Abstract
Chromatin modifications and epigenetic regulation are critical for sustained and abnormal inflammatory response seen in lungs of patients with chronic obstructive pulmonary disease (COPD) because the activities of enzymes that regulate these epigenetic modifications are altered in response to cigarette smoke. Cigarette smoke induces chromatin modifications and epigenetic changes by causing post-translational modifications of histone acetyltransferases, and histone/non-histone deacetylases (HDACs), such as HDAC2 and sirtuin 1 (SIRT1), which leads to chromatin remodeling. In this review, we discussed the current knowledge on cigarette smoke/oxidants-induced post-translational modifications of deacetylases (HDAC2 and SIRT1), disruption of HDAC2/SIRT1-RelA/p65 corepressor complex associated with acetylation of RelA/p65, and chromatin modifications (histone H3 phospho-acetylation) leading to sustained pro-inflammatory gene transcription. Knowledge on molecular mechanisms of epigenetic changes in abnormal lung inflammation will help in understanding the pathophysiology of COPD which may lead to the development of novel epigenetic therapies in the near future.
INTRODUCTION
During acute and chronic inflammation, reactive oxygen species (ROS) is produced at rates that overwhelm the capacity of endogenous antioxidant defense system to neutralize them, resulting in oxidative stress. Cigarette smoke which contains more than 1014 free radicals/oxidants and 4,700 chemical compounds is the main cause for the pathogenesis of chronic obstructive pulmonary disease (COPD). Cigarette smoke oxidatively challenges the lung directly via the presence of ROS as well as via the generation and release of ROS by inflammatory/structural cells endogenously and thereby activating various redox sensitive transcription factors, such as nuclear factor kappaB (NF-κ B), leading to increased expression of a range of pro-inflammatory cytokines and chemokines in COPD (Citation[1], Citation[2]). In light of the recent research, we provide current perspectives on the cigarette smoke-induced oxidative modifications of histone/protein deacetylases and chromatin remodeling which are involved in mediating sustained lung inflammation in COPD ().
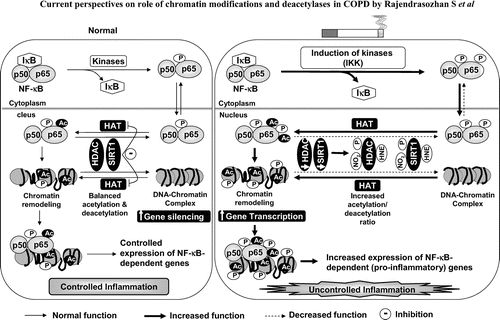
Figure 1 Regulation of NF-κ B-dependent gene expression by histone acetyltransferases (HATs) and histone deacetylases (HDACs). DNA is coiled around the histone proteins and form chromatin structure. Acetylation of histones leads to opening of the chromatin and increasing the accessibility of transcription factors, such as NF-κ B. HATs acetylate histones and RelA/p65 (subunit of NF-κ B) which facilitate the binding RelA/p65-p50 heterodimer onto the pro-inflammatory gene promoters and thereby increasing gene transcription. Deacetylation results in DNA rewinding around histone proteins, and decreasing gene transcription. Thus, HDACs are involved in the maintenance of histone acetylation and deacetylation balance thereby control the transcription of gene. In response to cigarette smoke exposure, deacetylases (especially HDAC2 and SIRT1) are post-translationally modified by phosphorylation, carbonylation, aldehyde adducts formation and/or nitration/nitrosylation leading to increased ubiquitination and subsequent degradation of deacetylases. Decrease in deacetylases results in increased histone acetylation/deacetylation ratio resulting in increased acetylation of histones (chromatin remodeling) and NF-κ B subunits leading to increased transcription of pro-inflammatory mediators. Dotted lines indicate less function (less phosphorylation and acetylation) whereas solid lines indicate increased function (more phosphorylation and acetylation). HAT: Histone acetyltransferase; HDAC: Histone deacetylase; P: Phosphorylation; Ac: Acetylation; 4-HNE: 4-hydroxy-2-nonenal; NO2: Nitrogen dioxide.
EPIGENETIC ALTERATIONS IN LUNG INFLAMMATION AND COPD
Genetically imprintable and heritable alterations in gene expression/function (protein-DNA modifications) induced by mechanisms other than changes in the underlying DNA base sequence termed epigenetics. Epigenetic events include post-translational modifications of the highly conserved core histone proteins (H2A, H2B, H3, and H4) via phosphorylation, acetylation, and ubiquitination (Citation[3], Citation[4]). Histone and DNA methylation can also modulate gene expression without altering the primary gene sequence. Due to availability of very limited information on histone (H3 and H4) and DNA methylation (CpG islands) in COPD, we have focused this perspective on other key aspects of epigenetic phenomena (histone phosphoacetylation and deacetylation) which are shown to be altered in response to oxidants and cigarette smoke in macrophages and lung epithelial cells, and in lungs of smokers and patients with COPD.
Histone acetylation and deacetylation. The ε -amino groups of conserved lysine residues present in histone tails can be subjected to reversible acetylation and deacetylation, which plays an important role in regulating gene transcription (Citation[4]). Acetylation of histones facilitates gene expression by augmenting the recruitment of transcription factors, such as NF-κ B, thereby allowing the access of transcriptional machinery to DNA. Deacetylation of histones suppresses gene expression by forming heterochromatin (tightly packed form of DNA). Cigarette smoke increased the acetylation of histone H3 in macrophages, and human and rodent lungs suggesting that histone acetylation and deacetylation play an important role in chromatin remodeling and consequently in increased and sustained inflammatory response in COPD (5–9).
The enzymes that regulate histone acetylation/deacetylation balance include histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs and HDACs also target non-histone proteins particularly transcription factors to modify their activity and subsequent gene expression. HAT activity is not altered in lungs of COPD, but there is a marked reduction of levels/activities of deacetylases [HDAC2 and sirtuin 1 (SIRT1)] in the lung and alveolar macrophages of patients with COPD (8–10). Despite there is no change in global HAT activity, histones H3 and H4 are acetylated on specific lysine residues in rodent lungs in response to cigarette smoke (Citation[6], Citation[7]), and in lungs of smokers/patients with COPD (Citation[8], Citation[9]). Thus, the acetylation/deacetylation balance is shifted towards acetylation possibly via histone phosphorylation facilitating gene transcription, such as genes for NF-κ B-dependent pro-inflammatory mediators in the lungs of patients with COPD.
Histone phosphorylation. Several HATs have the ability to increase the activity on Ser10-phosphorylated substrates (Citation[11], Citation[12]). It has been shown that phosphorylation of histone H3 at Ser10 correlates with gene transcription associated with activation of rapidly inducible pro-inflammatory gene promoters (Citation[13], Citation[14]). Furthermore, phosphorylation of histone H3 facilitates its interaction with CREB-binding protein (CBP) leading to subsequent acetylation of histone H3 (Citation[15]). Recently, Yang et al. have shown that the levels of phosphorylated (Ser10) and acetylated (Lys9) histone H3 are increased in response to cigarette smoke exposure, which corresponds with increased pro-inflammatory cytokine release in macrophages and mouse lungs (Citation[7]). However, the potential involvement of other serine sites in phospho-acetylation of histones H3 and H4 are not known.
Histone ubiquitination. Ubiquitination refers to the post-translational modification of proteins, including histones, by covalent attachment of one or more ubiquitin, a highly conserved regulatory protein. SUMOylation is an opposite of ubiquitination where SUMO proteins target lysine residues (which are subjected to ubiquitination), thereby hindering ubiquitination (Citation[16]). Acetylation of histone H3/H4 is reported to stimulate de-ubiquitination of histone H2A, which is related to enhanced gene expression (Citation[17]). Not much information is currently available regarding ubiquitination or SUMOylation of histones on any gene promoters in response to oxidants and cigarette smoke though it is conceived that cigarette smoke may trigger ubiquitination and inhibition of SUMOylation on various deacetylases.
HATs AND HDACs IN LUNG INFLAMMATION
Over 30 HATs including transcription factors, co-activators and other signaling molecules are discovered to date, which display distinct substrate specificities for histone and non-histone proteins (Citation[18]). CBP/p300 is the most extensively studied among the HATs, and it is vital for the co-activation of several transcription factors, including NF-κ B and AP-1. Increased acetylation of histones (H3/H4) and NF-κ B by CBP/p300 is associated with cigarette smoke-mediated pro-inflammatory cytokines release (Citation[5], Citation[7], Citation[19]), which is responsible for the sustained pro-inflammatory response seen in COPD.
So far, 18 isoforms of HDACs are identified, and they are grouped into four classes (Citation[20]). (i) Class I members: HDAC-1, 2, 3 and 8, (ii) Class II members: HDAC-4, 5, 6, 7, 9 and 10, (iii) Class III members: Sirtuin-1 (SIRT1), 2, 3, 4, 5, 6 and 7, and use NAD+ as a co-factor, and (iv) Class IV member: HDAC11. The function of HDACs in suppressing genes transcription is mainly associated with their ability to remove acetyl moieties from the ε -acetamido group on lysine residues within histones leading to rewinding of DNA. HDACs not only deacetylate histones but also have the ability to deacetylate non-histone proteins, such as NF-κ B and thereby have the ability to regulate NF-κ B-dependent pro-inflammatory gene transcription (Citation[5], Citation[20]). Among the different HDACs, HDAC2 and SIRT1 are of great interest in regulation of lung inflammation and in pathogenesis of COPD, because of (i) their interaction with NF-κ B and regulation of pro-inflammatory genes, (ii) significant reduction in lungs of smokers and in patients with COPD, (iii) involvement of SIRT1 in regulation of accelerated aging of the lung (rapid decline in lung function) and apoptosis/senescence in the pathogenesis of COPD and iv) requirement of HDAC2 for the anti-inflammatory effects of glucocorticoids (Citation[9], Citation[10], Citation[21], Citation[22], Citation[23], Citation[24]).
OXIDATIVE ACTIVATION OF HATs
CBP and p300 (referred to CBP/p300 because of their mutual interaction) are transcriptional co-activators with intrinsic HAT activity, and are regulated by MAP kinase (Citation[13]). Specific core histone lysine residues can be acetylated by CBP/p300 co-activator. Both p300 and CBP are also known to involve in the regulation of various DNA-binding transcriptional factors. For example, lysine acetylation of histones by CBP/p300-HAT causes DNA uncoiling, and allows accessibility of NF-κ B (RelA/p65) to bind the promoters of genes (Citation[25]). Thus, histone acetylation via CBP/p300 has a significant role in the activation of NF-κ B-mediated pro-inflammatory gene expression.
It has been shown that CBP can be phosphorylated by Iκ B kinase (IKK), particularly IKKα, which is translocated into nucleus (Citation[7], Citation[14], Citation[15]). IKKα phosphorylates histone H3 at Ser10 and RelA/p65 leading to acetylation of histone H3 and RelA/p65 by its interaction with CBP/p300 (Citation[7], Citation[14]). For example, phosphorylation of RelA/p65 at Ser276 and Ser311 facilitates its interaction with CBP/p300, which is able to acetylate RelA/p65 at Lys310 and other lysine residues. Acetylation of RelA/p65 at Lys310 increases its transactivation potential i.e. transcriptional activation of NF-κ B-dependent pro-inflammatory genes. We have recently shown that IKKα mediates chromatin remodeling (by increasing instrinsic HAT activity) via the activation of NF-κ B inducing kinase (NIK) in response to cigarette smoke in human lung epithelial cells, macrophages and mouse lungs (Citation[7]). Therefore, investigation of NIK-IKKα signaling pathway will further unveil the mechanism of chromatin remodeling observed in lungs of patients with COPD.
In addition to IKK, NF-κ B activation is also regulated by mitogen- and stress-activated protein kinase-1 (MSK1) by phosphorylation of RelA/p65 (Ser276) and histone H3, to establish a transcription activation complex (Citation[26]). Cigarette smoke extract increases the level of MSK1 in macrophages (MonoMac6 cells) suggesting MSK1 may play important role in IKKα -mediated NF-κ B activation and chromatin modification on pro-inflammatory gene promoters (Citation[7]). Oxidants are involved in the activation of kinases (IKKα, NIK and MSK1), which mediates HAT phosphorylation and perhaps CBP acetylation (27–29). Thus, cigarette smoke activates HATs by inducing various kinases directly or indirectly through oxidative stress.
OXIDATIVE MODIFICATIONS OF HDAC2
HDAC2, an important co-repressor protein, is a redox-sensitive protein and prone to alteration by oxidants and/or free radicals, which is associated with the reduced levels/activities of these deacetylases in lungs of smokers and patients with COPD compared to healthy non-smokers (Citation[9], Citation[21], Citation[30]). The decreased level/activity of HDAC2 is also observed in response to cigarette smoke extract in vitro exposure to macrophage-monocytes (MonoMac6 cells), human airway and bronchial epithelial cells, and in lungs of mice exposed to cigarette smoke suggesting the posttranslational modifications of HDAC2 (Citation[22], Citation[31], Citation[32]). It is not known whether a similar HDAC2 reduction occurs in other cell types (e.g. memory CD8+ T cells) involved in pathogenesis of COPD. Nitration of tyrosine residues on HDAC2 by nitric oxide (NO)/peroxynitrite donors also decreases their deacetylase activity in monocytes, epithelial cells and rodent lungs (Citation[6], Citation[32]). Moreover, the level of nitrated HDAC2 is significantly increased in alveolar macrophages from patients with COPD (Citation[33]). Furthermore, NO-mediated nitrosylation (reaction of nitric oxide with cysteine) of HDAC2 occurs on Cys262 and Cys274 residues in neuronal cells highlighting the involvement of several post-translational modifications on HDAC2 (Citation[34]). The oxidative post-translational modifications on HDAC2 is restored by pre-treatment of monocytes and epithelial cells with antioxidants, such as GSH monoethyl ester or polyphenol-curcumin in response to cigarette smoke extract treatment (Citation[22], Citation[35]). However, the site of HDAC2 modifications by cigarette smoke extract/ROS/reactive nitrogen species (RNS) is still elusive. Nevertheless, redox-dependent post-translational modifications occur on HDAC2 via nitrosylation and α - β unsaturated aldehyde-adduct formation (protein carbonyl-adducts) which are implicated in the loss of its deacetylase activity.
Oxidative stress plays an important role in reducing the activity of HDAC2 by post-translational modifications and kinase-dependent signaling mechanisms. Oxidatively/ post-translationally modified HDAC2 can be a target of proteasomal degradation. However, the mechanism underlying reduced level/activity of HDAC2 by oxidative modification is not clearly understood. It has been postulated that the ubiquitin-proteasomal pathway contributes to HDAC2 degradation. A recent study has shown that HDAC2 is a phosphoprotein and basal phosphorylation is required for its deacetylase activity (Citation[36]). However, hyperphosphorylation of HDAC2 will lead to a dissociation of the co-repressor complex, and increase its ubiquitination and eventual degradation (Citation[31], Citation[37]). Cigarette smoke is shown to cause increased phosphorylation of HDAC2 by kinase-dependent mechanisms (Citation[19], Citation[31]).
Treatment of macrophages with a casein kinase 2 (CK2) inhibitor restored the cigarette smoke-induced degradation of HDAC2 suggesting that HDAC2 is regulated by activation of kinases [CK2 is downstream of phosphatidylinositol 3-kinase (PI3K)] (Citation[31]). This observation was validated by Marwick and colleagues who showed HDAC2 stability and lack of HDAC2 phosphorylation in phosphoinositide-3 kinase (PI3K)δ knockout mice in response to cigarette smoke (Citation[38]). It is likely that inhibition of HDAC2 phosphorylation, and subsequent oxidative degradation (stabilization and buffering of HDAC2 levels) will lead to designing of therapeutic agents for reversal of steroid resistance in COPD (Citation[22], Citation[30], Citation[32], Citation[35]) as HDAC2 protein is recruited in corepressor complex to mediate the anti-inflammatory effect of glucocorticoid (Citation[30], Citation[39]). Genetic deletion of PI3Kδ, but not PI3Kγ, restored the steroid sensitivity in response to cigarette smoke exposure in mouse lung (Citation[38]). Since proteasomal inhibitors attenuated cigarette smoke-mediated degradation of HDAC2, it is thought that cigarette smoke might induce degradation of HDAC2 via the ubiquitin-proteasome pathway and that hyperphosphorylation might be the trigger for this event.
It is evident that both phosphorylation and dephosphorylation events are involved in cigarette smoke-mediated HDAC2 degradation. Hence, apart from kinase inhibitors, activation or restoration of inactive protein phosphatases, such as the MAPK phosphatase-1 (MKP-1) could prove a potential mechanism of restoring HDAC2 level/activity. The PP1/2A inhibitor, okadaic acid, induced a very similar response to cigarette smoke extract treatment in cells and cigarette smoke exposure is known to decrease expression of lung tissue protein phosphatases such as MKP-1 in vivo (Citation[40]). Furthermore, HDAC2 is phosphorylated in presence of okadaic acid, a phosphatase inhibitor, in monocyte-macrophage cell line (MonoMac6), suggesting that MKP-1 activators might be useful in attenuating the cigarette smoke-induced HDAC2 degradation with a long-term goal of restoring steroid function in patients with COPD and asthmatics who smoke.
OXIDATIVE MODIFICATIONS OF SIRT1
SIRT1 is a protein/histone deacetylase, which has anti-aging, anti-apoptotic, and anti-inflammatory properties. It deacetylases histones (H3 and H4) and non-histone proteins including transcription factors, co-activators and other signaling molecules (e.g. FOXO, p53 and RelA/p65). We and others have shown that SIRT1 deacetylases RelA/p65 subunit of NF-κ B Lys310 residue, a site that is critical for NF-κ B transcriptional activity (Citation[10], Citation[23], Citation[41], Citation[42], Citation[43]). Furthermore, the level of SIRT1 is reduced in rat lungs and MonoMac6 cells by cigarette smoke (Citation[23]) and in lungs of smokers and patients with COPD (Citation[10]), implicating an important role of SIRT1 in the pathogenesis of COPD. SIRT1 plays a key role in regulation of NF-κ B-dependent pro-inflammatory cytokines release as shown by pharmacological, and genetic loss and gain approaches (Citation[10], Citation[23], Citation[24], Citation[42], Citation[44], Citation[45]). This suggests that pharmacological induction of SIRT1 may be a strategy to ameliorate abnormal inflammation in lungs of patients with COPD.
SIRT1 undergoes oxidative/nitrosative post-translational modifications mediated by cigarette smoke as shown by increased nitration of tyrosine residues and carbonylation (4-hydroxy-2-nonenal-adducts formation) with cysteine, histidine and lysine residues in lungs of smokers and patients with COPD compared with nonsmokers (Citation[10]), and in human monocyte-macrophage (MonoMac6) (Citation[23]) as well as bronchial epithelial cell lines (Beas-2B) (Citation[46], Citation[47]). Furthermore, SIRT1 is shown to be a redox-sensitive molecule since intracellular modulation of thiols regulated its level and activity in these cells (Citation[46], Citation[47]). Recently, it has also been shown that phosphorylation plays an important role in SIRT1 regulation (Citation[48]), and cigarette smoke-mediated oxidants cause SIRT1 phosphorylation (via activation of kinases and inhibition of phosphatases) in macrophages and epithelial cells as well as in mouse lungs after cigarette smoke exposure (Citation[46], Citation[47]).
Proteasome inhibitors inhibited phosphorylation of SIRT1 suggesting that phosphorylation in addition to covalent oxidative/nitrosative modifications of SIRT1 cause irreversible modifications of SIRT1 and subsequent proteasomal degradation. Taken together, it may be surmised here that SIRT1 is a novel redox-sensitive protein which plays an important role in regulation of various key signaling molecules and it undergoes post-translational modifications. It remained to be determined whether SIRT1 can attenuate chronic lung inflammatory response or has any role in reversal of steroid resistance which is caused by cigarette smoke exposure.
OXIDATIVE DISRUPTION OF SIRT1/HDAC2 COREPRESSOR COMPLEX
HDACs and SIRT1 form corepressor complex with CBP/p300 and RelA/p65 and inhibit pro-inflammatory gene transcription. Cigarette smoke/oxidants-induced reduction in HDAC2 is associated with sustained activation of RelA/p65 in the nucleus via disruption of the corepressor complex associated with reduction of HDAC2 culminating in pro-inflammatory gene transcription (Citation[22], Citation[45], Citation[49], Citation[50]). HDAC inhibitor trichostatin A enhanced the NF-κ B-driven inflammatory gene transcription in various cells (Citation[25]). Therefore, alteration of HDACs by cigarette smoke/oxidants leads to acetylation of histones and disruption of co-repressor complex leading to the activation of transcription factors, such as NF-κ B resulting in the increased transcription of pro-inflammatory genes (Citation[22], Citation[31], Citation[45], Citation[49], Citation[50], Citation[51]).
HDAC2 is required for the anti-inflammatory effects of glucocorticoids, and therefore reduced level/activity of HDAC2 leads to steroid resistance as seen in patients with COPD (Citation[2], Citation[9], Citation[30], Citation[35], Citation[38]). It is possible that theophylline and curcumin which increase HDAC2 may have a role in reversal of steroid resistance by suppressing cigarette smoke/oxidant-induced IL-8 release in alveolar macrophage from patients with COPD as well as in the MonoMac6 cells (Citation[33], Citation[35], Citation[52], Citation[53]). It has also been shown that HDAC2 can deacetylate glucocorticoid receptor (GR) and hence increasing the association of GR with corepressor complex and subsequently attenuating the release of pro-inflammatory mediators (Citation[39]).
SIRT1 protein associates directly with RelA/p65 subunit of NF-κ B and deacetylates Lys310 of RelA/p65, a site that is critical for NF-κ B transcriptional activity, and thereby inhibits the activity of NF-κ B (Citation[23], Citation[42], Citation[43]). SIRT1 is known to regulate NF-κ B-dependent transcription and cell survival in response to TNF-α (Citation[41], Citation[42], Citation[54]). Similarly, a similar role of inhibition of NF-κ B and histone acetylation is ascribed for SIRT6 (Citation[55]). Furthermore, it has been shown that genetic loss of SIRT1 leads to dramatic increase in neutrophil infiltration in mouse lung suggesting that SIRT1 regulates lung inflammation (Citation[56]). Inhibition of SIRT1 by inhibitor or siRNA increased cigarette smoke-mediated NF-κ B activation and pro-inflammatory mediator release in human monocytic cell lines (Citation[10], Citation[23]).
This is explained by the finding that cigarette smoke-derived oxidants (ROS/RNS) caused post-transcriptional modifications of SIRT1 and subsequent degradation thereby disrupting SIRT1-RelA/p65 complex and increased the release of pro-inflammatory mediators (Citation[23]). Such oxidative/aldehyde/nitrosative modifications may in turn render SIRT1 ineffective for interaction with other signaling components. Further research is required to elucidate the mechanisms of oxidative stress-mediated regulation of HDAC2 and SIRT1 degradation and its association with regulatory/transcription factors in pro-inflammatory gene expression in COPD.
CONCLUSIONS AND FUTURE PERSPECTIVES
Oxidative stress plays a major role in the pro-inflammatory response through the activation of redox-sensitive transcription factors such as NF-κ B, and alteration in the redox-sensitive deacetylases (HDAC2 and SIRT1) involved in chromatin remodeling. Cigarette smoke-derived ROS and reactive aldehydes cause post-translational modifications, such as nitration/nitrosylation, carbonylation, adduct formation and/or phosphorylation of HDAC2 and SIRT1, and direct them towards proteasomal degradation. Decreased deacetylase/acetylase ratio leads to disruption of deacetylase-NF-κ B interaction, acetylation of NF-κ B, and opening of chromatin culminating in NF-kB-dependent gene transcription. The signaling mechanism underlying reduced level/activity of HDAC2 and SIRT1 by oxidants/cigarette smoke is not known. Furthermore, it remains to be known whether post-translational modifications of acetylases and deacetylases occur on various pro- and anti-inflammatory, pro- and anti-apoptotic, protease and anti-protease, antimicrobial and antioxidant genes in response to cigarette smoke and in lungs of patients with COPD.
We postulate that a ‘histone code’ exists that determines the pattern of acetylation and deacetylation on promoters of a variety of genes involved in pathogenesis of COPD. In this respect, it will be interesting to determine the involvement of deacetylases and histone modifications in phenotypic characterization of different cellular and inflammatory response in COPD based on epigenetics. It also remains to be determined whether other environmental factors, circadian rythm and diet play an important role in epigenetic modifications in pathogenesis of COPD and its exacerbations. It is known that only about 15 to 20% smokers suffer from COPD, in this perspective it will be interesting to explore the pattern of histone code, and histone acetylation/deacetylation pattern of various proteins in smokers with and without COPD. Such a study will provide insight as to what type of histone modifications and deacetylases offer resistance or lead to abnormal inflammation in COPD in some smokers and accordingly design new therapeutic strategies against COPD. It will also be interesting to find out whether there is a role of memory CD8+ cells or other cells in imprinting the epigenetic modification in COPD pathology. Future studies on oxidative modifications in NF-κ B pathway and their upstream regulatory kinases, and mechanisms of epigenetic changes (specific phospho-acetylation of histones and deacetylases) will lead to identification of novel epigenetic therapeutic targets for halting the progression of inflammation in this debilitating disease.
Declaration of interest
The authors report no conflicts of interest. The authors are responsible for the content and writing of this perspective article.
ACKNOWLEDGMENTS
Supported by the NIH-NHLBI Grant RO1-HL-085613 and NIEHS Environmental Health Sciences Center Grant ES-01247.
REFERENCES
- Rahman I. Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanisms. Cell Biochem Biophys 2005; 43: 167–188
- Rahman I, Adcock I M. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J 2006; 28: 219–242
- Gosden R G, Feinberg A P. Genetics and epigenetics—nature's pen-and-pencil set. N Engl J Med 2007; 356: 731–733
- Li B, Carey M, Workman J L. The role of chromatin duringtranscription. Cell 2007; 128: 707–719
- Rahman I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. J Biochem Mol Biol 2003; 36: 95–109
- Marwick J A, Kirkham P A, Stevenson C S, Danahay H, Giddings J, Butler K, Donaldson K, Macnee W, Rahman I. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol 2004; 31: 633–642
- Yang S R, Valvo S, Yao H, Kode A, Rajendrasozhan S, Edirisinghe I, Caito S, Adenuga D, Henry R, Fromm G, Maggirwar S, Li J D, Bulger M, Rahman I. IKK alpha causes chromatin modification on pro-inflammatory genes by cigarette smoke in mouse lung. Am J Respir Cell Mol Biol 2008; 38: 689–698
- Szulakowski P, Crowther A J, Jimenez L A, Donaldson K, Mayer R, Leonard T B, MacNee W, Drost E M. The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006; 174: 41–50
- Ito K, Ito M, Elliott W M, Cosio B, Caramori G, Kon O M, Barczyk A, Hayashi S, Adcock I M, Hogg J C, Barnes P J. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med 2005; 352: 1967–1976
- Rajendrasozhan S, Yang S R, Kinnula V L, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008; 177: 861–870
- Lo W S, Trievel R C, Rojas J R, Duggan L, Hsu J Y, Allis C D, Marmorstein R, Berger S L. Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol Cell 2000; 5: 917–926
- Clayton A L, Rose S, Barratt M J, Mahadevan L C. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J 2000; 19: 3714–3726
- Thomson S, Mahadevan L C, Clayton A L. MAP kinase-mediated signalling to nucleosomes and immediate-early gene induction. Semin Cell Dev Biol 1999; 10: 205–214
- Gloire G, Horion J, El Mjiyad N, Bex F, Chariot A, Dejardin E, Piette J. Promoter-dependent effect of IKKalpha on NF-kappaB/p65 DNA binding. J Biol Chem 2007; 282: 21308–21318
- Yamamoto Y, Verma U N, Prajapati S, Kwak Y T, Gaynor R B. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature 2003; 423: 655–659
- Di Fiore P P, Polo S, Hofmann K. When ubiquitin meets ubiquitin receptors: a signalling connection. Nat Rev Mol Cell Biol 2003; 4: 491–497
- Vissers J H, Nicassio F, van Lohuizen M, Di Fiore P P, Citterio E. The many faces of ubiquitinated histone H2A: insights from the DUBs. Cell Div 2008; 3: 8
- Spange S, Wagner T, Heinzel T, Kramer O H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 2009; 41: 185–198
- Rajendrasozhan S, Yang S R, Edirisinghe I, Yao H, Adenuga D, Rahman I. Deacetylases and NF-kappaB in redox regulation of cigarette smoke-induced lung inflammation: epigenetics in pathogenesis of COPD. Antioxid Redox Signal 2008; 10: 799–811
- de Ruijter A J, van Gennip A H, Caron H N, Kemp S, van Kuilenburg A B. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003; 370: 737–749
- Ito K, Lim S, Caramori G, Chung K F, Barnes P J, Adcock I M. Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. FASEB J 2001; 15: 1110–1112
- Yang S R, Chida A S, Bauter M R, Shafiq N, Seweryniak K, Maggirwar S B, Kilty I, Rahman I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol 2006; 291: L46–57
- Yang S R, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol 2007; 292: L567–576
- Milne J C, Denu J M. The Sirtuin family: therapeutic targets to treat diseases of aging. Curr Opin Chem Biol 2008; 12: 11–17
- Chen L, Fischle W, Verdin E, Greene W C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001; 293: 1653–1657
- Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). Embo J 2003; 22: 1313–1324
- Kamata H, Manabe T, Oka S, Kamata K, Hirata H. Hydrogen peroxide activates IkappaB kinases through phosphorylation of serine residues in the activation loops. FEBS Lett 2002; 519: 231–237
- Zhang J, Johnston G, Stebler B, Keller E T. Hydrogen peroxide activates NFkappaB and the interleukin-6 promoter through NFkappaB-inducing kinase. Antioxid Redox Signal 2001; 3: 493–504
- Kefaloyianni E, Gaitanaki C, Beis I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal 2006; 18: 2238–2251
- Barnes P J. Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol 2008, (In press)
- Adenuga D, Yao H, March T H, Seagrave J, Rahman I. Histone deacetylase 2 is phosphorylated, ubiquitinated and degraded by cigarette smoke. Am J Respir Cell Mol Biol 2008; 40: 464–473
- Ito K, Hanazawa T, Tomita K, Barnes P J, Adcock I M. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun 2004; 315: 240–245
- Cosio B G, Tsaprouni L, Ito K, Jazrawi E, Adcock I M, Barnes P J. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med 2004; 200: 689–695
- Nott A, Watson P M, Robinson J D, Crepaldi L, Riccio A. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 2008; 455: 411–415
- Meja K K, Rajendrasozhan S, Adenuga D, Biswas S K, Sundar I K, Spooner G, Marwick J A, Chakravarty P, Fletcher D, Whittaker P, Megson I L, Kirkham P A, Rahman I. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am J Respir Cell Mol Biol 2008; 39: 312–323
- Tsai S C, Seto E. Regulation of histone deacetylase 2 by protein kinase CK2. J Biol Chem 2002; 277: 31826–31833
- Galasinski S C, Resing K A, Goodrich J A, Ahn N G. Phosphatase inhibition leads to histone deacetylases 1 and 2 phosphorylation and disruption of corepressor interactions. J Biol Chem 2002; 277: 19618–19626
- Marwick J A, Stevenson C S, Barnes P J, Ito K, Adcock I M, Kirkham P A. Cigarette smoke reduces steroid sensitivity by reducing glucocorticoid receptor (GR) and GR co-repressor expression. Am J Respir Crit Care Med 2008; 177: A333
- Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes P J, Adcock I M. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med 2006; 203: 7–13
- Liu C, Russell R M, Wang X D. Low dose beta-carotene supplementation of ferrets attenuates smoke-induced lung phosphorylation of JNK, p38 MAPK, and p53 proteins. J Nutr 2004; 134: 2705–2710
- Yeung F, Hoberg J E, Ramsey C S, Keller M D, Jones D R, Frye R A, Mayo M W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 2004; 23: 2369–2380
- Chen J, Zhou Y, Mueller-Steiner S, Chen L F, Kwon H, Yi S, Mucke L, Gan L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem 2005; 280: 40364–40374
- Chen L F, Mu Y, Greene W C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J 2002; 21: 6539–6548
- Milne J C, Lambert P D, Schenk S, Carney D P, Smith J J, Gagne D J, Jin L, Boss O, Perni R B, Vu C B, Bemis J E, Xie R, Disch J S, Ng P Y, Nunes J J, Lynch A V, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair D A, Olefsky J M, Jirousek M R, Elliott P J, Westphal C H. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007; 450: 712–716
- Rajendrasozhan S, Yang S R, Caito S, Rahman I. Nucleocytoplasmic shuttling and post-translational modifications of sirtuin in response to cigarette smoke lead to increased acetylation of NF-kappaB and FOXO3. Am J Respir Crit Care Med 2008; 177: A266
- Caito S, Cook S, Yang S R, Rajendrasozhan S, Rahman I. Redox regulation and post-translational modification of sirtuin in response to cigarette smoke in lung epithelial cells. Am J Respir Crit Care Med 2008; 177: A348
- Caito S, Cook S, Yang S R, Rajendrasozhan S, Rahman I. Sirtuin 1, an oxidant sensitive deacetylase, is posttranslationally modified and degraded by proteasome in reponse to cigarette smoke in lung epithelial cells. FASEB J 2008; 22: 747–741
- Sasaki T, Maier B, Koclega K D, Chruszcz M, Gluba W, Stukenberg P T, Minor W, Scrable H. Phosphorylation regulates SIRT1 function. PLoS ONE 2008; 3: e4020
- Yao H, Edirisinghe I, Rajendrasozhan S, Yang S R, Caito S, Adenuga D, Rahman I. Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am J Physiol Lung Cell Mol Physiol 2008; 294: L1174–1186
- Yao H, Yang S R, Kode A, Rajendrasozhan S, Caito S, Adenuga D, Henry R, Edirisinghe I, Rahman I. Redox regulation of lung inflammation: role of NADPH oxidase and NF-kappaB signalling. Biochem Soc Trans 2007; 35: 1151–1155
- Ito K, Barnes P J, Adcock I M. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 2000; 20: 6891–6903
- Adenuga D, March T H, Seagrave J, Rahman I. Cigarette smoke-induced loss of nuclear HDAC2 is associated with chronic inflammation and emphysema in A/J mice. Am J Respir Crit Care Med 2008; 177: A866
- Marwick J A, Wallis G, Meja K, Kuster B, Bouwmeester T, Chakravarty P, Fletcher D, Whittaker P A, Barnes P J, Ito K, Adcock I M, Kirkham P A. Oxidative stress modulates theophylline effects on steroid responsiveness. Biochem Biophys Res Commun 2008; 377: 797–802
- Vaziri H, Dessain S K, Ng Eaton E, Imai S I, Frye R A, Pandita T K, Guarente L, Weinberg R A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001; 107: 149–159
- Kawahara T LA, Michishita E, Adler A S, Damian M, Berber E, Lin M, McCord R A, Ongaigui K CL, Boxer L D, Chang H Y, Chua K F. SIRT6 Links histone H3 Lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 2009; 136: 62–74
- McBurney M W, Yang X, Jardine K, Hixon M, Boekelheide K, Webb J R, Lansdorp P M, Lemieux M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol 2003; 23: 38–54