraformaldehyde for 0.5 h, and stained with crystal violet staining solution for 0.5 h. Finally, the results were photographed on phones.
Western blot
The cell lysate was mixed with a non-reducing lane marker sample buffer, followed by heat denaturation. A Bradford assay was performed to quantify the protein concentration, and 20 µg of protein was extracted from each sample for western blot analysis. The protein was applied to a 10% SDS-polyacrylamide gel, transferred to polyvinylidene fluoride (PVDF) membranes (TermoFisher), and then detected by proper primary and secondary antibodies before visualization by the ECL western blot kit. The visualized bands were exposed using a gel imaging system, and the results were analysed using ImageJ software.
Confocal microscopy
A549 cells were cultured on coverslips at 75% confluence and incubated with 0.5 and 1.0 µM E5, 1.0 µM colchicine, or 1.0 µM etoposide for 24 h. Cells were washed with PBS twice, fixed with 4% paraformaldehyde for 15 min, and permeabilized with 0.5% Triton X-100 for another 10 min. Subsequently, cells were blocked with 3% BSA for 1 h, washed once with PBS, and treated with an anti-tubulin antibody (1:300, Cytoskeleton, Inc.) in 3% BSA at 48 °C. Then 200 µL of Cy3-labeled goat anti-mouse IgG (H + L) (1:500, Cytoskeleton, Inc.) was added to each coverslip, which had been washed with 0.5% Triton X-100 (incubation for 5 min), in 5% BSA, and incubated at room temperature, followed by DAPI at 37 °C for 5 min. Finally, cells were visualized under an Olympus confocal microscope, and data were analyzed using FV-10-ASW 1.7 viewer software.
Animal studies
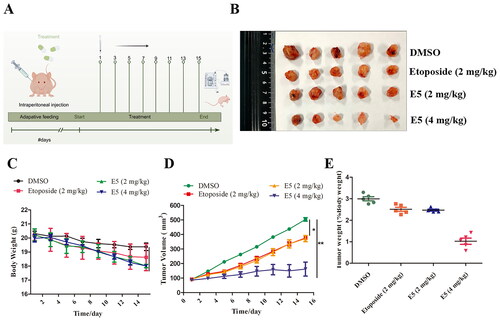
Twenty 5-week-old female BALB/c nude mice were procured from Jinan Pengyue Experimental Animal Breeding Co., Ltd., China. Mice were subcutaneously injected with A549 cells after 1 week of adaptive feeding. Cell density was adjusted to 5.0 × 106 cells/0.1 mL using a serum-free culture medium and placed on ice to prepare for inoculation. Subsequently, 100 μL of the prepared cell suspension was injected into the left axilla of the nude mice. After the tumours reached an average size of 90 mm3, the mice were randomly divided into control (DMSO), positive control drug (etoposide, 2 mg/kg), and low-dose E5 (2 mg/kg), and high-dose E5 (4 mg/kg) groups. Each group had 5 mice. Treatments were administered once every two days, eight times by intraperitoneal injection (i.p.). Tumour size was measured with calipers, and volume was calculated using the previously reported method: 0.5 × length ×width2. At the end of the experiment, the mice were euthanized (i.p., 0.3% pentobarbital sodium, 0.1 mL/10 g), and the tumours were isolated and weighed. All animal studies were conducted following the National Institutes of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Ethics Committee of Jining First People′s Hospital, Shandong First Medical University (JNRM-2022-DW-012).
Docking simulations
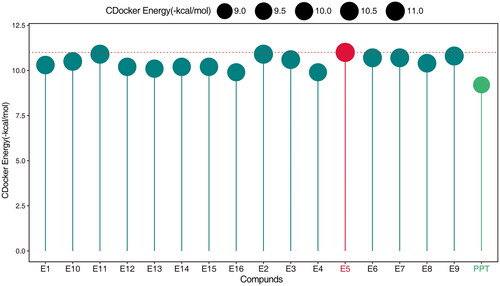
The molecular docking of compound E5 into the X-ray crystal structure of tubulin (PDB code: 1SA0) and Akt (PDB code: 3cqw) was conducted using Discovery Studio software (version 3.5) with the DS-CDOCKER protocol through the graphical user interface. The binding energy with tubulin is shown in .
Figure 3. Interaction binding energy between compounds E1-E16 and PPT.
Results
Chemistry
The structures of PPT derivatives E1-E16 are presented in .
Table 2. The structure of the compound E1-E16.
All compounds are reported here for the first time and were characterized using 1HNMR,13C NMR, and melting point tests.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro [3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl 4-(4-(phenylsulfonamido)phenyl)butanoate (E1):
Yellow powder, yield: 74.5%, Mp: 103.4-110.2 °C. 1H NMR (600 MHz, CDCl3) δ 8.04-7.98 (m, 1H), 7.78-7.74 (m, 2H), 7.55-7.50 (m, 1H), 7.43 (t, J = 7.8 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 6.97 (d, J = 8.4 Hz, 2H), 6.71 (s, 1H), 6.54 (s, 1H), 6.36 (s, 2H), 5.99 (d, J = 5.3 Hz, 2H), 5.86 (d, J = 9.2 Hz, 1H), 4.61 (d, J = 4.4 Hz, 1H), 4.35 (dd, J = 9.2, 7.2 Hz, 1H), 4.22–4.18 (m, 1H), 3.82 (s, 3H), 3.70 (s, 6H), 3.49–3.40 (m, 1H), 2.92 (dd, J = 14.6, 4.5 Hz, 1H), 2.66–2.55 (m, 2H), 2.44–2.38 (m, 1H), 2.36–2.30 (m, 1H), 1.94 (tt, J = 13.5, 6.7 Hz, 2H); ESI-TOF, calcd for C38H37NO11SH ([M + H]+) 715.21; Anal. calcd for C38H37NO11S: C, 63.77%; H, 5.21%; N, 1.96%; O, 24.59%; S, 4.48%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro [3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl 4-(4-((2-bromophenyl)sulfonamido)phenyl) butanoate (E2):
Yellow powder, yield: 72.5%, Mp: 107.5-110.3 °C. 1H NMR (600 MHz, CDCl3) δ 8.03-8.00 (m, 1H), 7.71 (dt, J = 7.3, 3.1 Hz, 1H), 7.37 (d, J = 2.0 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 7.06 (d, J = 8.4 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 6.70 (s, 1H), 6.54 (s, 1H), 6.37 (s, 2H), 6.01–5.97 (m, 2H), 5.86 (d, J = 9.3 Hz, 1H), 4.60 (d, J = 4.4 Hz, 1H), 4.33 (dd, J = 9.1, 7.3 Hz, 1H), 4.21–4.16 (m, 1H), 3.81 (s, 3H), 3.71 (s, 6H), 3.45 (t, J = 10.2 Hz, 1H), 2.83–2.74 (m, 1H), 2.57 (tt, J = 8.2, 4.1 Hz, 2H), 2.37 (ddd, J = 21.1, 10.7, 5.4 Hz, 2H), 1.94–1.87 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 173.76 (s, 12-C), 173.67 (s, 14-C), 152.64 (s, 3′, 5′-C), 148.16 (s, 7-C), 147.59 (s, 6-C), 138.62 (s, 24-C), 137.90 (s, 4′-C), 137.09 (s, 1′-C), 135.06 (s, 21-C), 134.83 (s, 18-C), 134.07 (s, 27-C), 133.86 (s, 9-C), 132.36 (s, 26-C), 132.26 (s, 29-C), 129.29 (s, 19, 23-C), 128.22 (s, 28-C), 127.82 (s, 10-C), 122.25 (s, 20, 22-C), 122.09 (s, 25-C), 109.76 (s, 8-C), 108.04 (s, 2′, 6′-C), 106.99 (s, 5-C), 101.66 (s, 13-C), 73.58 (s, 4-C), 71.39 (s, 11-C), 60.79 (s, 4′-OCH3), 56.12 (s, 3′, 5′-OCH3), 45.60 (s, 2-C), 43.70 (s, 1-C), 38.77 (s, 3-C), 34.34 (s, 17-C), 33.46 (s, 15-C), 26.20 (s, 16-C); ESI-TOF, calcd for C38H36BrNO11SH ([M + H]+) 793.12; Anal. calcd for C38H36BrNO11S: C, 57.44%; H, 4.57%; Br, 10.06%; N, 1.76%; O, 22.15%; S, 4.03%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro [3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((4-propylphenyl)sulfonamido)phenyl)butanoate (E3):
Yellow powder, yield: 59.7%, Mp: 94.7-101.3 °C. 1H NMR (600 MHz, CDCl3) δ 7.94-7.87 (m, 1H), 7.66 (d, J = 8.3 Hz, 2H), 7.23 (d, J = 8.2 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 6.95 (d, J = 8.3 Hz, 2H), 6.71 (s, 1H), 6.54 (s, 1H), 6.36 (s, 2H), 5.99 (d, J = 5.5 Hz, 2H), 5.86 (d, J = 9.2 Hz, 1H), 4.61 (d, J = 4.3 Hz, 1H), 4.36 (dd, J = 9.2, 7.2 Hz, 1H), 4.22–4.19 (m, 1H), 3.82 (s, 3H), 3.70 (s, 6H), 3.45 (s, 1H), 2.92 (dd, J = 14.6, 4.5 Hz, 1H), 2.65–2.56 (m, 4H), 2.44–2.32 (m, 2H), 1.95 (dd, J = 14.8, 7.4 Hz, 2H), 1.64–1.60 (m, 2H), 0.90 (dd, J = 7.3, 4.4 Hz, 3H); ESI-TOF, calcd for C41H43NO11SH ([M + H]+) 757.26; Anal. calcd for C41H43NO11S: C, 64.98%; H, 5.72%; N, 1.85%; O, 23.22%; S, 4.23%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro [3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((4-bromophenyl)sulfonamido)phenyl)butanoate (E4):
Yellow powder, yield: 75.5%, Mp: 110.4–118.3 °C. 1H NMR (600 MHz, CDCl3) δ 8.17 (s, 1H), 7.62 (d, J = 8.6 Hz, 2H), 7.56 (d, J = 8.6 Hz, 2H), 7.01 (d, J = 8.3 Hz, 2H), 6.96 (d, J = 8.4 Hz, 2H), 6.71 (s, 1H), 6.54 (s, 1H), 6.36 (s, 2H), 5.99 (d, J = 5.3 Hz, 2H), 5.86 (d, J = 9.2 Hz, 1H), 4.61 (d, J = 4.3 Hz, 1H), 4.36 (dd, J = 9.1, 7.3 Hz, 1H), 4.23–4.18 (m, 1H), 3.83 (s, 3H), 3.70 (s, 6H), 3.46 (t, J = 10.1 Hz, 1H), 2.93 (dd, J = 14.6, 4.4 Hz, 1H), 2.69–2.55 (m, 2H), 2.45–2.39 (m, 1H), 2.37–2.28 (m, 1H), 1.73–1.66 (m, 2H); ESI-TOF, calcd for C38H36BrNO11SH ([M + H]+) 793.12,; Anal. calcd for C38H36BrNO11S: C, 57.44%; H, 4.57%; Br, 10.06%; N, 1.76%; O, 22.15%; S, 4.03%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro
[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((3-bromophenyl)sulfonamido)phenyl)butanoate (E5):
Faint yellow powder, yield: 79.6%, Mp: 103.8-108.9 °C. 1H NMR (600 MHz, CDCl3) δ 8.34 (s, 1H), 7.89 (t, J = 1.6 Hz, 1H), 7.68 (d, J = 7.9 Hz, 1H), 7.65 − 7.62 (m, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.45 (d, J = 11.0 Hz, 1H), 7.36 (s, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.71 (s, 1H), 6.54 (s, 1H), 6.36 (s, 2H), 6.00–5.98 (m, 2H), 5.86 (d, J = 9.2 Hz, 1H), 4.61 (d, J = 4.4 Hz, 1H), 4.36 (dd, J = 9.1, 7.3 Hz, 1H), 4.23 − 4.19 (m, 1H), 3.83 (s, 3H), 3.69 (s, 6H), 3.46 (t, J = 10.4 Hz, 1H), 2.81–2.73 (m, 1H), 2.66–2.59 (m, 2H), 2.45–2.39 (m, 1H), 2.35–2.30 (m, 1H), 1.84–1.76 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 173.75 (s, 12-C), 173.69 (s, 14-C), 152.64 (s, 3′, 5′-C), 148.19 (s, 7-C), 147.61 (s, 6-C), 141.06 (s, 24-C), 138.57 (s, 4′-C), 136.93 (s, 1′-C), 135.89 (s, 21-C), 134.89 (s, 27-C), 134.28 (s, 18-C), 130.47 (s, 9-C), 130.14 (s, 28-C), 129.31 (s, 25-C), 129.14 (s, 19, 23-C), 128.19 (s, 10-C), 125.78 (s, 29-C), 124.01 (s, 26-C), 122.81 (s, 20, 22-C), 109.70 (s, 8-C), 107.91 (s, 2′, 6′-C), 107.15 (s, 5-C), 101.66 (s, 13-C), 73.57 (s, 4-C), 71.50 (s, 11-C), 60.84 (s, 4′-OCH3), 56.06 (s, 3′, 5′-OCH3), 45.74 (s, 2-C), 43.67 (s, 1-C), 38.89 (s, 3-C), 34.89 (s, 17-C), 33.70 (s, 15-C), 26.02 (s, 16-C); ESI-TOF, calcd for C38H36BrNO11SH ([M + H]+) 793.12; Anal. calcd for C38H36BrNO11S: C, 57.44%; H, 4.57%; Br, 10.06%; N, 1.76%; O, 22.15%; S, 4.03%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro [3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((3-fluorophenyl)sulfonamido)phenyl)butanoate (E6):
Yellow powder, yield: 74.2%, Mp: 99.8-107.1 °C. 1H NMR (600 MHz, CDCl3) δ 8.21 (d, J = 47.5 Hz, 1H), 7.82-7.79 (m, 1H), 7.56–7.52 (m, 2H), 7.45 (d, J = 8.1 Hz, 1H), 7.42 (dd, J = 10.7, 5.3 Hz, 1H), 7.36 (s, 1H), 7.01 (d, J = 8.3 Hz, 2H), 6.71 (s, 1H), 6.54 (s, 1H), 6.36 (s, 2H), 6.01–5.96 (m, 2H), 5.86 (d, J = 9.2 Hz, 1H), 4.61 (d, J = 4.3 Hz, 1H), 4.35 (dt, J = 18.1, 9.1 Hz, 1H), 4.21 (t, J = 10.0 Hz, 1H), 3.83 (s, 3H), 3.69 (s, 6H), 3.45 (t, J = 10.1 Hz, 1H), 2.93 (dd, J = 14.6, 4.4 Hz, 1H), 2.68–2.60 (m, 2H), 2.44–2.39 (m, 1H), 2.35-2.29 (m, 1H), 1.81-1.75 (m, 2H); ESI-TOF, calcd for C38H36FNO11SH ([M + H]+) 733.20; Anal. calcd for C38H36FNO11S: C, 62.20%; H, 4.95%; F, 2.59%; N, 1.91%; O, 23.98%; S, 4.37%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro [3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((2-fluorophenyl)sulfonamido)phenyl)butanoate (E7):
Yellow powder, yield: 70.9%, Mp: 100.3-108.8 °C. 1H NMR (600 MHz, CDCl3) δ 8.17 (s, 1H), 7.83-7.79 (m, 1H), 7.55–7.50 (m, 1H), 7.36 (d, J = 7.7 Hz, 1H), 7.19 (dd, J = 13.5, 5.9 Hz, 2H), 7.14–7.12 (m, 1H), 7.04 (d, J = 8.5 Hz, 2H), 6.70 (s, 1H), 6.54 (s, 1H), 6.37 (s, 2H), 6.00–5.97 (m, 2H), 5.87 (d, J = 9.3 Hz, 1H), 4.60 (t, J = 6.8 Hz, 1H), 4.33 (dt, J = 14.9, 7.5 Hz, 1H), 4.19 (t, J = 10.0 Hz, 1H), 3.81 (s, 3H), 3.71 (s, 6H), 3.45 (t, J = 10.2 Hz, 1H), 2.92 (dd, J = 14.6, 4.5 Hz, 1H), 2.62–2.55 (m, 2H), 2.42–2.33 (m, 2H), 1.72 (ddd, J = 14.1, 13.4, 7.4 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 173.79 (s, 12-C), 173.70 (s, 14-C), 157.88 (s, 25-C), 152.64 (s, 3′, 5′-C), 148.17 (s, 7-C), 147.59 (s, 6-C), 138.50 (s, 4′-C), 137.07 (s, 1′-C), 134.84 (s, 21-C), 134.02 (s, 18-C), 132.37 (s, 27-C), 130.93 (s, 9-C), 129.30 (s, 19,23,29-C), 128.21 (s, 10-C), 126.85 (s, 24-C), 124.00 (s, 28-C), 122.01 (s, 20, 22-C), 116.82 (s, 26-C), 109.75 (s, 8-C), 108.02 (s, 2′, 6′-C), 107.02 (s, 5-C), 101.66 (s, 13-C), 73.58 (s, 4-C), 71.41 (s, 11-C), 60.79 (s, 4′-OCH3), 56.11 (s, 3′, 5′-OCH3), 45.62 (s, 2-C), 43.69 (s, 1-C), 38.79 (s, 3-C), 34.30 (s, 17-C), 33.41 (s, 15-C), 26.17 (s, 16-C); ESI-TOF, calcd for C38H36FNO11SH ([M + H]+) 733.20; Anal. calcd for C38H36FNO11S: C, 62.20%; H, 4.95%; F, 2.59%; N, 1.91%; O, 23.98%; S, 4.37%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro [3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((4-(trifluoromethyl)phenyl)sulfonamido)phenyl)butanoate (E8):
Yellow powder, yield: 69.4%, Mp: 97.5-105.8 °C. 1H NMR (600 MHz, CDCl3) δ 8.07 (dd, J = 13.1, 6.4 Hz, 1H), 7.80 (d, J = 8.8 Hz, 2H), 7.53 (d, J = 8.6 Hz, 1H), 7.36 (s, 1H), 7.01 (d, J = 8.4 Hz, 3H), 6.94 (d, J = 8.3 Hz, 2H), 6.71 (s, 1H), 6.55 (s, 1H), 6.36 (s, 2H), 5.99 (d, J = 5.1 Hz, 2H), 5.86 (d, J = 9.1 Hz, 1H), 4.61 (d, J = 4.3 Hz, 1H), 4.36 (dd, J = 9.1, 7.3 Hz, 1H), 4.25–4.16 (m, 1H), 3.83 (s, 3H), 3.69 (s, 6H), 3.42 (d, J = 39.1 Hz, 1H), 2.92 (dd, J = 14.6, 4.4 Hz, 1H), 2.69–2.63 (m, 1H), 2.58 (dt, J = 19.5, 6.6 Hz, 1H), 2.42 (dt, J = 14.2, 7.2 Hz, 1H), 2.35–2.28 (m, 1H), 1.94 (ddd, J = 28.0, 13.9, 7.0 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 173.68 (s, 12-C), 173.63 (s, 14-C), 152.62 (s, 3′, 5′-C), 148.18 (s, 7-C), 147.59 (s, 6-C), 147.10 (s, 24-C), 138.63 (s, 4′-C), 137.48 (s, 1′-C), 136.88 (s, 21-C), 134.88 (s, 27-C), 134.18 (s, 18-C), 132.42 (s, 9-C), 129.41 (s, 25, 29-C), 129.31 (s, 19, 23-C), 128.15 (s, 10-C), 124.48 (s, 28-C), 123.99 (s, 26-C), 122.83 (s, 30-C), 120.71 (s, 18, 20-C), 109.68 (s, 8-C), 107.86 (s, 2′, 6′-C), 107.17 (s, 5-C), 101.64 (s, 13-C), 73.57 (s, 4-C), 71.51 (s, 11-C), 60.82 (s, 4′-OCH3), 56.01 (s, 3′, 5′-OCH3)), 45.77 (s, 2-C), 43.64 (s, 1-C), 38.92 (s, 3-C), 34.06 (s, 17-C), 33.09 (s, 15-C), 25.91 (s, 16-C); ESI-TOF, calcd for C39H36F3NO11SH ([M + H]+) 783.20; Anal. calcd for C39H36F3NO11S: C, 59.77%; H, 4.63%; F, 7.27%; N, 1.79%; O, 22.45%; S, 4.09%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro
[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((2,4-difluorophenyl)sulfonamido)phenyl)butanoate (E9):
Yellow powder, yield: 66.3%, Mp: 115.2-120.4 °C. 1H NMR (600 MHz, CDCl3) δ 8.22-8.17 (m, 1H), 7.83 (dd, J = 14.3, 8.0 Hz, 1H), 7.13 (d, J = 7.0 Hz, 1H), 7.03 (s, 3H), 6.92 (q, J = 8.1 Hz, 2H), 6.70 (s, 1H), 6.54 (s, 1H), 6.37 (s, 2H), 5.99 (d, J = 6.7 Hz, 2H), 5.87 (d, J = 9.2 Hz, 1H), 4.61 (d, J = 4.3 Hz, 1H), 4.33 (dt, J = 19.2, 9.7 Hz, 1H), 4.20 (t, J = 10.0 Hz, 1H), 3.82 (s, 3H), 3.71 (s, 6H), 3.43 (d, J = 26.9 Hz, 1H), 2.92 (dd, J = 14.6, 4.5 Hz, 1H), 2.64–2.53 (m, 2H), 2.44–2.32 (m, 2H), 1.93 (dd, J = 14.2, 6.8 Hz, 2H); ESI-TOF, calcd for C38H35F2NO11SH ([M + H]+) 751.19; Anal. calcd for C38H35F2NO11S: C, 60.71%; H, 4.69%; F, 5.05%; N, 1.86%; O, 23.41%; S, 4.26%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro
[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((2,6-difluorophenyl)sulfonamido)phenyl)butanoate (E10):
Yellow powder, yield: 62.9%, Mp: 105.9-113.7 °C. 1H NMR (600 MHz, CDCl3) δ 7.50–7.44 (m, 1H), 7.10 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H), 6.98 (t, J = 8.8 Hz, 2H), 6.71 (s, 1H), 6.54 (s, 1H), 6.37 (s, 2H), 5.99 (dd, J = 8.0, 0.9 Hz, 2H), 5.87 (d, J = 9.2 Hz, 1H), 4.61 (d, J = 4.4 Hz, 1H), 4.34 (dd, J = 9.1, 7.3 Hz, 1H), 4.22–4.18 (m, 1H), 3.81 (s, 3H), 3.71 (s, 6H), 3.45 (t, J = 10.3 Hz, 1H), 2.92 (dd, J = 14.6, 4.5 Hz, 1H), 2.64–2.56 (m, 2H), 2.44–2.33 (m, 2H), 1.96–1.90 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 173.77 (s, 12-C), 173.69 (s, 14-C), 160.55 (s, 29-C), 158.83 (s, 25-C), 152.61 (s, 3′, 5′-C), 148.15 (s, 7-C), 147.58 (s, 6-C), 138.66 (s, 4′-C), 137.03 (s, 1′-C), 134.88 (s, 21-C), 134.82 (s, 18-C), 133.79 (s, 27-C), 132.34 (s, 9-C), 129.42 (s, 19, 23-C), 128.17 (s, 10-C), 121.61 (s, 20, 22-C), 113.18 (s, 24-C), 113.02 (s, 28-C), 113.00 (s, 26-C), 109.72 (s, 8-C), 107.99 (s, 2′, 6′-C), 107.01 (s, 5-C), 101.64 (s, 13-C), 73.56 (s, 4-C), 71.40 (s, 11-C), 60.77 (s, 4′-OCH3), 56.08 (s, 3′, 5′-OCH3), 45.61 (s, 2-C), 43.67 (s, 1-C), 38.78 (s, 3-C), 34.27 (s, 17-C), 33.38 (s, 15-C), 26.13 (s, 16-C); ESI-TOF, calcd for C38H35F2NO11SH ([M + H]+) 751.19; Anal. calcd for C38H35F2NO11S: C, 60.71%; H, 4.69%; F, 5.05%; N, 1.86%; O, 23.41%; S, 4.26%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro [3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((2-nitrophenyl)sulfonamido)phenyl)butanoate (E11):
Yellow powder, yield: 69.1%, Mp: 98.6-107.8 °C. 1H NMR (600 MHz, CDCl3) δ 7.86-7.81 (m, 2H), 7.69 (td, J = 7.8, 1.1 Hz, 1H), 7.59 (dd, J = 11.2, 4.2 Hz, 1H), 7.33 (s, 1H), 7.14–7.10 (m, 2H), 7.08 (d, J = 8.4 Hz, 2H), 6.70 (s, 1H), 6.54 (s, 1H), 6.37 (s, 2H), 6.00 (d, J = 7.5 Hz, 2H), 5.88 (d, J = 9.3 Hz, 1H), 4.60 (d, J = 4.4 Hz, 1H), 4.33 (dd, J = 9.1, 7.2 Hz, 1H), 4.22–4.18 (m, 1H), 3.80 (s, 3H), 3.72 (s, 6H), 3.60 (s, 1H), 2.93 (dd, J = 14.6, 4.5 Hz, 1H), 2.66–2.59 (m, 2H), 2.46–2.37 (m, 2H), 1.98–1.91 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 173.73 (s, 12-C), 173.66 (s, 14-C), 152.60 (s, 3′, 5′-C), 148.18 (s, 25-C), 148.15 (s, 7-C), 147.56 (s, 6-C), 139.54 (s, 4′-C), 137.08 (s, 1′-C), 134.81 (s, 28-C), 133.97 (s, 21-C), 133.58 (s, 24-C), 132.58 (s, 18-C), 132.34 (s, 27-C), 132.27 (s, 9-C), 131.78 (s, 29-C), 129.40 (s, 19, 23-C), 128.17 (s, 10-C), 125.29 (s, 26-C), 123.71 (s, 20, 22-C), 109.75 (s, 8-C), 108.05 (s, 2′, 6′-C), 106.94 (s, 5-C), 101.66 (s, 13-C), 73.58 (s, 4-C), 71.34 (s, 11-C), 60.76 (s, 4′-OCH3), 56.11 (s, 3′, 5′-OCH3), 45.55 (s, 2-C), 43.66 (s, 1-C), 38.74 (s, 3-C), 34.41 (s, 17-C), 33.48 (s, 15-C), 26.19 (s, 16-C); ESI-TOF, calcd for C38H36N2O13SH ([M + H]+) 760.19; Anal. calcd for C38H36N2O13S: C, 59.99%; H, 4.77%; N, 3.68%; O, 27.34%; S, 4.21%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro
[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((4-methylphenyl)sulfonamido)phenyl)butanoate (E12):
Yellow powder, yield: 68.8%, Mp: 91.4-99.5 °C. 1H NMR (600 MHz, CDCl3) δ 7.89 (t, J = 8.5 Hz, 1H), 7.65 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 6.98–6.95 (m, 2H), 6.71 (s, 1H), 6.54 (s, 1H), 6.36 (s, 2H), 5.99 (dd, J = 6.4, 1.0 Hz, 2H), 5.87 (d, J = 9.2 Hz, 1H), 4.61 (d, J = 4.4 Hz, 1H), 4.35 (dd, J = 9.2, 7.2 Hz, 1H), 4.23-4.19 (m, 1H), 3.82 (s, 3H), 3.70 (s, 6H), 3.46 (s, 1H), 2.93 (dd, J = 14.6, 4.5 Hz, 1H), 2.65–2.56 (m, 2H), 2.45–2.39 (m, 2H), 2.37 (s, 3H), 1.93 (dt, J = 19.9, 6.3 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 173.77 (s, 12-C), 173.68 (s, 14-C), 152.61 (s, 3′, 5′-C), 148.15 (s, 7-C), 147.58 (s, 6-C), 143.73 (s, 27-C), 137.99 (s, 4′-C), 136.96 (s, 24-C), 136.23 (s, 1′-C), 134.84 (s, 21-C), 134.75 (s, 18-C), 132.35 (s, 9-C), 129.60 (s, 26, 28-C), 129.16 (s, 19, 23-C), 128.18 (s, 10-C), 127.24 (s, 25, 29-C), 122.21 (s, 20, 22-C), 109.69 (s, 8-C), 107.92 (s, 2′, 6′-C), 107.07 (s, 5-C), 101.63 (s, 13-C), 73.54 (s, 4-C), 71.44 (s, 11-C), 60.78 (s, 4′-OCH3), 56.05 (s, 3′, 5′-OCH3), 45.66 (s, 2-C), 43.66 (s, 1-C), 38.82 (s, 3-C), 34.20 (s, 17-C), 33.28 (s, 15-C), 26.11 (s, 16-C), 21.54 (s, 30-C). 13C NMR (151 MHz, CDCl3) δ 174.47 (s, 12-C), 173.67 (s, 14-C), 152.61 (s), 148.18 (s, 7-C), 147.59 (s, 6-C), 147.13 (s, 28-C), 145.94 (s, 25-C), 144.84 (s, 24-C), 139.37 (s), 138.61 (s), 136.79 (s, 21-C), 134.90 (s, 18-C), 133.71 (s, 9-C), 132.40 (s, 26-C), 129.42 (s), 128.12 (s, 10-C), 127.11 (s, 27-C), 125.05 (s, 29-C), 122.27 (s), 109.64 (s), 107.80 (s), 107.22 (s), 101.65 (s), 73.53 (s), 71.54 (s), 60.81 (s), 55.98 (s), 45.78 (s), 43.63 (s), 38.94 (s), 34.87 (s), 33.96 (s), 25.75 (s), 24.54 (s); ESI-TOF, calcd for C39H39NO11SH ([M + H]+) 729.22; Anal. calcd for C39H39NO11S: C, 64.19%; H, 5.39%; N, 1.92%; O, 24.11%; S, 4.39%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro
[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((3-nitrophenyl)sulfonamido)phenyl)butanoate (E13):
Yellow powder, yield: 72.5%, Mp: 103.4-109.8 °C. 1H NMR (600 MHz, CDCl3) δ 8.56 (t, J = 1.8 Hz, 1H), 8.38 (dd, J = 8.2, 1.3 Hz, 1H), 8.05 (d, J = 7.8 Hz, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.19 (s, 1H), 6.98 (d, J = 8.3 Hz, 2H), 6.90 (d, J = 8.3 Hz, 2H), 6.70 (s, 1H), 6.54 (s, 1H), 6.33 (s, 2H), 5.98 (dd, J = 6.1, 5.1 Hz, 2H), 5.84 (d, J = 9.0 Hz, 1H), 4.61 (d, J = 4.2 Hz, 1H), 4.38 (dd, J = 9.2, 7.3 Hz, 1H), 4.24-4.19 (m, 1H), 3.85 (s, 3H), 3.64 (s, 6H), 3.44 (s, 1H), 2.92 (dd, J = 14.6, 4.3 Hz, 1H), 2.73–2.66 (m, 1H), 2.61–2.55 (m, 1H), 2.43–2.38 (m, 1H), 2.26–2.20 (m, 1H), 1.91 (ddd, J = 21.6, 14.2, 7.8 Hz, 2H); ESI-TOF, calcd for C38H36N2O13SH ([M + H]+) 760.19; Anal. calcd for C38H36N2O13S: C, 59.99%; H, 4.77%; N, 3.68%; O, 27.34%; S, 4.21%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro
[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((4-nitrophenyl)sulfonamido)phenyl)butanoate (E14):
Yellow powder, yield: 75.5%, Mp: 110.5-118.6 °C. 1H NMR (600 MHz, CDCl3) δ 8.38 (d, J = 8.8 Hz, 1H), 8.26 (t, J = 8.4 Hz, 2H), 7.93 (dd, J = 9.0, 2.2 Hz, 2H), 6.98 (t, J = 6.5 Hz, 2H), 6.92 (t, J = 5.8 Hz, 2H), 6.69 (s, 1H), 6.55 (s, 1H), 6.34 (s, 2H), 6.00–5.98 (m, 2H), 5.84 (d, J = 9.0 Hz, 1H), 4.61 (d, J = 4.3 Hz, 1H), 4.37 (dd, J = 9.2, 7.3 Hz, 1H), 4.24–4.19 (m, 1H), 3.85 (s, 3H), 3.66 (s, 6H), 3.45 (t, J = 10.1 Hz, 1H), 2.92 (dd, J = 14.6, 4.3 Hz, 1H), 2.73–2.66 (m, 1H), 2.57 (dt, J = 16.2, 5.0 Hz, 1H), 2.47-2.38 (m, 1H), 2.30–2.22 (m, 1H), 1.96–1.89 (m, 2H); ESI-TOF, calcd for C38H36N2O13SH ([M + H]+) 760.19; Anal. calcd for C38H36N2O13S: C, 59.99%; H, 4.77%; N, 3.68%; O, 27.34%; S, 4.21%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro
[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((2-methyl-5-nitrophenyl)sulfonamido)phenyl)butanoate (E15):
Yellow powder, yield: 66.3%, Mp: 97.6-102.5 °C. 1H NMR (600 MHz, CDCl3) δ 8.75 (d, J = 2.3 Hz, 1H), 8.25 (dd, J = 8.3, 2.4 Hz, 1H), 7.48 − 7.45 (m, 1H), 6.98 (t, J = 7.7 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 6.70 (s, 1H), 6.54 (s, 1H), 6.35 (s, 2H), 5.99 (d, J = 5.5 Hz, 2H), 5.84 (d, J = 9.1 Hz, 1H), 4.60 (d, J = 4.2 Hz, 1H), 4.35 (dt, J = 12.7, 6.4 Hz, 1H), 4.21 (dd, J = 13.1, 7.0 Hz, 1H), 3.83 (s, 3H), 3.67 (s, 6H), 3.45 (s, 1H), 2.92 (dd, J = 14.6, 4.4 Hz, 1H), 2.73 (s, 3H), 2.68–2.60 (m, 1H), 2.60–2.52 (m, 1H), 2.40 (dt, J = 16.3, 6.9 Hz, 1H), 2.31–2.25 (m, 1H), 1.98–1.89 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 174.47 (s, 12-C), 173.67 (s, 14-C), 152.61 (s, 3′, 5′-C), 148.18 (s, 7-C), 147.59 (s, 6-C), 147.13 (s, 28-C), 145.94 (s, 25-C), 144.84 (s, 24-C), 139.37 (s, 4′-C), 138.61 (s, 1′-C), 136.79 (s, 21-C), 134.90 (s, 18-C), 133.71 (s, 9-C), 132.40 (s, 26-C), 129.42 (s, 19, 23-C), 128.12 (s, 10-C), 127.11 (s, 27-C), 125.05 (s, 29-C), 122.27 (s, 20, 22-C), 109.64 (s, 8-C), 107.80 (s, 2′, 6′-C), 107.22 (s, 5-C), 101.65 (s, 13-C), 73.53 (s, 4-C), 71.54 (s, 11-C), 60.81 (s, 4′-OCH3), 55.98 (s, 3′, 5′-OCH3), 45.78 (s, 2-C), 43.63 (s, 1-C), 38.94 (s, 3-C), 34.87 (s, 17-C), 33.96 (s, 15-C), 25.75 (s, 16-C), 24.54 (s, 30-C); ESI-TOF, calcd for C39H38N2O13SH ([M + H]+) 774.21; Anal. calcd for C39H38N2O13S: C, 60.46%; H, 4.94%; N, 3.62%; O, 26.84%; S, 4.14%.
(5R,5aR,8aR,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro
[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-5-yl4-(4-((4-methoxyphenyl)sulfonamido)pheny)butanoate (E16):
Yellow powder, yield: 64.2%, Mp: 102.7-110.2 °C. 1H NMR (600 MHz, CDCl3) δ 7.94 (d, J = 8.9 Hz, 1H), 7.72–7.68 (m, 2H), 7.01 (d, J = 8.5 Hz, 2H), 6.99–6.96 (m, 2H), 6.88 (t, J = 5.9 Hz, 2H), 6.71 (s, 1H), 6.54 (s, 1H), 6.37 (s, 2H), 5.98 (dd, J = 5.2, 4.2 Hz, 2H), 5.86 (d, J = 9.2 Hz, 1H), 4.60 (d, J = 4.4 Hz, 1H), 4.35 (dd, J = 9.1, 7.2 Hz, 1H), 4.22–4.17 (m, 1H), 3.81 (d, J = 1.9 Hz, 6H), 3.70 (s, 6H), 3.50–3.42 (m, 1H), 2.93 (dd, J = 14.6, 4.5 Hz, 1H), 2.67–2.55 (m, 2H), 2.46–2.39 (m, 1H), 2.38–2.32 (m, 1H), 1.94–1.90 (m, 2H); ESI-TOF, calcd for C39H39NO12SH ([M + H]+) 745.22; Anal. calcd for C39H39NO12S: C, 62.81%; H, 5.27%; N, 1.88%; O, 25.74%; S, 4.30%.
The results obtained align consistently with the depicted structures (Supplemental Figure 1).
Biological activity
Antiproliferative activity and structure-activity relationship of E1-E16
Initial experiments, including studies of antiproliferative effect and toxicity, were conducted for E1–E16 against four human cancer cell lines: H446, MCF-7, Hela, and A549. In addition, two non-cancer cell lines, L02, and BEAS-2B cell lines, were included in the study. The evaluation was performed using the CCK-8 assay in vitro. PPT was the lead compound, and etoposide was used as a positive control. The resulting IC50 values are calculated and presented in .
Table 3. Antiproliferative activity in vitro.
Most compounds demonstrated significant antiproliferation activity against the four cancer cell lines. In particular, for A549 cells, compounds E5 (IC50 = 0.35 ± 0.13 µM), E9 (IC50 = 0.55 ± 0.17 µM), and E10 (IC50 = 0.95 ± 0.22 µM) exhibited markedly superior antiproliferation effects compared to the parent compound PPT (IC50 = 14.74 ± 1.14 µM) and the positive drug etoposide (IC50 = 9.32 ± 0.55 µM). Furthermore, E5 (IC50 = 2.42 ± 0.30 µM) demonstrated activity comparable to etoposide (IC50 = 2.21 ± 0.42 µM), outperforming PPT (IC50 = 3.69 ± 0.38 µM) against H446 cells. Although the antiproliferation effects against MCF-7 cells were better than PPT, the difference was not as pronounced. For A549 and H466 cells, compounds E5, E6, E9, E10, and E13 with substitutions (F, Br, NO2) on the benzene ring showed superior antiproliferation activity compared to compounds E3 and E12 with donor substituents.
Remarkably, as expected, the lead compound PPT demonstrated outstanding antiproliferative activity against all human cancer and normal cell lines. In contrast, the antiproliferation effects of E1–E16 on noncancer cells were significantly less toxic than those on cancer cell lines, particularly for L02.
Based on these findings, we hypothesize that the intervention of the p-aminophenylbutyric acid group enhanced antiproliferation activity while simultaneously reducing cytotoxicity toward noncancer cells. To investigate the underlying anticancer mechanisms, E5 was chosen for further investigation against A549 cells.
Compound E5 induced A549 cell cycle arrest
Cell cycle progression analysis was used to detect E5′s inhibitory effect. First, to better understand E5′s potential, a molecular docking of E5 with tubulin (PDB code: 1SA0) was conducted. The results revealed that compound E5 docked well with 1SA0 at the colchicine site, forming various interactions such as four hydrogen bonds, pi-alkyl, and carbon-hydrogen interactions (). The flat scaffold structure of the E5 group is flexibly inserted into a hydrophobic pocket of tubulin.
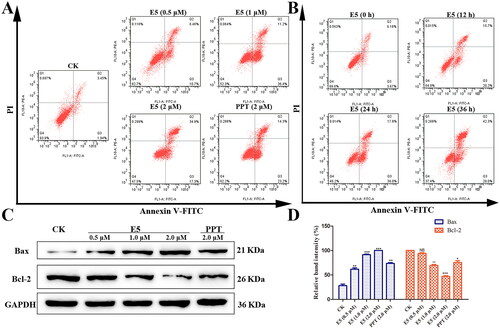
Figure 4. Compound E5 binding mode at the tubulin colchicine site (PDB code: 1SA0) and compound E5 induced arrest of S and G2/M in A549 cells. (A) 2D image of the interaction between E5 and amino acid residues of the nearby active site. (B) 3D image of E5 was inserted into the tubulin binding site. (C and D) Flow cytometry analysis of E5 in a dose-dependent and time-dependent manner. (E) quantitative analysis. *p < 0.05 and **p < 0.01 vs. vehicle group.
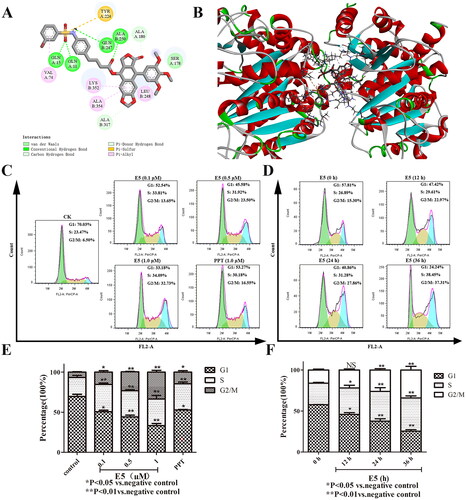
Next, to determine whether the cytotoxic potency of compound E5 resulted from cell cycle progression, the cell cycle distribution in A549 cells was further assessed by flow cytometry after labeling with propidium iodide. As shown in , the population of cells in phases S and G2/M increased dramatically with the elevated concentration of compound E5. Compared to the vehicle, A549 cells incubated with an increased concentration of E5 accumulated primarily in the S and G2/M phases, with the percentage rising from 23.47% to 34.09% and from 6.50% to 32.73%, respectively, along with concomitant losses in the G1 phase. PPT could induce cell cycle arrest as a positive control, although not as significantly (). Furthermore, 0.5 µM of compound E5 could arrest the cell cycle in the G2/M phase in a time-dependent manner (). In summary, E5-induced cell cycle arrest could be responsible for cytotoxicity in cancer cells.
Compound E5-induced A549 cell apoptosis
The study′s subsequent focus was determining whether compound E5 induced apoptotic effects in A549 cells. An apoptosis assay was conducted using Annexin V/PI double staining. The results revealed that E5 treatment increased the ratio of early to late apoptotic cells in a time-dependent manner, particularly at a concentration of 1.0 µM (). Subsequently, we assessed the levels of cleaved Bax and Bcl-2 by Western blotting. Analysis showed that E5 significantly decreased the expression of the antiapoptotic protein Bcl-2 and increased Bax expression. These findings suggest that E5 treatment enhanced early apoptosis in A549 cells at lower concentrations. As E5 concentration increases, it is more likely to induce late apoptosis in A549 cells.
Figure 5. Compound E5 induced A549 cell apoptosis. Cells were incubated with PPT (2.0 µM) and E5 (0.5, 1.0, and 2.0 µM) for 24 h, and also incubated with E5 (1.0 µM) for 0, 12, 24, and 36 h. (A and B) Flow cytometry analysis of E5 in a dose- and time-dependent manner using Annexin V/PI double staining. (C) Western blot for cell apoptosis-related proteins (Bax and Bcl-2) separated by SDS-PAGE, and GAPDH was used as the internal control. Data are expressed as mean ± SD. (D) quantitative analysis. *p < 0.05 and **p < 0.01 vs. vehicle group.
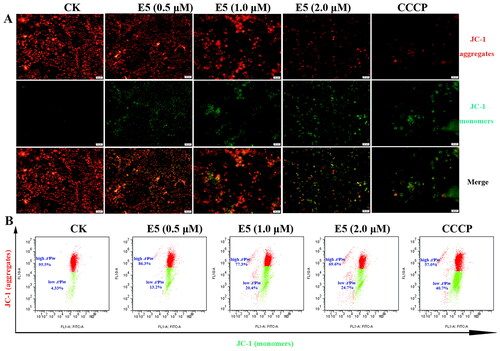
Compound E5 reduced mitochondrial transmembrane potential in A549 cells
Mitochondria play a critical role in the function of cancer cells, and mitochondrial depolarization serves as a precursor in the early stages of apoptosis and proliferation. To further validate the effects of compound E5 on MMP, the fluorescent dye JC-1 was used to detect MMP (ΔΨm). Red fluorescence indicates the formation of J-aggregates with high ΔΨm in normal polarized mitochondria, while green fluorescence indicates the formation of J-monomers with low ΔΨm in depolarized mitochondria. As shown in , a dose-dependent loss of ΔΨm was observed in A549 cells treated with compound E5 compared to the control group. The ratio of cells with low membrane potential exposed to E5 (2.0 µM) increased from 4.33% to 24.7% after 12 h of treatment ().
Figure 6. The mitochondrial transmembrane potential was analysed in E5-treated A549 cells by JC-1 staining. (A) Luminescence microscope analysis. (B) Flow cytometry analysis.
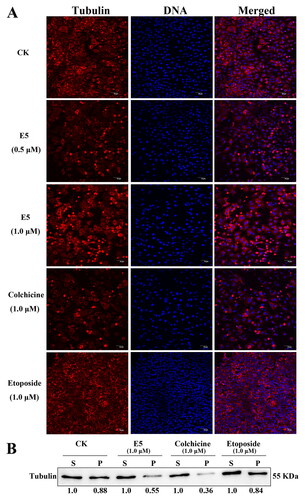
Compound E5 enhanced tubulin depolymerization and disrupted cell division
In conjunction with previous reports, disrupting the dynamic balance between microtubulin depolymerization and polymerization can impede the normal progression of cell division, ultimately leading to cell death. To validate the impact of drugs on tubulin, the disaggregation or aggregation of microtubules was observed by confocal laser scanning microscopy after treatment with the compound. As illustrated in , the results for compound E5 at concentrations of 0.5 and 1.0 µM mirrored those of the colchicine treatment group (1.0 µM), inducing significant tubulin depolymerization. In contrast, the etoposide (1.0 µM) group did not show such an effect, a finding further supported by additional western blot experiments ().
Figure 7. Effects of E5 (0.5 and 1.0 µM), etoposide (1.0 µM), and colchicine (1.0 µM) on interphase microtubules of A549 cells. (A) Tubulin was tagged with Cy3-labeled goat anti-mouse IgG (H + L) (red) and DAPI-stained cell nuclei (blue). Images were captured using a confocal fluorescence microscope. (B) E5 affected the assembly of microtubules in vitro. A549 cells were treated with E5 (1.0), colchicine (1.0 μM), and etoposide (1.0 μM) for 24 h, cytosolic (S, soluble) and cytoskeletal (P, polymerized tubulin) tubulin fractions were separated and immunoblotted with antibody against β-tubulin.
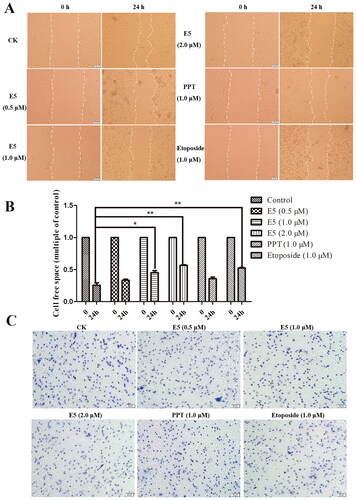
Effect of E5 on cell migration and invasion of A549 cells
We further investigated the impact of E5 on A549 cell migration through wound healing and transwell invasion experiments. Cells were treated with the positive drugs PPT (1.0 µM), etoposide (1.0 µM), and E5 (0.5, 1.0, and 2.0 µM) for 24 h. In the cell migration assay, images were captured at 0 and 24 h. In the wound healing test (), cells in the vehicle group gradually occupied the cell-free space within the channel over time, and the cell channel almost completely healed after 24 h. In contrast, in the E5 treatment group, cell scratch healing was significantly slowed dose dependent, especially at a concentration of 2.0 µM. Meanwhile, the etoposide group also exhibited a strong inhibitory effect. The results of the transwell experiment also demonstrated that E5 effectively inhibited cell invasion, the effect being significantly better than the positive drugs PPT and etoposide at a concentration of 1.0 µM ().
Figure 8. Wound healing migration assay and transwell invasion assay of A549 cells. (A) Cells were incubated with positive drugs PPT (1.0 µM), etoposide (1.0 µM), and E5 (0.5, 1.0, and 2.0 µM) for 24 h. The images were photographed at 0 and 24 h. (B) Relative inhibition of cell migration. The percentage of inhibition was calculated relative to the control group. (C) Representative images of the transwell assay at 24 h.
Effect of E5 on PI3K/Akt expression in A549 cells
Observing the ability of compound E5 to induce apoptosis in A549 cells, we explored its potential mechanisms. The PI3K/Akt pathway controls cancer cell growth, development, and movement. In , molecular docking of E5 with Akt (PDB code: 3cqw) was conducted. The results revealed that compound E5 docked well with 3cqw through interactions, such as two hydrogen bonds, pi-alkyl, and carbon-hydrogen interactions. The colony formation assay showed that E5-treated A549 cells exhibited a lower clone formation rate and a smaller clone size than the control and PPT-treated group cells (). Furthermore, to investigate the correlation between cell apoptosis and the PI3K/Akt pathway, A549 cells were treated with LY294002 (1.0 µM, a PI3K inhibitor), PPT (1.0 µM), and E5 (0.1, 1.0, and 2.0 µM) for 24 h. As shown in , the expression of pPI3K and pAkt was down-regulated dose-dependent after E5 treatment. LY294002 treatment also resulted in a notable reduction in the pPI3K/PI3K and pAkt/Akt ratio.
Figure 9. Binding mode of compound E5 at the colchicine site of Akt (PDB code: 3cqw) and effects of compound E5 on the PI3K/Akt signalling pathway in A549 cells. Cells were incubated with the positive drugs PPT (1.0 µM), LY294002 (1.0 µM), and E5 (0.1, 0.5, 1.0, and 2.0 µM) for 24 h. (A) 2D image of the interaction between E5 and amino acid residues of the nearby active site. (B) Relative inhibition of cell colony. (C) 3D image of E5 was inserted into the tubulin binding site. The images were photographed at 0 and 24 h. (D) Western blot analyses of PI3K, pPI3K, Akt, and pAkt proteins in A549 cells separated by SDS-PAGE and GAPDH were used as an internal control.
Compound E5 exhibited antitumour activity in vivo
Finally, we used a xenograft tumour model in nude mice to assess the anticancer properties of compound E5 through the transplantation of A549 cells. As illustrated in , on days 7, 9, 11, 13, and 15, xenograft tumour growth was significantly inhibited by high-dose E5 (4 mg/kg) compared to the control (DMSO) group. However, there were no significant changes in the body weight of the mice (). Furthermore, both compound E5 and etoposide, at the same dose, exhibited a specific inhibitory effect on tumour growth ().
Figure 10. Compound E5 suppresses tumour growth of A549 xenografts in BALB/c nude mice. (A) Diagram of the administration pattern. (B) Tumour images were recorded after 15 days of treatment. (C and D) Body weight and tumour size were measured every two days. (E) The percentage of tumor weight was calculated.
Discussion
Cancer remains a formidable global health challenge, with high incidence and mortality rates contributing to nearly 10 million deaths in 2020 (Ferlay et al. Citation2021). Natural compounds have proven effective in identifying potential treatments for this deadly disease. However, the lack of tumour selectivity and significant toxic side effects in many natural compounds hinder their clinical development (Zi et al. Citation2019). In recent decades, molecularly targeted drug therapy has emerged as a promising approach to cancer treatment, with particular emphasis on microtubule protein inhibitors (Khwaja et al. Citation2021). PPT, a representative natural product, has not been used in clinical settings due to its high toxicity. However, studies suggest that rational modifications at the C-4 position could yield PPT derivatives with reduced toxicity and increased efficacy (Bromberg et al. Citation2003; Zi et al. Citation2019). In this context, we designed and synthesized 16 PPT derivatives with modifications at the C-4 position to enhance their antiproliferative activity while minimizing toxicity. Almost all compounds exhibited robust antiproliferative effects against four cancer cell lines while showing lower toxicity to normal cells. This suggests the potential of these modified derivatives as promising candidates for further development in cancer treatment.
Microtubules, consisting of microtubule protein dimers, are tubular organelles constituting a polar cytoskeleton crucial for maintaining cell shape, intracellular transportation, and mitosis (Gudimchuk et al. Citation2020). Microtubule protein inhibitors play a pivotal role in cancer treatment by influencing the structure and polymerization state. This selective targeting of rapidly proliferating cancer cells reduces the toxicity of normal cells. In particular, compounds ID09 and ID33, have demonstrated their effectiveness in targeting the tubulin colchicine binding site (Cui et al. Citation2020). These inhibitors induce apoptosis in tumour cells by impeding tubulin polymerization and disrupting microtubule and microfilament formation.
Similarly, in our study, the optimized PPT derivative E5 could induce S and G2/M phase cell cycle arrest in a dose- and time-dependent manner, acting as an accelerator of tubulin depolymerization. Further investigation revealed that compound E5 effectively competes for the colchicine site in tubulin. This mechanism underscores the potential of E5 as a microtubule protein inhibitor with implications for cancer therapy.
In recent decades, significant progress has been made in understanding the molecular mechanisms underlying various genetic factors in human cancer. Essential cell processes such as metabolism, cell cycle regulation, differentiation, and survival are heavily influenced by protein kinases (Jiang et al. Citation2020). Among these, Akt kinase stands out as a critical molecule in the PI3K signalling pathway, playing a pivotal role in controlling various cellular activities, including cell apoptosis, viability, growth, gene expression, glucose utilization, ribosomal activity, and cell movement (Shtilbans et al. Citation2008; Ma et al. Citation2021).
In this study, we investigated the antitumor mechanism of E5 and observed its ability to induce cell apoptosis and depolarize cell mitochondria. Given the crucial role of the PI3K/Akt pathway in controlling cancer cell growth, development, and movement (Jiang et al. Citation2020), we further explored the regulatory effect of E5 on this signalling pathway. Encouragingly, compound E5 down-regulated the expression of PI3K and AKT. This observation provides insight into the molecular basis for the inhibition of lung cancer cell growth by E5, shedding light on its potential as an effective agent in cancer therapy. Last, given that the in vivo study in this study remains preliminary, further research is required for its enhancement. This includes investigating whether E5 possesses favourable hemocompatibility, animal tolerance, and appropriate pharmacokinetic properties.
Conclusions
Sixteen PPT derivatives were synthesized, and their cytotoxicity against human cancer cell lines was assessed. Among them, compound E5 showed significant antitumor activity, particularly against A549 cells, surpassing the efficacy of PPT. All derivatives showed low cytotoxicity to normal cells. Further studies revealed that E5 induced cell cycle arrest and inhibited tubulin polymerization. Molecular docking confirmed the competition of E5 with colchicine for tubulin binding. E5 also induced early apoptosis, inhibited migration and invasion, and down-regulated PI3K/Akt signalling pathway proteins in A549 cells. These positions compound E5 as a potential therapeutic agent for lung cancer, pending further mechanistic exploration.
Authors′ contributions
W.X.S., and P.J. contributed to the study′s conception and design. W.X.S., Y.J.G., B.B.C., and J.H.W. performed compound synthesis and identification. J.X.G., B.B.C. and J.H.W. performed cell culture and western blots. Y.J.G., B.B.C., and J.X.G. analyzed and interpreted the data. W.X.S., Y.J.G., and P.J. wrote the first draft of the manuscript. The revised manuscript was written through the contributions of all authors. All authors have read and approved the final version of the manuscript.
Supplemental Material
Download TIFF Image (2.5 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data supporting the findings of this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Abd Emoniem N, Mukhtar RM, Ghaboosh H, Elshamly EM, Mohamed MA, Elsaman T, Alzain AA. 2023. Turning down PI3K/AKT/mTOR signalling pathway by natural products: an in silico multi-target approach. SAR QSAR Environ Res. 34(2):163–182. doi: 10.1080/1062936X.2023.2181392.
- Aggarwal C, Somaiah N, Simon G. 2012. Antiangiogenic agents in the management of non-small cell lung cancer: where do we stand now and where are we headed? Cancer Biol Ther. 13(5):247–263. doi: 10.4161/cbt.19594.
- Bai G, Zhao D, Ran X, Zhang L, Zhao D. 2020. Novel hybrids of podophyllotoxin and coumarin inhibit the growth and migration of human oral squamous carcinoma cells. Front Chem. 8:626075–626087. doi: 10.3389/fchem.2020.626075.
- Bromberg KD, Burgin AB, Osheroff N. 2003. A two-drug model for etoposide action against human topoisomerase II alpha. J Biol Chem. 278(9):7406–7412. doi: 10.1074/jbc.M212056200.
- Chang TH, Szabo E. 2002. Enhanced growth inhibition by combination differentiation therapy with ligands of peroxisome proliferator-activated receptor-gamma and inhibitors of histone deacetylase in adenocarcinoma of the lung. Clin Cancer Res. 8(4):1206–1212.
- Cui YJ, Liu C, Ma CC, Ji YT, Yao YL, Tang LQ, Zhang CM, Wu JD, Liu ZP. 2020. SAR investigation and discovery of water-soluble 1-methyl-1,4-dihydroindeno[1,2-c]pyrazoles as potent tubulin polymerization inhibitors. J Med Chem. 63(23):14840–14866. doi: 10.1021/acs.jmedchem.0c01345.
- Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M, Znaor A, Bray F. 2021. Cancer statistics for the year 2020: an overview. Int J Cancer. 149(4):778–789. doi: 10.1002/ijc.33588.
- Gordaliza M, Castro MA, del Corral JM, Feliciano AS. 2000. Antitumor properties of podophyllotoxin and related compounds. Curr Pharm Des. 6(18):1811–1839. doi: 10.2174/1381612003398582.
- Gudimchuk NB, Ulyanov EV, O’Toole E, Page CL, Vinogradov DS, Morgan G, Li G, Moore JK, Szczesna E, Roll-Mecak A, et al. 2020. Mechanisms of microtubule dynamics and force generation examined with computational modeling and electron cryotomography. Nat Commun. 11(1):3765–3779. doi: 10.1038/s41467-020-17553-2.
- Guo JX, Zhou YH, Huang YQ, Liu GJ, Pan JX. 2017. Synergistic effects of chlorambucil and TRAIL on apoptosis and proliferation of Raji cells. Eur Rev Med Pharmacol Sci. 21(20):4703–4710.
- Jiang N, Dai Q, Su X, Fu J, Feng X, Peng J. 2020. Role of PI3k/Akt pathway in cancer: the framework of malignant behavior. Mol Biol Rep. 47(6):4587–4629. doi: 10.1007/s11033-020-05435-1.
- Khwaja S, Kumar K, Das R, Negi AS. 2021. Microtubule associated proteins as targets for anticancer drug development. Bioorg Chem. 116:105320–105335. doi: 10.1016/j.bioorg.2021.105320.
- Ma Y, Yang X, Han H, Wen Z, Yang M, Zhang Y, Fu J, Wang X, Yin T, Lu G, et al. 2021. Design, synthesis and biological evaluation of anilide (dicarboxylic acid) shikonin esters as antitumor agents through targeting PI3k/Akt/mTor signaling pathway. Bioorg Chem. 111:104872–104883. doi: 10.1016/j.bioorg.2021.104872.
- Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. 2017. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull. 7(3):339–348. doi: 10.15171/apb.2017.041.
- Mouna R, Broisat A, Ahmed A, Debiossat M, Boumendjel A, Ghezzi C, Kabouche Z. 2022. Antiproliferative activity, cell-cycle arrest, apoptotic induction and LC-HRMS/MS analyses of extracts from two Linum species. Pharm Biol. 60(1):1491–1501. doi: 10.1080/13880209.2022.2102196.
- Panda M, Biswal BK. 2019. Cell signaling and cancer: a mechanistic insight into drug resistance. Mol Biol Rep. 46(5):5645–5659. doi: 10.1007/s11033-019-04958-6.
- Pitts SL, Jablonksy MJ, Duca M, Dauzonne D, Monneret C, Arimondo PB, Anklin C, Graves DE, Osheroff N. 2011. Contributions of the d-ring to the activity of etoposide against human topoisomerase II alpha: potential interactions with DNA in the ternary enzyme-drug-DNA complex. Biochem. 50(22):5058–5066. doi: 10.1021/bi200531q.
- Salem FS, Badr MO, Neamat-Allah AN. 2011. Biochemical and pathological studies on the effects of levamisole and chlorambucil on Ehrlich ascites carcinoma-bearing mice. Vet Ital. 47(1):89–95.
- Shah Z, Gohar UF, Jamshed I, Mushtaq A, Mukhtar H, Zia-Ui-Haq M, Toma SI, Manea R, Moga M, Popovici B. 2021. Podophyllotoxin: history, recent advances and future prospects. Biomol. 11(4):603–629. doi: 10.3390/biom11040603.
- Shtilbans V, Wu M, Burstein DE. 2008. Current overview of the role of Akt in cancer studies via applied immunohistochemistry. Ann Diagn Pathol. 12(2):153–160. doi: 10.1016/j.anndiagpath.2007.12.001.
- Siegel RL, Miller KD, Fuchs HE, Jemal A. 2022. Cancer statistics, 2022. CA Cancer J Clin. 72(1):7–33. doi: 10.3322/caac.21708.
- Sk UH, Dixit D, Sen E. 2013. Comparative study of microtubule inhibitors-estramustine and natural podophyllotoxin conjugated PAMAM dendrimer on glioma cell proliferation. Eur J Med Chem. 68:47–57. doi: 10.1016/j.ejmech.2013.07.007.
- Tshering Vogel DW, Zbaeren P, Thoeny HC. 2010. Cancer of the oral cavity and oropharynx. Cancer Imaging. 10(1):62–72. doi: 10.1102/1470-7330.2010.0008.
- Xiao J, Gao M, Sun Z, Diao Q, Wang P, Gao F. 2020. Recent advances of podophyllotoxin/epipodophyllotoxin hybrids in anticancer activity, mode of action, and structure-activity relationship: an update (2010-2020). Eur J Med Chem. 208:112830–112853. doi: 10.1016/j.ejmech.2020.112830.
- Zhao W, Cong Y, Li HM, Li S, Shen Y, Qi Q, Zhang Y, Li YZ, Tang YJ. 2021. Challenges and potential for improving the druggability of podophyllotoxin-derived drugs in cancer chemotherapy. Nat Prod Rep. 38(3):470–488. doi: 10.1039/d0np00041h.
- Zi CT, Gao YS, Yang L, Feng SY, Huang Y, Sun L, Jin Y, Xu FQ, Dong FW, Li Y, et al. 2019. Design, synthesis, and biological evaluation of novel biotinylated podophyllotoxin derivatives as potential antitumor agents. Front Chem. 7:434–447. doi: 10.3389/fchem.2019.00434.