Abstract
Chronic obstructive pulmonary disease (COPD) is characterized by inflammatory immune response, emphysematous destruction of alveolar structures and obstruction in the small conducting airways. Transforming growth factor (TGF)-β is involved in the maintenance of normal lung tissue homeostasis as a regulator of extracellular proteolysis, tissue repair and inflammatory functions. This study was undertaken to characterize TGF-β signaling in pathologically distinct areas of COPD lungs. Using Smad2 phosporylation (P-Smad2) as an indicator of TGF-β signaling activity we analyzed COPD patient tissues and controls by immunohistochemistry. Emphysematous lung showed significantly reduced P-Smad2 immunoreactivity both in the alveolar and bronchiolar epithelium, which is evidence of reduced TGF-β signaling activity. On the contrary, in the thickened peribronchial areas there was an increase in the amount of P-Smad2 positive cells. Isolated COPD lung fibroblasts also displayed increased TGF-β signaling and target gene expression suggesting that the fibroblasts are characteristic to the small airway disease phenotype. Our results indicate that TGF-β signaling activity is differentially regulated in distinct areas of COPD lung and likely contributes to both emphysematous development and small airway obstruction.
INTRODUCTION
The World Health Organization predicts that chronic obstructive pulmonary disease (COPD) will become the third-leading cause of death worldwide by 2030 (WHO, WHO/MNC/CRA/02.1). Cigarette smoke causes a dual response in the pulmonary tissue: airway obstruction via fibrosis and mucus production, and cell/tissue loss in the alveolar space, emphysema. Genetic factors may determine the different disease manifestations (obstruction or emphysema) in different patients. Most patients with COPD have emphysema, and the extent of emphysema increases with disease severity (Citation[1]). The emphysematous lung has diminished elasticity and decreased alveolar area for gas exchange, which account for the clinical picture: severe dyspnea and hypoxemia and/or hypercapnia. Tissue loss in emphysema is believed to be due to an increased activation of inflammatory mediators and proteolytic enzymes and a decreased ability to repair the alveolar tissue (Citation[2]).
Transforming growth factor (TGF)-β isoforms play important roles during lung development (Citation[3], Citation[4]). TGF-β is also considered an important factor in the maintenance of normal tissue structure in the alveolar wall as a regulator of extracellular matrix production. In addition, TGF-β participates in tissue repair processes through modulation of inflammatory, proteolytic and antioxidant pathways (Citation[5]). Increased production and activation of TGF-β 1 is strongly associated with the pathogenesis of fibrotic lung diseases (Citation[6]). Genetic association studies and analyses of alterations in TGF-β expression levels suggest the involvement of TGF-β also in the development of COPD phenotypes (7–10). Many experimental mouse models with impaired TGF-β system develop pulmonary emphysema, although the severity and onset of the pathologic alterations vary (11–14). Interestingly, mice impaired for TGF-β activation (β 6 knockout) or signaling (Smad3 knockout) develop age related emphysema but are protected from bleomycin induced fibrosis (Citation[15], Citation[16]). This suggests that the correct balance of TGF-β activity is important for lung homeostasis (Citation[5], Citation[17]).
There are 3 mammalian TGF-β isoforms (TGF-β 1-3), which are all expressed in the lung (Citation[18]). TGF-β is produced by cells in latent complexes consisting of the mature growth factor associated non-covalently with its prodomain as well as latent TGF-β binding protein (LTBP). LTBPs target the complexes into extracellular matrix structures, where TGF-β can be stored in an inactive form. Activation of these latent complexes is one of the key steps in the regulation of TGF-β function (Citation[19], Citation[20]). Polymorphisms in LTBP-4 gene have been associated with COPD (Citation[8]).
Previous analyses of TGF-β expression levels in patient biopsies suggest that aberrant growth factor levels are associated with COPD (Citation[9], Citation[21], Citation[22]). However, there are somewhat conflicting results implicating either decreased or increased levels of TGF-β 1. This is likely a result of the heterogeneous nature of the disease itself as well as the technical differences. It is also clear that analyses of TGF-β expression levels, whether mRNA of protein, do not necessarily reflect ongoing TGF-β signaling in the lung. The signaling is initiated by binding of activated TGF-β 1, -β 2 or -β 3 to the cell surface heteromeric receptor complex followed by phosphorylation and nuclear transport of Smad2/3 transcription factors (Citation[23]). This study was carried out to compare TGF-β signaling activity in pathologically distinct areas of COPD lungs by using phosphorylated Smad2 (P-Smad2) as an indicator of signaling activity.
MATERIALS AND METHODS
Patient material
Normal and COPD lung tissue biopsies were collected from either lobectomy due to a local solitary tumour or hamartoma, or lung transplantations at the Department of Pathology, Helsinki University Central Hospital. COPD was defined according to the GOLD criteria (FEV1 < 80% of reference, FEV1/FVC < 70% and bronchodilatation effect < 12%). Altogether 9 non-smoking controls with normal lung functions, 7 COPD patients with Stage II-III COPD and 9 with Stage IV (very severe) COPD were used (summarized in ). The study protocol was approved by the Helsinki University Central Hospital ethical board and registered at www.hus.fi/clinicaltrials.
Table 1 Characteristics of the patients
Immunohistochemistry
Paraffin-embedded tissue sections from lung biopsies were deparaffinized in xylene and rehydrated in graded alcohol. Antigens were retrieved by heating the sections in 10 mM citrate buffer (pH 6.0) and endogenous peroxidase activity was neutralized with 0.3% hydrogen peroxide. Stainings were performed with Histostain Plus Broad Spectrum Kit (Zymed, San Francisco, CA) according to the manufacturer's instructions. The primary antibody used was rabbit anti-phospho (Ser465/467)-Smad2 (Millipore). The sections were exposed to the antibody o/n at +4°C. The bound antibodies were visualized using peroxidase substrate 3-amino-9-ethylcarbazole (AEC; Zymed) and counterstained with Mayer's haematoxylin. In control sections, primary antibody was replaced by PBS or rabbit isotype control (Zymed). Representative images were taken with Olympus U-CMAD3 camera (Olympus, Japan) and QuickPHOTO CAMERA 2.1 software (Promicra, Czech Republic).
Quantification of P-Smad2 positive cells
From control and COPD samples positive (stained reddish-brown) and negative (stained blue) nuclei were counted under the microscope using 40x magnification. A hundred nuclei were counted from at least 2 different parts of each tissue specimen, and the average percentage of positive nuclei in each tissue sample was used for statistics. For alveolar wall quantification, normal alveolar structures from control samples and areas of emphysematous alveolar structures from COPD samples were selected.
Quantification of peribronchiolar fibrosis
Masson's Trichrome stained paraffin sections were used for the quantification of peribronchiolar thickening in lung biopsies. Masson's Trichrome is a standard histological staining protocol, which stains collagen blue and cellular components red. Photomicrographs of bronchioles (1–4/section) were taken with Olympus BX51 microscope, saved as JPG image files and analyzed using Image-Pro digital image processing software (Media Cybernetics, USA). The proportion of blue staining from the total area of bronchiolar tissue was measured. The results are expressed as percent blue staining in the bronchiolar wall ± standard error of the mean.
Statistics
Data were analyzed with SPSS 15.0 for Windows (SPSS) using the non-parametric Kruskal-Wallis test. A p-value of < 0.05 was considered to be statistically significant.
Cell culture and transient transfection
Primary lung fibroblasts were isolated from the explanted lung at the time of transplantation from patients with COPD as described (Citation[24]). CCL-190 normal adult lung fibroblasts were obtained from American Type Culture Collection (ATCC). The cells were used between passages 4–10 and cultured in DMEM supplemented with 10% fetal calf serum (PromoCell) and antibiotics.
Cells to be transiently transfected were plated in 24-well plates (Greiner). On the next day, the cells were co-transfected with the (CAGA)12-luciferase promoter construct [kindly provided by Professor Peter ten Dijke, the Netherlands Cancer Institute, Amsterdam (Citation[25])] together with pRL-TK (Renilla luciferase control, Promega) plasmid using the FuGENE HD transfection reagent (Roche). Subsequently, 48 h after transfection, the cells were lysed and subjected to luciferase activity measurements by a Dual Luciferase Kit (Promega) and Digene DCR-1 luminometer (MGM Instruments).
RNA isolation and real-time RT-PCR
Total cellular RNA was isolated using RNeasy Mini kit (Qiagen). RNA from cell lysates or homogenized (Lysin Matrix D, Q BIOgene) snap-frozen lung biopsy samples was prepared according to the manufacturer's instructions. RNA concentration and purity was determined using Agilent 2100 Bioanalyzer at the Biomedicum Biochip Center (Helsinki). Reverse transcription was carried out with Random hexamer primers (Invitrogen) and Superscript III reverse transcriptase (Life Technologies) using 1.0 μ g of total RNA according to manufacturer's instructions.
The cDNAs were amplified using TaqMan Assays-on-Demand gene expression products (Applied Biosystems) and GeneAmp 7500 Sequence Detector thermal cycler (Applied Biosystems). Control amplifications directly from RNA were performed in order to rule out DNA contamination. The levels of gene expression were determined using the Ct method and the results have been expressed as mRNA expression levels normalized to the levels of a gene with a constant expression (TBP, tata binding protein).
RESULTS
Expression of TGF-β isoforms and target genes in lung tissue
To analyze whether there are alterations in the overall expression levels of the different TGF-β isoforms we performed quantitative real-time RT-PCR gene expression analyses. Total cellular RNA was isolated from control and COPD IV lung tissues (5 samples of each) from which TGF-β 1-3 mRNA levels were analyzed. All TGF-β isoforms were frequently expressed in lung tissue; however, TGF-β 1 was clearly the most abundantly expressed isoform [, (Citation[24])]. In the current study no differences in TGF-β expression levels were noted between normal and COPD lungs.
Figure 1 Expression of TGF-β isoforms in COPD lung tissue. Relative mRNA expression levels of TGF-β 1-3 (A), CTGF and PAI-1 (B) in control and COPD IV lung tissues as analyzed by quantitative real-time RT-PCR. The mRNA levels were normalized to the expression levels of a control gene (TBP, tata binding protein) and are expressed relative to control-1 (set to 1). The error bars represent standard error of the mean of the samples (n = 5).*, p < 0.05.
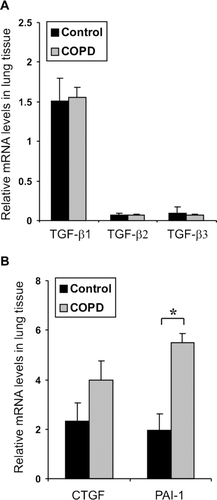
To characterize overall alterations in TGF-β signaling, the gene expression levels of a few known target genes were analyzed from lung tissues. Both CTGF and PAI-1 are directly regulated by TGF-β activity in the lung, although other inducers have also been described (Citation[26], Citation[27]). Slightly increased CTGF mRNA expression levels were found in COPD lungs, although the difference did not reach statistical significance (p = 0.1). PAI-1 levels were, however, significantly up-regulated (∼ 3-fold) in COPD lungs (). In the absence of changes in TGF-β expression levels, these results may suggest increased TGF-β activation and/or signaling activity in COPD lungs.
Decreased P-Smad2 levels in emphysematous lung tissue
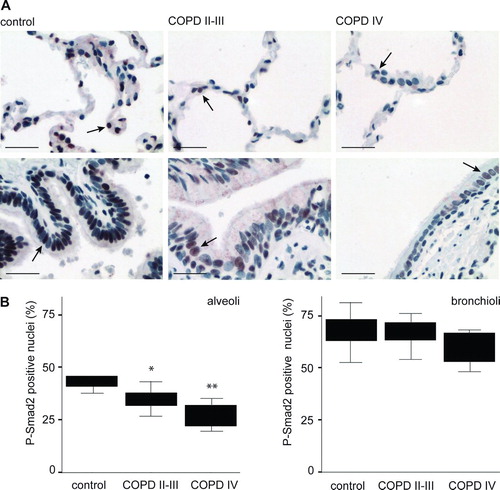
To specifically localize TGF-β signaling activity in the lung tissue, sections from control, COPD II-III and COPD IV lungs were stained with antibodies detecting specifically the phosphorylated form of Smad2 (P-Smad2). As expected, P-Smad2 immunoreactivity was predominantly observed in the nucleus (). There was abundant TGF-β signaling activity in control lungs implying that TGF-β participates in the maintenance of normal lung structure. Positive cells were observed in the bronchiolar epithelium, the bronchiolar adventitial layer i.e., the tissue surrounding small bronchioli, and to a lesser extent in the alveolar wall. Quantification of P-Smad2 positive nuclei was then performed from emphysematous areas of COPD lung identified by experienced lung pathologist. Unexpectedly, the emphysematous tissue showed significantly fewer positive nuclei in the alveolar epithelium (). In addition, in bronchiolar epithelium a small decrease in the number of positive nuclei was observed in COPD IV lungs, but this was not statistically significant. Importantly, P-Smad2 staining intensity was remarkably decreased both in alveolar and bronchiolar epithelium suggesting down-regulation of TGF-β signaling activity in the emphysematous alveolus.
Figure 2 P-Smad2 staining of emphysematous alveolar septal areas and bronchiolar epithelium. Paraffin sections from control adult, COPD II-III and COPD IV patient lungs were immunostained for P-Smad2. A. Emphysematous alveolar septal (upper panels) and bronchiolar (lower panels) areas are shown. Positive staining is reddish-brown. Scale bar = 100 μ m. B. Quantification of P-Smad2 positive nuclei. The results are presented as boxplots.*, p = 0.028;**, p = 0.006.
P-Smad2 levels in peribronchiolar areas of COPD lung
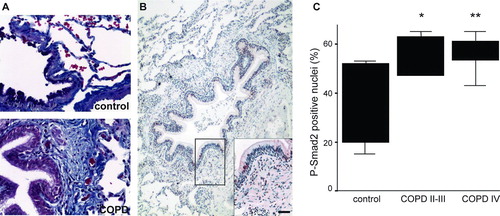
TGF-β activation is increased in squamous metaplasia in COPD patients (Citation[28]), which may be related to the development of peribronchial fibrotic alterations. Therefore we quantified P-Smad2 positive nuclei also from peribronchial regions displaying small airway thickening in COPD. Peribronchial fibrosis of small airways was first analyzed using digital morphometry from Masson's Trichrome stained lung biopsies (, collagen fibers stain blue). As expected, fibrotic tissue was found to be increased significantly (p = 0.003) in bronchioles from COPD patient biopsies (ctrl 6.53 ± 1.95 versus COPDII-IV 15.52 ± 4.00). The proportion of P-Smad2 positive nuclei in these thickened peribronchiolar areas was increased (, ).
Figure 3 P-Smad2 positive nuclei in peribronchiolar lung. A. Representative Masson's Trichrome staining of control adult and COPD lungs. Collagen fibers stain blue. B. Representative P-Smad2 staining of a patient with stage II COPD. A small airway with peribronchiolar thickening and inflammation is shown. The positive nuclear staining is reddish-brown. Scale bar = 100 μ m. C. Quantification of P-Smad2 positive nuclei in the peribronchiolar area of small airways from the different patient groups. The results are presented as boxplots.*, p = 0.025;**, p = 0.01.
TGF-β signaling in isolated lung fibroblasts
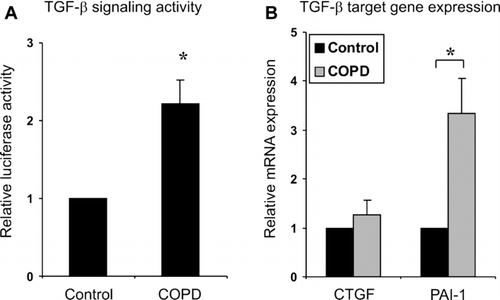
The current studies suggest decreased TGF-β signaling in emphysematous lung and increased signaling in areas displaying small airway fibrosis. Although COPD patients can be classically divided into two distinct phenotypes with either the emphysematous or airway component dominating, patients usually represent a combination of these two phenotypes. We analyzed which disease phenotype is reflected in isolated lung fibroblasts. TGF-β signaling activity was analyzed using transient transfection of TGF-β responsive promoter [(CAGA)12-luciferese, see Methods] followed by measurement of luciferase activity. TGF-β signaling activity was significantly increased in COPD fibroblasts (). In addition, PAI-1 mRNA expression levels were up-regulated suggesting that cultured COPD fibroblasts represent a fibrotic phenotype at least in terms of the TGF-β system.
Figure 4 Lung fibroblasts (CCL-190 normal adult and COPD fibroblasts) were transiently transfected with a TGF-β responsive (CAGA)12-luciferase promoter construct and cultured for two days. Luciferase activities were measured and normalized by comparing them with the activities of co-transfected Renilla luciferase activities. The results are expressed as relative luciferase activities. B. Relative mRNA expression levels of CTGF and PAI-1 in control and COPD fibroblasts as analyzed by quantitative real-time RT-PCR. The mRNA expression levels were normalized to the expression levels of a control gene (TBP, tata binding protein) and are expressed relative to control (set to 1). The error bars represent the standard error of the mean of the samples (COPD fibroblast, n = 3).*, p < 0.05.
DISCUSSION
COPD is a complex disease mainly comprised of emphysema, chronic bronchitis and small airway disease. The molecular mechanisms leading to these disease phenotypes are only partially understood. Emphysema is the most frequent manifestation of COPD and the extent of emphysematous areas in the lung correlate with disease severity. In experimental mouse models abnormalities in the TGF-β signaling system frequently lead to the development of emphysema.
This has been linked to the role of TGF-β as a regulator of extracellular proteolysis, tissue repair and inflammatory functions. Human genetic studies have revealed that COPD is associated with polymorphisms in TGF-β 1 as well as in several genes that regulate TGF-β availability, activation or signaling (Citation[7], Citation[8]). These genes include LTBP-4, which is an extracellular regulator of TGF-β targeting and activation. This highlights the specific role of the activation process in the control of TGF-β function in the lung.
Only a few studies have revealed alterations in the expression levels of TGF-β 1 and target genes such as CTGF in COPD patient lungs. Increased levels have been observed in small airway epithelium (Citation[21]) as well as in whole lung tissue (Citation[9]), while in other studies decreased expression has been reported (Citation[10], Citation[29]). There are also reports suggesting that TGF-β receptor levels as well as expression of the inhibitory Smad7 are aberrant in COPD lungs (Citation[29], Citation[30]). Decreased levels of the inhibitory Smads 6 and 7 may be a consequence of reduced TGF-β signaling since both are regulated by TGF-β as an inhibitory feedback mechanism (Citation[31], Citation[32]). On the other hand, elevated expression may cause reduced TGF-β signaling. Since all these alterations affect cellular responsiveness to TGF-β it is unclear how they contribute to TGF-β function. To specifically characterize alterations in the TGF-β signaling activity in COPD lung we quantified the levels of phosphorylated Smad2 in specific areas in the lung. Smad2 is phosphorylated directly by TGF-β RI in response to TGF-β, after which it is complexed with Smad4 and transported into the nucleus (Citation[33]). Using P-Smad2 levels as a direct indicator of TGF-β signaling activity we found that in pathologically distinct areas of COPD lung TGF-β signaling was regulated in an opposite manner. This suggests that the balance of TGF-β activity is important in COPD pathobiology and that complex mechanisms regulate lung homeostasis.
Emphysema is described as airspace enlargement due to loss of alveolar structure. There is a progressive destruction of alveolar-capillary cells as well as loss of parenchymal elastic recoil. Inflammatory mediators are thought to play a central role in extracellular matrix proteolysis, especially in the destruction of the elastin network. Matrix metalloproteinases, such as MMPs-2, −9, −12 and neutrophil elastase produced by epithelial cells, macrophages and dendritic cells are strongly associated with the development of emphysema in COPD patients as well in experimental animal models (Citation[15], Citation[34]). TGF-β down-regulates macrophage production of proteolytic enzymes, which is exemplified by MMP-12-dependent emphysema development in mice lacking the TGF-β activating epithelial integrin α vβ 6 (Citation[15]). We have analyzed for the first time TGF-β signaling activity directly in the alveolar and bronchiolar epithelium in emphysematous areas of COPD lungs. A dramatic decrease in signaling activity was observed compared to the abundant epithelial signaling in normal lungs. This likely contributes to the reduced repair capacity of the emphysematous lung as well as to local production of proteases. Consistent with reduced repair capacity COPD fibroblasts also manifest impaired migration and collagen contraction in response to TGF-β (Citation[35]).
In addition to emphysema, COPD pathogenesis involves airway disease characterized by obstruction of the small airways. Inflammatory processes are involved also in the thickening of the airway wall and accumulation of extracellular matrix components such as collagens. The role of increased TGF-β activation and signaling is known to mediate fibrotic processes in the lung (Citation[6]). Accordingly, we detected increased levels of P-Smad2 positive cells in the thickened peribronchiolar regions of COPD lungs. This is in agreement with studies suggesting increased TGF-β expression in small airways (Citation[21], Citation[22], Citation[28]). The work by Araya et al. suggests that IL-1β induced alterations in the TGF-β activating integrin α vβ 8 lead to increased activation of the growth factor followed by accumulation of fibroblasts and extracellular matrix in these fibrotic areas (Citation[28]). Fibroblasts are central in this process and display increased activation of TGF-β. We observed increased TGF-β signaling in isolated COPD fibroblasts using TGF-β responsive promoter as an indicator of the signaling activity. In addition, the expression of the TGF-β target gene, PA1-1, was significantly up-regulated in cultured COPD fibroblasts. Analyses of expression levels of TGF-β target genes in whole lung tissue homogenates replicated PAI-1 induction implying that the observed alterations reflect changes in fibroblasts around small airways. Specific alterations in emphysematous tissue were masked by the low abundance of partially destructed alveolar tissue in the lung biopsies.
Both emphysema and small airway obstruction lead to airflow limitation. However, the mechanisms leading to these two manifestations of the same disease differ although the growth factors involved may partly be common. TGF-β is one of the important regulators of lung repair functions and a delicate balance of growth factor activation contributes to whether emphysema or fibrosis occurs (Citation[17]). It has been suggested that TGF-β activation mechanisms differ in small airways and alveolar structures, different integrins being involved in these crucial steps (Citation[5], Citation[36]). It appears that different areas in the COPD lung show either decreased or increased TGF-β activity/signaling and thus the targeting of TGF-β directly may not prove to be a good approach for the treatment of COPD patients. Future studies on specific mechanisms of TGF-β activation and signaling cascades may be the key to finding new therapeutic targets.
Declaration of interest
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
ACKNOWLEDGMENTS
We are grateful to the patients who consented to participate in our study. We thank Tiina Marjomaa, Anitra Ahonen and Eva Sutinen for excellent technical assistance. Funding was provided by the Academy of Finland (JK-O, KK), Finnish Cancer Foundation (JK-O), Finnish Cultural Foundation (KK), Sigrid Juselius Foundation (JK-O), Biocentrum Helsinki (JK-O, MM, KK), Helsinki University Hospital Fund (VLK, JK-O, MM), the University of Helsinki (JK-O), The Yrjö Jahnsson Foundation (MM, VLK), the Finnish Antituberculosis Association Foundation (VLK, MM), the Finnish Medical Foundation (MM), the Jalmari and Rauha Ahokas Foundation (MM).
REFERENCES
- Shaker S B, Dirksen A, Bach K S, Mortensen J. Imaging in chronic obstructive pulmonary disease. COPD 2007; 4(2)143–161
- Taraseviciene-Stewart L, Voelkel N F. Molecular pathogenesis of emphysema. J Clin Invest 2008; 118(2)394–402
- Bartram U, Speer C P. The role of transforming growth factor β in lung development and disease. Chest 2004; 125(2)754–765
- Chen H, Zhuang F, Liu Y H, Xu B, Del Moral P, Deng W, Chai Y, Kolb M, Gauldie J, Warburton D, Moses H L, Shi W. TGF-β receptor II in epithelia versus mesenchyme plays distinct roles in the developing lung. Eur Respir J 2008; 32(2)285–295
- Sheppard D. Transforming growth factor β : a central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc 2006; 3(5)413–7
- Branton M H, Kopp J B. TGF-β and fibrosis. Microbes Infect 1999; 1(15)1349–1365
- Celedon J C, Lange C, Raby B A, Litonjua A A, Palmer L J, DeMeo D L, Reilly J J, Kwiatkowski D J, Chapman H A, Laird N, Sylvia J S, Hernandez M, Speizer F E, Weiss S T, Silverman E K. The transforming growth factor-β 1 (TGFB1) gene is associated with chronic obstructive pulmonary disease (COPD). Hum Mol Genet 2004; 13(15)1649–1656
- Hersh C P, Demeo D L, Lazarus R, Celedon J C, Raby B A, Benditt J O, Criner G, Make B, Martinez F J, Scanlon P D, Sciurba F C, Utz J P, Reilly J J, Silverman E K. Genetic association analysis of functional impairment in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006; 173(9)977–984
- Ning W, Li C J, Kaminski N, Feghali-Bostwick C A, Alber S M, Di Y P, Otterbein S L, Song R, Hayashi S, Zhou Z, Pinsky D J, Watkins S C, Pilewski J M, Sciurba F C, Peters D G, Hogg J C, Choi A M. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci USA 2004; 101(41)14895–14900
- Golpon H A, Coldren C D, Zamora M R, Cosgrove G P, Moore M D, Tuder R M, Geraci M W, Voelkel N F. Emphysema lung tissue gene expression profiling. Am J Respir Cell Mol Biol 2004; 31(6)595–600
- Sterner-Kock A, Thorey I S, Koli K, Wempe F, Otte J, Bangsow T, Kuhlmeier K, Kirchner T, Jin S, Keski-Oja J, von Melchner H. Disruption of the gene encoding the latent transforming growth factor-β binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev 2002; 16(17)2264–2273
- Colarossi C, Chen Y, Obata H, Jurukovski V, Fontana L, Dabovic B, Rifkin D B. Lung alveolar septation defects in Ltbp-3-null mice. Am J Pathol 2005; 167(2)419–428
- Neptune E R, Frischmeyer P A, Arking D E, Myers L, Bunton T E, Gayraud B, Ramirez F, Sakai L Y, Dietz H C. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat Genet 2003; 33407–33411
- Wang X, Inoue S, Gu J, Miyoshi E, Noda K, Li W, Mizuno-Horikawa Y, Nakano M, Asahi M, Takahashi M, Uozumi N, Ihara S, Lee S H, Ikeda Y, Yamaguchi Y, Aze Y, Tomiyama Y, Fujii J, Suzuki K, Kondo A, Shapiro S D, Lopez-Otin C, Kuwaki T, Okabe M, Honke K, Taniguchi N. Dysregulation of TGF-β 1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc Natl Acad Sci USA 2005; 102(44)15791–15796
- Morris D G, Huang X, Kaminski N, Wang Y, Shapiro S D, Dolganov G, Glick A, Sheppard D. Loss of integrin α vβ 6-mediated TGF-β activation causes Mmp12-dependent emphysema. Nature 2003; 422(6928)169–173
- Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, Lavery C, Margetts P J, Roberts A B, Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to TGF-β -mediated pulmonary fibrosis. J Immunol 2004; 173(3)2099–2108
- Gauldie J, Kolb M, Ask K, Martin G, Bonniaud P, Warburton D. Smad3 signaling involved in pulmonary fibrosis and emphysema. Proc Am Thorac Soc 2006; 3(8)696–702
- Pelton R W, Johnson M D, Perkett E A, Gold L I, Moses H L. Expression of transforming growth factor-β 1, -β 2, and -β 3 mRNA and protein in the murine lung. Am J Respir Cell Mol Biol 1991; 5(6)522–530
- Hyytiäinen M, Penttinen C, Keski-Oja J. Latent TGF-β binding proteins: extracellular matrix association and roles in TGF-β activation. Crit Rev Clin Lab Sci 2004; 41(3)233–264
- Rifkin D B. Latent transforming growth factor-β (TGF-β) binding proteins: orchestrators of TGF-β availability. J Biol Chem 2005; 280(9)7409–7412
- Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, Satoh M, Okada Y, Yamasawa F, Nakahara K, Umeda A. Increased expression of transforming growth factor-β 1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med 2001; 163(6)1476–1483
- de Boer W I, van Schadewijk A, Sont J K, Sharma H S, Stolk J, Hiemstra P S, van Krieken J H. Transforming growth factor β 1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 158(6)1951–1957
- Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J, Heldin C H, Miyazono K, ten Dijke P. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J 1997; 16(17)5353–5362
- Koli K, Myllärniemi M, Vuorinen K, Salmenkivi K, Ryynänen M J, Kinnula V L, Keski-Oja J. Bone morphogenetic protein-4 inhibitor gremlin is overexpressed in idiopathic pulmonary fibrosis. Am J Pathol 2006; 169(1)61–71
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier J M. Direct binding of Smad3 and Smad4 to critical TGF β -inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 1998; 17(11)3091–3100
- Kucich U, Rosenbloom J C, Herrick D J, Abrams W R, Hamilton A D, Sebti S M, Rosenbloom J. Signaling events required for transforming growth factor-β stimulation of connective tissue growth factor expression by cultured human lung fibroblasts. Arch Biochem Biophys 2001; 395(1)103–112
- Keski-Oja J, Raghow R, Sawdey M, Loskutoff D J, Postlethwaite A E, Kang A H, Moses H L. Regulation of mRNAs for type-1 plasminogen activator inhibitor, fibronectin, and type I procollagen by transforming growth factor-β. Divergent responses in lung fibroblasts and carcinoma cells. J Biol Chem 1988; 263(7)3111–3115
- Araya J, Cambier S, Markovics J A, Wolters P, Jablons D, Hill A, Finkbeiner W, Jones K, Broaddus V C, Sheppard D, Barzcak A, Xiao Y, Erle D J, Nishimura S L. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest 2007; 117(11)3551–3562
- Zandvoort A, Postma D S, Jonker M R, Noordhoek J A, Vos J T, van der Geld Y M, Timens W. Altered expression of the Smad signalling pathway: implications for COPD pathogenesis. Eur Respir J 2006; 28(3)533–541
- Springer J, Scholz F R, Peiser C, Groneberg D A, Fischer A. SMAD-signaling in chronic obstructive pulmonary disease: transcriptional down-regulation of inhibitory SMAD 6 and 7 by cigarette smoke. Biol Chem 2004; 385(7)649–653
- Nakao A, Afrakhte M, Moren A, Nakayama T, Christian J L, Heuchel R, Itoh S, Kawabata M, Heldin N E, Heldin C H, ten Dijke P. Identification of Smad7, a TGF-β -inducible antagonist of TGF-β signalling. Nature 1997; 389(6651)631–635
- Afrakhte M, Moren A, Jossan S, Itoh S, Sampath K, Westermark B, Heldin C H, Heldin N E, ten Dijke P. Induction of inhibitory Smad6 and Smad7 mRNA by TGF-β family members. Biochem Biophys Res Commun 1998; 249(2)505–511
- Massague J. TGF-β signal transduction. Annu Rev Biochem 1998; 67753–67791
- Ohnishi K, Takagi M, Kurokawa Y, Satomi S, Konttinen Y T. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab Invest 1998; 78(9)1077–1087
- Togo S, Holz O, Liu X, Sugiura H, Kamio K, Wang X, Kawasaki S, Ahn Y, Fredriksson K, Skold C M, Mueller K C, Branscheid D, Welker L, Watz H, Magnussen H, Rennard S I. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am J Respir Crit Care Med 2008; 178(3)248–260
- Araya J, Cambier S, Morris A, Finkbeiner W, Nishimura S L. Integrin-mediated transforming growth factor-β activation regulates homeostasis of the pulmonary epithelial-mesenchymal trophic unit. Am J Pathol 2006; 169(2)405–415