Abstract
Dendritic cells are professional antigen presenting cells linking innate and adaptive immune responses. Different dendritic cell subsets were identified in human lung, each with their own functional characteristics. As innate and adaptive immune responses are activated in Chronic Obstructive Pulmonary Disease (COPD), dendritic cells could play a role in the pathogenesis of this disease. Indeed, cigarette smoke appears to modulate dendritic cell function in vitro and alters dendritic cell numbers and function in cigarette smoke exposed mice. The number of pulmonary dendritic cells differs between COPD patients, smokers and non-smokers. Moreover, the number of Langerhans-type dendritic cells increases with the severity of the disease. In this review we will discuss the scientific evidence regarding the role of dendritic cells in COPD and we will put forward the concept of modulation of dendritic cell differentiation and function as a crucial step in the pathogenesis of COPD.
INTRODUCTION
Chronic Obstructive Pulmonary Disease (COPD) is an inflammatory disease of the airways and lung parenchyma due to the inhalation of noxious particles and gases, and is associated with an abnormal systemic inflammatory response. Tobacco smoke and pollutants originating from biomass combustion are the most important environmental risk factors. COPD is characterized by an airflow limitation that is not fully reversible, and that is usually progressive, even after smoking cessation (Citation[1]). The airflow limitation is caused by an inflammation of the small airways (i.e., bronchiolitis) combined with a destruction of the alveolar septa (i.e., emphysema), the latter resulting in enlargement of the alveolar air spaces and an impaired gas exchange. The exact pathogenetic mechanisms of the ongoing pulmonary inflammation and damage in COPD, even after the initial inciting agent has disappeared (i.e. smoking cessation), are poorly understood. Latent viral respiratory infections, chronic bacterial colonization of the lower airways, repetitive infectious exacerbations, auto-immune responses against changed epitopes in the lung and certain genetic predispositions are proposed as important driving mechanisms of the perpetuating inflammation in patients with COPD (Citation[2]).
The lungs of COPD patients are infiltrated with cells of the innate immune system such as neutrophils and macrophages (Citation[3], Citation[4]), but there is also evidence for an activated adaptive immune response with accumulation of CD8+ T cells, B cells and the presence of lymphoid follicles (5–7).
Dendritic cells (DCs) are professional antigen presenting cells, originating from the hematopoietic system. In general, DCs are divided in two main populations: myeloid DCs (mDCs) and plasmacytoid DCs (pDCs). mDCs are thought to originate from myeloid precursors, whereas pDCs originate from lymphoid progenitors, although there is evidence that bone marrow pDCs could also differentiate into mDCs (Citation[8]).
DCs are present in the skin and mucosal surfaces, where they continuously sample antigens. Simultaneously, they sense the environment for microbial compounds and danger signals, using pathogen recognition receptors (Toll-like receptors (TLR), Nod-Like receptors and C-type lectins) and damage associated pattern recognition receptors. DCs integrate the information from the antigens and danger signals and subsequently migrate towards the draining lymph nodes, whilst processing the antigen and upregulating the expression of co-stimulatory molecules (i.e., maturation) (Citation[9]).
In the lymph nodes, mature DCs present the antigen on Major Histocompability (MHC) class II molecules to naïve CD4+ T lymphocytes. In addition, they can present and cross-present antigens to CD8+ T cells using MHC class I molecules. This immunological synapse allows for the selection of lymphocytes with the antigen-matched T cell receptor. Depending on the degree of expression of the co-stimulatory molecules and the cytokines produced by the DC, the lymphocytes can undergo clonal expansion. Moreover, the co-stimulatory process can steer the expanding T cells towards a T-helper (Th) 1, Th 2, Th 17 or regulatory T cell response. Thus, DCs are important orchestrators of immunity, linking innate and adaptive immune responses (Citation[10]).
Human pulmonary dendritic cell subsets
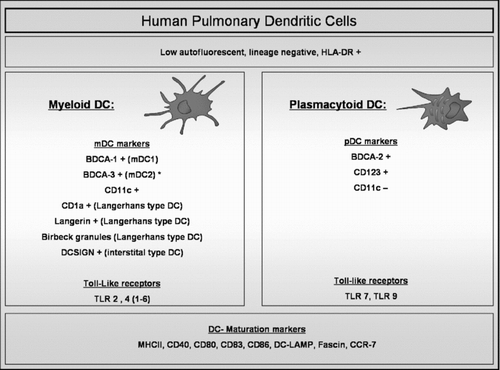
The exact definition of the different DC subtypes in the human lung is incomplete. Two groups identified myeloid DCs and plasmacytoid DCs in mononuclear single cell suspensions from digested human lung tissue by flowcytometry () (Citation[11], Citation[12]). Both groups defined DCs within the low autofluorescent, lineage negative (−ve), HLA-DR positive (+ve) cell population. Demedts et al used Blood Dendritic Cell Antigen (BDCA) 1, 3, and 2 to identify mDC type 1, mDC type 2 and pDCs respectively. In contrast, Masten et al. defined mDCs as CD11c +ve cells that were CD1a +ve or BDCA1 +ve, and pDCs as CD11c −ve, CD123 +ve cells. mDCs and pDCs were also identified in human broncho-alveolar lavage (BAL) fluid. In BAL, BDCA-3 was also present on pDCs, suggesting that this marker does not delineate a separate mDC2 subset in all compartments of the lung (Citation[13]). mDCs and mature DCs were identified in induced sputum (Citation[14], Citation[15]). In addition, Langerhans-type DCs (defined as langerin +ve or Birbeck granule +ve DCs) (Citation[11], Citation[16], Citation[17]) and DC-SIGN (CD209) +ve interstitial-type DCs (Citation[18]) were identified in the human lung by electron microscopy or immunohistochemistry.
Figure 1 Human Pulmonary Dendritic cells. Pulmonary dendritic cells (DCs) are identified by flow cytometry within the cell population that is low autofluorescent (to distinguish them from macrophages which are high autofluorescent), HLA-DR +ve and that does not express lineage markers (CD3, CD14, CD16, CD19, CD20 and CD56). Within this cell population, different markers identify plasmacytoid (pDC) and different myeloid DC (mDC) subsets. mDC and pDC express different types of Toll-like receptors (TLR). Mature DCs are identified by upregulation of specific maturation markers such as co-stimulatory molecules and the chemokine receptor CCR7. (BDCA: Blood Dendritic Cell Antigen; DC-SIGN: Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin; MHC: Major Histocompability Complex; DC-LAMP: Dendritic Cell Lysosome-Associated Membrane glycoprotein). (*): BDCA-3 is also expressed on pDCs in broncho-alveolar lavage fluid.
Evidence from in vitro studies suggests that monocytes are important precursors of DCs. (Citation[19], Citation[20]) When studying human pulmonary mDCs, one should keep in mind that antigen presenting cells partially consist of a spectrum of specialized differentiated monocytes ranging from DCs to macrophages (Citation[21]). The exact interrelationship of the different markers of monocytes and DCs and how they reflect different subsets of pulmonary DC (precursors) largely remains to be elucidated.
It is tempting to speculate that, within these different DC subsets, carrying different surface markers, functional specialization does occur. Indeed, human pulmonary mDC type 1 and 2 were shown to release proinflammatory cytokines (TNF-α, IL-1β, IL-6 and IL-8) in response to Toll-like receptors (TLR)-2 and TLR-4 ligands, whereas pulmonary pDCs released high amounts of interferon-α in response to the TLR-9 ligand CpG oligonucleotides (Citation[22]). Moreover, mDCs derived from human lung digests or from BAL fluid were shown to be strong inducers of T cell proliferation in a mixed leukocyte reaction, while pDCs hardly induced any T cell proliferation (Citation[22], Citation[23]). Functional specialization of DC subsets has also been described in other organs such as the skin with for instance altered susceptibility to TLR ligands in Langerhans-type DC versus interstitial-type DC (Citation[24]).
Functional differences in dendritic cells exposed to cigarette smoke
Several in vitro studies have evaluated the effect of nicotine and cigarette smoke extract (CSE) on the maturation process and T cell stimulatory capacity of mDCs. Human monocyte-derived DCs, which have a predominant interstitial phenotype, showed increased maturation and increased IL-10 and IL-12 production upon exposure to a high dose nicotine (Citation[25]), whereas a low dose of nicotine resulted in a decreased immature DC dependent T- cell proliferation and a decreased IL-12 production after Lipopolysaccharide (LPS) stimulation (Citation[26]). These nicotine-exposed DCs were indeed less capable to induce differentiation of naïve T cells into T-helper 1 cells (Citation[27]). Adding CSE to monocyte-derived DCs resulted in an attenuation of the maturation process with reduced expression of costimulatory molecules and a reduced expression of CCR7, a chemokine receptor involved in DC trafficking towards lymphoid tissues. Moreover, the DC-induced T cell proliferation was significantly reduced when DCs were cultured in the presence of CSE. Maturing CSE exposed DC produced less IL-12 and IL-23 and skewed the T cell response towards a Th 2 profile (Citation[28], Citation[29]). In addition, the production of the neutrophil attracting chemokine IL-8 was upregulated in these cigarette smoke exposed maturing monocyte derived DC (Citation[30]). In summary, these in vitro data suggest that cigarette smoke directly inhibits the process of monocyte-derived DC maturation and skews the adaptive immune response away from Th1 towards a Th2 response.
In a mouse model of COPD, upregulation of pulmonary DC maturation markers was accompanied by the characteristic airway inflammation and emphysema (Citation[31]). In contrast, Robbins et al found, using another protocol of smoke exposure, decreased expression of the maturation marker CD80 on pulmonary DC in the absence of pulmonary inflammation (Citation[32]). Moreover, using the latter protocol, there was an impaired DC maturation in murine thoracic lymph nodes of smoke exposed mice compared to air exposed controls (Citation[33]). Interestingly, ex vivo data from human mDC in BAL showed an increased expression of maturation markers by mDC combined with a decrease of CCR7 expression in smokers compared to non-smokers (Citation[34]), whereas smoking reduced the number of mature DCs in bronchial biopsies of asthma patients (Citation[35]). The issue of altered DC maturation by smoking and in COPD is controversial due to these conflicting data and due to the fact that there is currently no single established marker that clearly defines the mature status of human pulmonary DCs.
Apart from the integration of danger signals and antigens, DCs are also capable of producing substantial amounts of proteinases, contributing to the proteinase-antiproteinase imbalance in COPD. Mouse DC showed indeed an increase of Matrix metalloproteinase-12 production upon cigarette smoke exposure, and could thereby contribute to the pathogenesis of emphysema (Citation[36]).
Dendritic cells in smokers and COPD patients
There have been only a few studies addressing the number and distribution of DCs in the lungs of smokers and patients with COPD (). Most of these descriptive studies have a cross-sectional design, implicating that the sequence of events can not be established and that any evidence for causality is weak. At first sight, some data in the literature do appear discrepant or even contradictory, but this may be due to differences in the area of interest (bronchial biopsies sampling large airways versus surgical resection specimens sampling small airways and parenchyma versus BAL sampling the alveolar lumen), differences in the examination techniques (electron microscopy, flowcytometry or immunohistochemistry) or differences in the immunohistochemical markers used to identify and enumerate the DCs. It is also critical to discriminate between the effects of smoking per se on DC numbers, phenotypic markers or functions versus the disease-specific effects of COPD on DCs, irrespective of the current smoking status.
Table 1 Studies of Pulmonary Dendritic cells in Smokers and COPD patients
In the large airways (trachea, bronchi), sampled by bronchoscopy with endobronchial biopsies, the number of CD1a+ve DCs was evaluated in healthy smoking controls and current smoking COPD patients, showing no significant differences between groups (Citation[37]). Others evaluated the number of DCs in large airways using electron microscopy, based upon morphologic criteria derived from cultured BDCA-1+ve DC, and showed a significant decrease in the number of DCs in the large airways of smoking COPD patients versus ex-smoking COPD patients and never smoking controls (Citation[38]).
In the small airways (bronchioli), which is the main location responsible for airway obstruction in COPD, the number of DC was evaluated using immunohistochemical staining for CD1a and BDCA-1, showing no difference between smokers and non-smokers (Citation[16]). In patients with COPD, however, a significant increase in the number of CD207 (langerin) positive cells was found compared to never smokers and healthy smokers (without airway obstruction), suggesting an accumulation of Langerhans-type DC in COPD. Moreover, the number of langerin +ve DC further increased with the severity of the disease (Citation[39]).
Studies sampling BAL fluid evaluated the number of DC between never-smokers and smokers without COPD, showing a significant increase in the expression of langerin and CD1a on myeloid DCs as well as an increase in Birbeck granule positive Langerhans-type DC in smokers versus non-smokers (Citation[17], Citation[34]). Finally, in the alveolar parenchyma, the number of CD1a +ve DCs was increased in smokers versus never smokers, whereas the number of BDCA-1 +ve DCs was not different (Citation[16]).
In summary, evidence points towards an accumulation of mDCs with Langerhans-type cell markers (langerin, CD1a and Birbeck granules) in the small airways and alveoli of smokers and COPD patients. These langerin +ve Langerhans-type DCs are mainly regarded as immature DCs (Citation[40]). This is in agreement with data from experimental models of COPD, in which mice are chronically exposed to cigarette smoke and develop manifest pulmonary inflammation and emphysema. A clear accumulation of mDCs was seen in the BAL fluid and in single cell suspensenions from digested lungs of these mice (Citation[31]). A recent immunohistochemical study confirmed the accumulation of DCs in cigarette smoke exposed mice (Citation[41]). A different protocol of smoke exposure resulted in a decrease of the number of DCs in the lung in the absence of pulmonary inflammation (Citation[32]). Evidence from the mouse model of COPD and from human lung tissue suggests that an activated macrophage inflammatory protein 3α (MIP3alpha)/CC-chemokine ligand (CCL20) – CC-chemokine receptor 6 (CCR6)- axis in COPD is responsible for the accumulation of mDCs in the lung (Citation[39], Citation[42]).
Modulation of DC differentiation and function in COPD pathogenesis
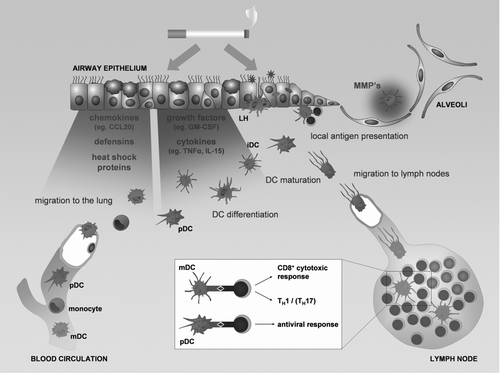
Dendritic cell precursors (blood DCs and monocytes) enter the lung from the blood stream under the influence of chemokines, defensins and heat shock proteins produced by the lung tissue (see ). Cigarette smoke could (directly or indirectly by inducing growth factor and cytokine production in the surrounding lung tissue) modulate the differentiation of DC precursors, leading to an altered composition of the DC population in the lungs of smokers and COPD patients. Current evidence shows an accumulation of Langerhans-type DCs in smokers and COPD patients, suggesting a stimulation of the differentiation process of DC precursors towards this DC subset. Interestingly, Transforming Growth Factor beta (TGFb) and Tumor Necrosis Factor alpha (TNFa), which are known to drive Langerhans differentiation in vitro are increased in the airways of COPD patients (Citation[43], Citation[44]). Whether other Langerhans promoting differentiaton factors—such as Notch ligand Delta-1, Interleukin-15, Activin-A and RANK-ligand - are involved in pulmonary Langerhans-type DC differentiation remains to be elucidated. It is currently unknown if other DC subsets such as interstitial-type DCs and pDCs accumulate in COPD.
Figure 2 The role of dendritic cells in the pathogenesis of COPD. Chemokines, defensins and heath shock proteins produced by the lung tissue in response to the damaging effect of cigarette smoke attract circulating dendritic cells precursors (blood myeloid dendritic cells (mDC), plasmacytoid dendritic cells (pDC) and monocytes). The differentiation of the DC precursors into different subsets will be modulated by the growth factors and cytokines released by the damaged lung tissue, resulting in an altered DC population in the lung with increased numbers of Langerhans type DCs (LH). Together with the interstitial type DCs (iDC) and the pDCs, they sample antigens and sense for danger signals in the mucosal area. Maturing DCs migrate towards the lymph nodes where they present antigens to lymphocytes and orchestrate the outcome of the adaptive immune response. DCs can also present their antigens locally in the airway mucosa. The process of maturation can be directly attenuated by cigarette smoke. The altered DC population can skew the adaptive immune response towards a T helper 1, CD8+ cytotoxic T cell or T helper 17 profile. In addition DCs can contribute to the protease-antiprotease imbalance by producing metalloproteinases (MMP's), involved in the development of emphysema.
At the epithelial surface, DCs are capable of sensing danger signals and take up antigens to process them. However, little is known about the influence of cigarette smoke on the expression and function of innate receptors of DCs, including TLR and lectin-like receptors. Once the DC has sampled the antigen, it will process the antigen, upregulate the expression of CC-chemokine receptor 7 (CCR7) and migrate towards the secondary lymphoid organs (i.e. mediastinal lymph nodes). As discussed above, in vitro data suggest that cigarette smoke reduces the ability of DCs to mature, giving a possible mechanism for higher susceptibility for microbial infection in smokers (Citation[45]) and the occurrence of low grade infection or colonization in smoking COPD patients (Citation[46]). This microbial stimulus can contribute towards a perpetuating damaging inflammation which will in turn release more danger signals, eventually increasing the number of mature DCs in COPD. In addition, the reduced expression of CCR7 combined with increased expression of maturation markers on BAL DCs of smokers (Citation[34]) suggest that activated DCs could be retained in the airway mucosa and present their antigens locally, inducing lymphocyte proliferation and lymphoid neogenesis in the lung and contributing to the ongoing inflammation (Citation[47]). However, the whole issue on DC maturation in COPD is controversial with conflicting data as discussed above and is an important focus for future research (Citation[48], Citation[49]).
Furthermore, the altered composition of the DC population, with functionally modified DC subsets, could skew the resulting adaptive immune response away from tolerance and towards ongoing Th1 and cytotoxic CD8+ T cell responses, which are the hallmarks of the chronic adaptive inflammation in COPD (Citation[49]). In accordance, the accumulating Langerhans-type DC subset is known to be a potent activator of these T cell responses (Citation[50], Citation[51]). Moreover, mouse DCs exposed to cigarette smoke extract potentiate CD8+ T cell proliferation in vitro (Citation[52]).
A recent publication suggests a role for Langerhans-type DCs in driving the Th17 response (Citation[53]), which could hypothetically be involved in COPD pathogenesis.
Finally, there are several reports on auto-immune features of COPD with a role for anti-elastin and anti-endothelial antibodies and a blunted regulatory T cell response in COPD patients (54–56). The functionally modified pulmonary DC population could play a crucial role in this process, as pulmonary DCs that are activated in situ by cigarette smoke induced tissue damage and inflammation could break tolerance by presenting and cross-presenting self-antigens, similar to the described role of in situ activated DCs in other auto-immune diseases (Citation[57]).
Perspectives
Further phenotypic evaluation of human pulmonary DCs needs to be carried out in order to identify different subsets of DC, which can be separate stages of differentiation from common blood DC precursors. Evaluating the effects of cigarette smoke on the differentiation of DCs and on the maturation of Langerhans-type DC compared to interstitial-type DC and plasmacytoid DC is mandatory to understand the role of the DC in smoking-related diseases. Moreover, the role of the plasmacytoid DC in COPD remains to be elucidated, both during stable disease and at exacerbations. Since plasmacytoid DC have important antiviral and tolerogenic properties, they could play a role in the pathogenesis of COPD.
Declaration of interest
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
This work was supported by the Fund for Scientific Research in Flanders (research projects G.0011.03 and G0343.01N) and by project grant 01251504 from the Concerted Research Initiative of the Ghent University. GRVP is a doctoral research fellow of the Fund for Scientific Research in Flanders
REFERENCES
- Rabe K F, Hurd S, Anzueto A, Barnes P J, Buist S A, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R, van Weel C, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease: GOLD Executive Summary. Am J Respir Crit Care Med 2007; 176: 532–555
- Curtis J L, Freeman C M, Hogg J C. The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc 2007; 4: 512–521
- Lapperre T S, Willems L N, Timens W, Rabe K F, Hiemstra P S, Postma D S, Sterk P J. Small airways dysfunction and neutrophilic inflammation in bronchial biopsies and BAL in COPD. Chest 2007; 131: 53–59
- Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P, Mapp C E, Fabbri L M, Donner C F, Saetta M. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med 1998; 158: 1277–1285
- Saetta M, Di Stefano A, Turato G, Facchini F, Corbino L, Mapp C, Maestrelli P, Ciaccia A, Fabbri L. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157: 822–826
- Hogg J C, Chu F, Utokaparch S, Woods R, Elliott W M, Buzatu L, Cherniack R M, Rogers R M, Sciurba F C, Coxson H O, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 2645–2653
- Gosman M M, Willemse B W, Jansen D F, Lapperre T S, van S A, Hiemstra P S, Postma D S, Timens W, Kerstjens H A. Increased Number of B-Cells in Bronchial Biopsies in COPD. Eur Respir J 2006; 27: 60–64
- Zuniga E I, McGavern D B, Pruneda-Paz J L, Teng C, Oldstone M B. Bone Marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat Immunol 2004; 5: 1227–1234
- Vermaelen K, Pauwels R. Pulmonary dendritic cells. Am J Respir Crit Care Med 2005; 172: 530–551
- Steinman R M, Banchereau J. Taking dendritic cells into medicine. Nature 2007; 449: 419–426
- Demedts I K, Brusselle G G, Vermaelen K Y, Pauwels R A. Identification and characterization of human pulmonary dendritic cells. Am J Respir Cell Mol Biol 2005; 32: 177–184
- Masten B J, Olson G K, Tarleton C A, Rund C, Schuyler M, Mehran R, Archibeque T, Lipscomb M F. Characterization of myeloid and plasmacytoid dendritic cells in human lung. J Immunol 2006; 177: 7784–7793
- Bratke K, Lommatzsch M, Julius P, Kuepper M, Kleine H D, Luttmann W, Christian Virchow J. Dendritic cell subsets in human bronchoalveolar lavage fluid after segmental allergen challenge. Thorax 2007; 62: 168–175
- McCarthy N E, Jones H A, Marks N A, Shiner R J, Ind P W, Al-Hassi H O, English N R, Murray C M, Lambert J R, Knight S C, et al. Inhaled allergen-driven CD1c up-regulation and enhanced antigen uptake by activated human respiratory-tract dendritic cells in atopic asthma. Clin Exp Allergy 2007; 37: 72–82
- Alexis N E, Lay J C, Almond M, Bromberg P A, Patel D D, Peden D B. Acute LPS Inhalation in healthy volunteers induces dendritic cell maturation in vivo. J Allergy Clin Immunol 2005; 115: 345–350
- Soler P, Moreau A, Basset F, Hance A J. Cigarette smoking-induced changes in the number and differentiated state of pulmonary dendritic cells/Langerhans cells. Am Rev Respir Dis 1989; 139: 1112–1117
- Casolaro M A, Bernaudin J F, Saltini C, Ferrans V J, Crystal R G. Accumulation of Langerhans' cells on the epithelial surface of the lower respiratory tract in normal subjects in association with cigarette smoking. Am Rev Respir Dis 1988; 137: 406–411
- Marchal-Somme J, Uzunhan Y, Marchand-Adam S, Kambouchner M, Valeyre D, Crestani B, Soler P. Dendritic cells accumulate in human fibrotic interstitial lung disease. Am J Respir Crit Care Med 2007; 176: 1007–1014
- Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med 1994; 179: 1109–1118
- Leon B, Ardavin C. Monocyte-Derived Dendritic Cells in Innate and Adaptive Immunity. Immunol Cell Biol 2008; 86: 320–324
- Hume D A. Macrophages As APC and the dendritic cell myth. J Immunol 2008; 181: 5829–5835
- Demedts I K, Bracke K R, Maes T, Joos G F, Brusselle G G. Different roles for human lung dendritic cell subsets in pulmonary immune defense mechanisms. Am J Respir Cell Mol Biol 2006; 35: 387–393
- Schaumann F, Muller M, Braun A, Luettig B, Peden D B, Hohlfeld J M, Krug N. Endotoxin augments myeloid dendritic cell influx into the airways in patients with allergic asthma. Am J Respir Crit Care Med 2008; 177: 1307–1313
- van d er, Aar A MG, Sylva-Steenland R MR, Bos J D, Kapsenberg M L, de Jong E C, Teunissen M BM. Cutting edge: Loss of TLR2, TLR4, and TLR5 on Langerhans cells abolishes bacterial recognition. J Immunol 2007; 178: 1986–1990
- Aicher A, Heeschen C, Mohaupt M, Cooke J P, Zeiher A M, Dimmeler S. Nicotine strongly activates dendritic cell-mediated adaptive immunity: potential role for progression of atherosclerotic lesions. Circulation 2003; 107: 604–611
- Nouri-Shirazi M, Guinet E. Evidence for the immunosuppressive role of nicotine on human dendritic cell functions. Immunology 2003; 109: 365–373
- Nouri-Shirazi M. A Possible mechanism linking cigarette smoke to higher incidence of respiratory infection and asthma. Immunol Lett 2006; 103: 167–176
- Vassallo R, Tamada K, Lau J S, Kroening P R, Chen L. Cigarette smoke extract suppresses human dendritic cell function leading to preferential induction of Th-2 priming. J Immunol 2005; 175: 2684–2691
- Kroening P R, Barnes T W, Pease L, Limper A, Kita H, Vassallo R. Cigarette smoke-induced oxidative stress suppresses generation of dendritic cell IL-12 and IL-23 through ERK-dependent pathways. J Immunol 2008; 181: 1536–1547
- Vassallo R, Kroening P R, Parambil J, Kita H. Nicotine and oxidative cigarette smoke constituents induce immune-modulatory and pro-inflammatory dendritic cell responses. Mol Immunol 2008; 45: 3321–3329
- D'hulst A I, Vermaelen K Y, Brusselle G G, Joos G F, Pauwels R A. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J 2005; 26: 204–213
- Robbins C S, Dawe D E, Goncharova S I, Pouladi M A, Drannik A G, Swirski F K, Cox G, Stampfli M R. Cigarette smoke decreases pulmonary dendritic cells and impacts antiviral immune responsiveness. Am J Respir Cell Mol Biol 2004; 30: 202–211
- Robbins C S, Franco F, Mouded M, Cernadas M, Shapiro S D. Cigarette smoke exposure impairs dendritic cell maturation and T cell proliferation in thoracic lymph nodes of mice. J Immunol 2008; 180: 6623–6628
- Bratke K, Klug M, Bier A, Julius P, Kuepper M, Virchow J C, Lommatzsch M. Function-associated surface molecules on airway dendritic cells in cigarette smokers. Am J Respir Cell Mol Biol 2008; 38: 655–660
- Tsoumakidou M, Elston W, Zhu J, Wang Z, Gamble E, Siafakas N M, Barnes N C, Jeffery P K. Cigarette smoking alters bronchial mucosal immunity in asthma. Am J Respir Crit Care Med 2007; 175: 919–925
- Bracke K, Cataldo D, Maes T, Gueders M, Noel A, Foidart J M, Brusselle G, Pauwels R A. Matrix metalloproteinase-12 and cathepsin D expression in pulmonary macrophages and dendritic cells of cigarette smoke-exposed mice. Int Arch Allergy Immunol 2005; 138: 169–179
- Verhoeven G T, Hegmans J PJJ, Mulder P GH, Bogaard J M, Hoogsteden H C, Prins J B. Effects of fluticasone propionate in COPD patients with bronchial hyperresponsiveness. Thorax 2002; 57: 694–700
- Rogers A V, Adelroth E, Hattotuwa K, Dewar A, Jeffery P K. Bronchial mucosal dendritic cells in smokers and ex-smokers with COPD: an electron microscopic study. Thorax 2008; 63: 108–114
- Demedts I K, Bracke K R, Van Pottelberge G R, Testelmans D, Verleden G M, Vermassen F E, Joos G F, Brusselle G G. Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007; 175: 998–1005
- McDermott R, Ziylan U, Spehner D, Bausinger H, Lipsker D, Mommaas M, Cazenave J P, Raposo G, Goud B, de la Salle H, et al. Birbeck granules are subdomains of endosomal recycling compartment in human epidermal langerhans cells, which form where langerin accumulates. Mol Biol Cell 2002; 13: 317–335
- Martorana P A, Lunghi B, Lucattelli M, De C G, Beume R, Lungarella G. Effect of roflumilast on inflammatory cells in the lungs of cigarette smoke-exposed mice. BMC Pulm Med 2008; 8: 17
- Bracke K R, D'hulst A I, Maes T, Moerloose K B, Demedts I K, Lebecque S, Joos G F, Brusselle G G. Cigarette smoke-induced pulmonary inflammation and emphysema are attenuated in CCR6-deficient mice. J Immunol 2006; 177: 4350–4359
- Keatings V M, Collins P D, Scott D M, Barnes P J. Differences in Interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996; 153: 530–534
- Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, Satoh M, Okada Y, Yamasawa F, Nakahara K, Umeda A. Increased expression of transforming growth factor-Beta1 in small airway epithelium from tobacco smokers and patients with Chronic Obstructive Pulmonary Disease (COPD). Am J Respir Crit Care Med 2001; 163: 1476–1483
- Stampfli M R, Anderson G P. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol 2009; 9: 377–384
- Sethi S, Murphy T F. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 2008; 359: 2355–2365
- Veres T Z, Shevchenko M, Krasteva G, Spies E, Prenzler F, Rochlitzer S, Tschernig T, Krug N, Kummer W, Braun A. Dendritic cell-nerve clusters are sites of T cell proliferation in allergic airway inflammation. Am J Pathol 2009; 174: 808–817
- Tsoumakidou M, Jeffery P K. Dendritic cell maturity and obstructive airway disease. Am J Respir Crit Care Med 2007; 176: 833–834
- Tsoumakidou M, Demedts I K, Brusselle G G, Jeffery P K. Dendritic cells in chronic obstructive pulmonary disease: new players in an old game. Am J Respir Crit Care Med 2008; 177: 1180–1186
- Ratzinger G, Baggers J, de Cos M A, Yuan J, Dao T, Reagan J L, Munz C, Heller G, Young J W. Mature human langerhans cells derived from CD34+ hematopoietic progenitors stimulate greater cytolytic t lymphocyte activity in the absence of bioactive IL-12p70, by either single peptide presentation or cross-priming, than do dermal-interstitial or monocyte-derived dendritic cells. J Immunol 2004; 173: 2780–2791
- Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol 2008; 8: 935–947
- Mortaz E, Kraneveld A D, Smit J J, Kool M, Lambrecht B N, Kunkel S L, Lukacs N W, Nijkamp F P, Folkerts G. Effect of cigarette smoke extract on dendritic cells and their impact on T-Cell proliferation. PLoS ONE 2009; 4: e4946
- Mathers A R, Janelsins B M, Rubin J P, Tkacheva O A, Shufesky W J, Watkins S C, Morelli A E, Larregina A T. Differential Capability of human cutaneous dendritic cell subsets to initiate Th17 responses. J Immunol 2009; 182: 921–933
- Lee S H, Goswami S, Grudo A, Song Lz, Bandi V, Goodnight-White S, Green L, Hacken-Bitar J, Huh J, Bakaeen F, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med 2007; 13: 567–569
- Barcelo B, Pons J, Ferrer J M, Sauleda J, Fuster A, Agusti A G. Phenotypic characterisation of T-lymphocytes in COPD: Abnormal CD4+CD25+ regulatory T-lymphocyte response to tobacco smoking. Eur Respir J 2008; 31: 555–562
- Taraseviciene-Stewart L, Douglas I S, Nana-Sinkam P S, Lee J D, Tuder R M, Nicolls M R, Voelkel N F. Is alveolar destruction and emphysema in chronic obstructive pulmonary disease an immune disease?. Proc Am Thorac Soc 2006; 3: 687–690
- Mehling A, Loser K, Varga G, Metze D, Luger T A, Schwarz T, Grabbe S, Beissert S. Overexpression of CD40 ligand in murine epidermis results in chronic skin inflammation and systemic autoimmunity. J Exp Med 2001; 194: 615–628