
Abstract
Chronic obstructive pulmonary disease (COPD) is a chronic respiratory condition characterized by persistent inflammation and oxidative stress, which ultimately leads to progressive restriction of airflow. Extensive research findings have cogently suggested that the dysregulation of essential transition metal ions, notably iron, copper, and zinc, stands as a critical nexus in the perpetuation of inflammatory processes and oxidative damage within the lungs of COPD patients. Unraveling the intricate interplay between metal homeostasis, oxidative stress, and inflammatory signaling is of paramount importance in unraveling the intricacies of COPD pathogenesis. This comprehensive review aims to examine the current literature on the sources, regulation, and mechanisms by which metal dyshomeostasis contributes to COPD progression. We specifically focus on iron, copper, and zinc, given their well-characterized roles in orchestrating cytokine production, immune cell function, antioxidant depletion, and matrix remodeling. Despite the limited number of clinical trials investigating metal modulation in COPD, the advent of emerging methodologies tailored to monitor metal fluxes and gauge responses to chelation and supplementation hold great promise in unlocking the potential of metal-based interventions. We conclude that targeted restoration of metal homeostasis represents a promising frontier for ameliorating pathological processes driving COPD progression.
Introduction
Chronic obstructive pulmonary disease (COPD) stands as a prominent global cause of morbidity and mortality, predicted to rank as the third leading cause of death by 2030 [Citation1–3]. Encompassing emphysema, chronic bronchitis, and small airways disease, COPD manifests through persistent respiratory symptoms, progressive and irreversible airflow limitation, and chronic inflammatory responses in the lungs triggered by inhaled irritants [Citation4–7]. Although smoking remains the primary risk factor [Citation8], the development of COPD among only a subset of smokers suggests the involvement of other mediators [Citation9], leaving the key drivers of disease onset and progression shrouded in uncertainty.
Central to the hallmark features of COPD are chronic airway inflammation, lung parenchyma inflammation, and pulmonary vascular inflammation. These processes are intricately intertwined with oxidative stress resulting from an imbalance between reactive oxygen species (ROS) and endogenous antioxidant defenses [Citation10–14]. ROS, encompassing oxygen radicals like superoxide and hydroxyl radicals, as well as non-radical species like hydrogen peroxide, inflict cellular injury by oxidizing DNA, proteins, and lipids when not adequately countered by antioxidants [Citation15].
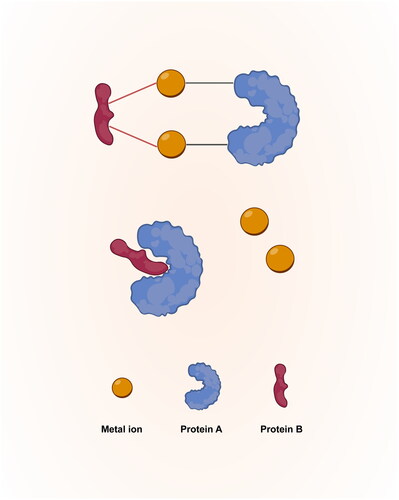
Metal ions, such as iron, copper, and zinc, are essential micronutrients that play critical roles in maintaining human health and lifespan [Citation16]. These metals are critical cofactors for an extensive array of enzymes and proteins involved in key biochemical processes [Citation17]. They also serve as matchmakers and coordinate the interaction of protein subunits [Citation18,Citation19] (). Optimal levels of these micronutrients are crucial for diverse activities ranging from oxygen transport to DNA synthesis and antioxidant defenses [Citation20–22]. Maintaining a delicate balance of these transition metals is imperative for normal biological functions, while disruptions in metal ion equilibrium have far-reaching detrimental consequences on systemic physiology and pose risks for the development of chronic diseases [Citation17,Citation23].
Figure 1. Metal ions serve as matchmakers and coordinate the interaction of protein subunits.
Recent evidence has highlighted the dysregulation of transition metal ions, specifically iron, copper, and zinc, as pivotal culprits that amplify both inflammation and oxidative damage within the lungs. This metal ion imbalance has emerged as a key driver of COPD pathogenesis [Citation24–27]. These redox-active transition metals catalyze the production of detrimental ROS through Fenton chemistry and related reactions [Citation28]. When present in excess or deficiency, these metals further augment cytokine production, immune cell activation, and tissue remodeling [Citation27,Citation29,Citation30]. Imbalances in the homeostasis and regulation of these metals appear to critically perpetuate the vicious cycle of inflammation, oxidative stress, and lung damage as COPD progresses [Citation31–33].
In this review, we delve into the current mechanistic insights surrounding the contributions of metal dyshomeostasis to COPD pathogenesis. Our primary focus lies on iron, copper, and zinc, given their extensive characterization and well-established roles as inflammatory mediators. By unraveling their impacts on inflammatory and oxidative pathways, we aim to provide strategies for preventing or mitigating disease progression through targeted metal modulation and monitoring. Ultimately, we conclude that restoring metal ion homeostasis represents a promising yet underutilized therapeutic avenue that warrants further clinical development.
The origins of metal metabolism alterations in COPD
The origins of metal metabolism alterations in COPD are multifactorial and not yet fully elucidated. Potential contributors include environmental exposures, ROS, inflammation, and nutritional influences. Environmental factors, especially cigarette smoke exposure, are well-established risk factors for COPD [Citation34–36]. Cigarette smoke contains a plethora of toxic substances, including various metals. These metals have the potential to disrupt the delicate equilibrium of metal ions within the pulmonary system [Citation37,Citation38]. Additionally, the high levels of ROS present in cigarette smoke can alter the expression and function of proteins involved in metal distribution [Citation39,Citation40], contributing to dysregulated metal homeostasis. The chronic inflammatory milieu in the lungs of COPD patients, mediated by increased levels of inflammatory cells and cytokines, appears to impact metal homeostasis as well. Notably, pro-inflammatory cytokines such as IL-1β and IL-6 have been shown to upregulate metal transporter proteins [Citation41,Citation42], potentially enhancing cellular metal uptake and retention. Moreover, nutritional factors, particularly dietary deficiencies, also play a key role in shaping metal metabolism and modulating COPD progression. For example, a correlation has been observed between low zinc intake and an increased prevalence of COPD [Citation43]. Overall, further research is needed to delineate the complex web of events leading to aberrant metal homeostasis. In particular, tracking metal regulation in the early stages of smoke exposure and COPD development will provide crucial insights into initiating factors and windows for targeted intervention.
Iron and COPD
Iron, a vital element involved in oxygen sensing, energy production, and DNA synthesis, also possesses redox reactivity that allows it to participate in the generation of reactive oxygen species (ROS) through Fenton chemistry [Citation44,Citation45]. This dual nature of iron, while essential, necessitates tight regulation of iron homeostasis to prevent oxidative stress-induced tissue damage [Citation46,Citation47].
In the lungs, the majority of iron is bound to hemoglobin within erythrocytes [Citation48]. Alveolar macrophages play a crucial role in recycling iron from aged erythrocytes through heme oxygenase-1 induction and other pathways [Citation49,Citation50]. These macrophages express ferritin for iron storage and the iron exporter ferroportin [Citation49,Citation51,Citation52]. Under normal circumstances, iron is sequestered within proteins, resulting in negligible free iron levels [Citation53]. However, disruption of these finely-tuned regulatory mechanisms in COPD leads to impaired iron excretion and accumulation of iron within the airspaces, driving oxidative stress and inflammation [Citation54,Citation55].
Notably, the levels of redox-active iron in the lungs of COPD patients are increased in proportion to disease severity [Citation56,Citation57]. This enhanced iron accumulation is associated with reduced activity of iron regulatory proteins (IRPs), which normally repress uptake mechanisms such as divalent metal transporter 1 (DMT1) [Citation58]. Combined with increased hemolysis caused by cigarette smoke exposure [Citation59], this altered environment facilitates pathological iron loading within the pulmonary airspaces.
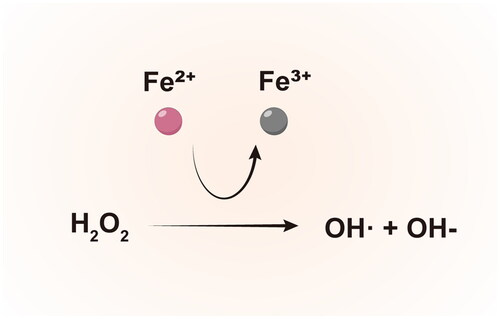
The excess iron in COPD pathogenesis exacerbates oxidative stress and inflammation through several key mechanisms. Redox cycling of ferrous iron with oxygen species catalyzes the formation of highly reactive hydroxyl radicals (OH•) through Fenton chemistry [Citation60] (). Hydroxyl radicals, due to their diffusion-limited reactivity, inflict oxidative damage upon proteins, lipids, carbohydrates, and DNA [Citation61]. This oxidative stress is strongly correlated with lung iron content, as evidenced by markers such as 8-isoprostane [Citation62].
Figure 2. Fenton reaction.
Iron also amplifies cytokine-mediated inflammation. Increased iron uptake leads to elevated production of inflammatory mediators such as tumor necrosis factor (TNF)-α in macrophages [Citation63,Citation64]. This effect is mediated by iron-induced stabilization of hypoxia-inducible factor (HIF)-1α and activation of nuclear factor kappa B (NF-κB), both of which are master transcriptional regulators of inflammatory mediators [Citation65–67]. HIF-1α further stimulates vascular remodeling, which has been implicated in the development of pulmonary hypertension in COPD [Citation68]. Furthermore, Iron can activate immune cells, including neutrophils, contributing to a self-propagating pro-inflammatory cycle that likely underlies the correlation between lung iron burden and COPD severity [Citation69,Citation70].
In light of these inherent properties, the strategic modulation of iron homeostasis emerges as an enticing avenue in the battle against COPD pathogenesis. Findings from animal models subjected to chronic cigarette smoke exposure underscore the potential benefits of iron chelation, most notably in attenuating oxidative stress, dampening inflammatory responses, mitigating mucus hypersecretion, and ameliorating emphysematous changes [Citation58]. Compellingly, preclinical studies have elucidated the protective attributes of the natural siderophore deferiprone in the context of COPD pathogenesis [Citation58,Citation71]. However, the application of iron chelation in clinical settings for COPD management remains comparatively understudied and unexplored.
The prospective surveillance of iron levels within exhaled breath condensate, sputum or bronchoalveolar lavage (BAL) fluid over time holds promise as a valuable tool for risk assessment and disease progression prediction [Citation55,Citation57,Citation72,Citation73]. Of particular significance is the potential for noninvasive tracking of iron flux within the pulmonary airspaces, attainable through measurements in induced sputum. These advances offer intriguing prospects for gaining critical insights into the dynamic interplay of iron dysregulation in COPD. Despite these encouraging strides, it is prudent to acknowledge that further translational research efforts are indispensable to fully harness the disease-modifying potential within the intricacies of iron dysregulation in COPD.
Copper and COPD
Copper, an essential micronutrient and transition metal, plays critical roles in various enzymes, including cytochrome c oxidase, superoxide dismutase (SOD), and ceruloplasmin [Citation74]. Copper is also an essential cofactor for lysyl oxidase, which crosslinks elastin and collagen fibers in the extracellular matrix of the lung parenchyma [Citation75]. Nevertheless, an excess of copper can lead to tissue injury [Citation76]. Notably, patients with COPD exhibit elevated levels of copper in serum and sputum compared to healthy individuals [Citation24,Citation77–80]. This pathological increase in copper likely drives the progression of COPD by exerting overlapping effects on inflammation and oxidative stress. In terms of inflammation, copper directly stimulates the production of interleukin (IL)-6 and IL-8 by bronchial epithelial cells in a dose-dependent manner [Citation81]. Furthermore, copper amplifies the activation of NF-κB, a central pro-inflammatory transcription factor that regulates various cytokines, chemokines, and adhesion molecules [Citation82]. Moreover, copper significantly exacerbates oxidative stress by acting as a catalyst for reactive oxygen species (ROS) production through Fenton-like chemistry, mirroring the role iron plays in the process [Citation83]. This elevation in oxidative stress can cause damage with proteins, lipids, and DNA, eventually triggering a cascade of events leading to airway inflammation and structural modifications in extracellular matrix components [Citation84].
However, an intriguing dichotomy emerges when the animal model of COPD is examined, revealing that dietary copper depletion leads to emphysema [Citation85]. This enigmatic observation indicates that optimal copper levels are required to maintain lung structure and function. The emphysema resulting from copper deficiency may stem from impaired activity of lysyl oxidase, a copper-dependent enzyme that crosslinks elastin and collagen fibers [Citation75]. Copper deficiency could also trigger alveolar cell death by anoikis through downregulation of hypoxia-inducible factor-1α (HIF-1α) and focal adhesion kinase (FAK) [Citation85]. Hence, it becomes evident that both excessive and deficient copper levels harbor the potential to contribute to COPD pathology, albeit through distinct mechanisms. More research is needed to fully elucidate the complex relationship between systemic copper status, tissue-specific copper levels, and emphysema development.
Taken together, these intricate facets of copper’s influence offer a compelling rationale for copper-targeted therapies in COPD. Similar to iron, monitoring copper fluxes across different lung compartments could emerge as a valuable tool in evaluating disease status and response to therapeutic interventions. Overall, the present body of evidence underscores the disrupted equilibrium of copper homeostasis as a pivotal factor in COPD progression.
Zinc and COPD
Zinc emerges as a distinctive player in COPD pathogenesis, standing in stark contrast to iron and copper. The presence of adequate zinc levels appears to be beneficial in countering inflammation and oxidative stress. Zinc possesses antioxidant and anti-inflammatory properties when maintained at sufficient levels [Citation31]. However, zinc deficiency is observed in bronchoalveolar lavage (BAL) fluid and serum of COPD patients, and its severity worsens as the disease progresses [Citation80,Citation86].
Zinc reaches the lungs through dietary uptake and circulating zinc-protein complexes like α2-macroglobulin, which can enter the airspaces [Citation87,Citation88]. Zinc serves as a necessary cofactor for over 300 enzymes and 2,000 transcription factors [Citation89]. It plays both structural and catalytic roles in antioxidant metalloproteins like Cu/Zn superoxide dismutase (SOD) [Citation90]. In COPD airspaces, zinc deficiency impairs defenses against oxidative stress through several mechanisms. Firstly, reduced zinc levels decrease the activity of SOD, hindering dismutation of superoxide into oxygen and hydrogen peroxide [Citation91]. Diminished SOD activity can directly contribute to unchecked oxidation in COPD lungs. Another impacted zinc-dependent oxidoreductase is glutathione peroxidase, responsible for degrading H2O2 and lipid peroxides. Zinc deficiency impairs this enzyme as well, contributing to oxidative damage [Citation92].
Zinc deficiency is also a critical factor exacerbating the inflammatory processes observed in COPD. Zinc inhibits NF-κB activity by preventing IKKβ-mediated phosphorylation and subsequent degradation of IκBα. This mechanism limits NF-κB nuclear translocation and DNA binding [Citation93], thereby curtailing the downstream production of pro-inflammatory cytokines, including TNF-α and IL-1β [Citation94]. Furthermore, zinc plays an instrumental role in enhancing the expression of A20, a zinc finger protein recognized for its anti-inflammatory properties, through epigenetic modifications at the A20 promoter [Citation95]. In the absence of this regulatory effect, the pro-inflammatory mediators that orchestrate the COPD inflammatory cascade will become highly upregulated. Zinc also regulates the activity of immune cells, most notably neutrophils. By suppressing the enzymatic activity of peptidylarginine deiminases (PADs), zinc effectively limits the citrullination of histone H3 96. This inhibition of histone H3 citrullination is a crucial step in mitigating the formation and release of neutrophil extracellular traps (NETs), which are integral to inflammatory responses [Citation97]. Conversely, low zinc levels lead to an elevated release of NETs and intensified neutrophil degranulation [Citation96]. Taken together, zinc acts as a brake on inflammation, and its deficiency paves the way for the unbridled acceleration of COPD progression.
As such, zinc supplementation holds promise as an intervention to counteract COPD pathogenesis. In patients, zinc supplementation has been shown to increase SOD activity, elevate glutathione levels, and reduce inflammatory markers like C-reactive protein (CRP) [Citation89,Citation98]. However, the effects zinc on lung function of COPD patients are varied, potentially due to heterogeneity in patient zinc status, dosing regimens, or study durations [Citation99]. Further investigation is needed to identify zinc deficient patients and optimize the dosing regimen. One approach to stratify patients is the measurement of the exchangeable zinc pool through oral zinc tolerance testing [Citation100]. Overall, zinc represents a crucial micronutrient deficiency in COPD pathogenesis, and targeted repletion could offer therapeutic antioxidant and anti-inflammatory benefits.
The impact of metal dyshomeostasis on metabolic reprogramming in COPD
Alterations in metal homeostasis in COPD extends beyond its conventional association with oxidative stress and inflammation. Notably, this dysregulation is also implicated in instigating metabolic shifts, which are increasingly recognized as hallmarks of COPD pathogenesis [Citation101,Citation102].
The dysregulation of iron homeostasis, as observed in COPD, initiates a cascade of events that modulate key metabolic pathways, particularly those governing glycolysis. Iron upregulates the expression of glucose transporter 1 (GLUT1), prompting a metabolic shift toward glycolysis [Citation103]. This phenomenon, known as the Warburg effect, is characterized by an increased reliance on glycolysis for energy production, even in the presence of oxygen [Citation104]. This metabolic adaptation not only fuels the energy demands of activated immune cells but also contributes to the inflammatory milieu in COPD [Citation105,Citation106]. The metabolites that accumulate in glycolytic conditions further amplify inflammatory processes [Citation107].
Disturbances in copper homeostasis also emerge as significant contributors to the metabolic alterations associated with COPD. As a key component of complex IV of the electron transport chain, copper availability regulates the delicate balance between glycolysis and oxidative phosphorylation (OXPHOS) [Citation108]. Copper deficiency impairs complex IV activity and mitochondrial respiration, driving a compensatory increase in glycolytic metabolism to meet cellular energy demands. This results in lactate accumulation and altered redox status, further amplifying inflammatory signaling [Citation74,Citation109,Citation110]. Conversely, copper overload enhances complex IV activity and mitochondrial function, steering cellular metabolism toward OXPHOS [Citation111]. Furthermore, an excess of copper contributes to lipid oxidation, which negatively impacts membrane integrity and function.
Zinc deficiency in COPD not only compromises antioxidant defenses but also disrupts metabolic homeostasis. Reduced zinc levels diminish the activity of vital metabolic enzymes, most notably carbonic anhydrase [Citation112]. This ubiquitous zinc metalloenzyme catalyzes the interconversion of carbon dioxide (CO2) and carbonic acid (H2CO3), impacting a vast array of physiological functions such as electrolyte secretion, gluconeogenesis, lipogenesis, and ureagenesis [Citation113,Citation114]. Moreover, zinc dyshomeostasis distorts lipid metabolism primarily through its effects on peroxisome proliferator-activated receptors (PPARs), which rely on zinc-finger structures to function properly [Citation115,Citation116].
Overall, the disruption of metal homeostasis in COPD alters cellular metabolism. Restoring metal equilibrium may therefore confer metabolic benefits in COPD alongside antioxidant and anti-inflammatory effects.
Potential implications of metal-associated redox changes in autoantibody production in COPD
A critical aspect of metal-driven oxidative stress that warrants consideration is the potential alteration of self-antigens and a resultant breach in immunological self-tolerance. It has been demonstrated that oxidative changes to proteins can greatly enhance their immunogenicity, enabling a modified antigen to elicit a more robust immune response [Citation117]. In COPD, it is plausible that an imbalance of redox-active metals may create an environment conducive to the oxidative modification of self-proteins in the lungs. These neoantigens could stimulate autoantibody production and lead to loss of self-tolerance, contributing to the autoimmune components of COPD pathogenesis. Intriguingly, COPD patients exhibit elevated levels of autoantibodies [Citation118,Citation119], lending clinical support to this theory. This may represent an additional pathway through which metal dyshomeostasis and the resulting oxidative stress may fuel autoantibody production in COPD, thereby perpetuating inflammatory cascades. Further exploration of the interplay between metal-driven oxidative modifications, breakdown of self-tolerance, and autoimmunity will provide valuable insights into the multifaceted immunological repercussions of metal imbalance. Unraveling these connections represents an important frontier in understanding the intricate role metals play in orchestrating immune dysregulation in COPD pathogenesis.
The impact of metal dyshomeostasis on infections in COPD
Infections represent major drivers of acute exacerbations and disease progression in COPD [Citation120–122]. Both viral and bacterial pathogens, especially nontypeable Haemophilus influenzae, Moraxella catarrhalis, and Streptococcus pneumoniae, are frequently isolated during exacerbation episodes [Citation123]. Intriguingly, accumulating evidence indicates that the pathogenesis of these infections is closely intertwined with the host’s metal status, with iron, copper, and zinc emerging as key players.
Iron serves as a key micronutrient for respiratory pathogens [Citation124]. It facilitates the growth and virulence of pathogens by acting as a crucial cofactor in metabolic processes and oxygen utilization [Citation125]. Therapeutic iron chelation restricts bacterial growth in vitro, while iron supplementation exhibits opposite effects [Citation126,Citation127], supporting the notion that iron availability directly impacts susceptibility to infection in COPD. Furthermore, intracellular iron accumulation in macrophages impairs their antimicrobial functions by inhibiting lysosomal acidification [Citation128]. This may result in reduced bacterial clearance and increased vulnerability to respiratory infections.
The role of copper in microbial infections in COPD is a multifaceted phenomenon with intricate implications. The extensive presence of genes encoding copper-dependent proteins in diverse bacterial genomes serves as a compelling indicator of the vital role copper plays in microbial growth [Citation129,Citation130]. Nonetheless, the dichotomy of copper emerges as it exhibits antimicrobial properties when harnessed by the host, acting as a formidable weapon to limit pathogen growth [Citation131,Citation132]. Copper deficiency in animals has been shown to impair critical immune functions, including antibody production and respiratory burst in neutrophils and macrophages [Citation133–136]. The susceptibility of individuals with Menkes disease, a lethal copper deficiency disorder, to microbial infections further emphasizes copper’s critical role in host defense [Citation137]. Conversely, pathogenic bacteria have evolved sophisticated strategies to withstand the toxic effects of elevated copper levels. Notably, bacteria like Haemophilus influenzae demonstrate elaborate copper-responsive systems to navigate the delicate balance between mitigating toxicity and ensuring a sufficient supply for vital cuproproteins [Citation138]. This adaptation empowers such microbes to thrive in the copper-rich inflammatory niche characteristic of COPD airways, shedding light on the interplay between copper, microbial pathogenesis, and host defense mechanisms in respiratory infections.
Zinc deficiency is linked to enhanced vulnerability to infections caused by Streptococcus pneumoniae [Citation139]. This elevated risk is likely due to zinc’s direct antimicrobial properties against pathogens [Citation140]. Research in murine models has shown that dietary zinc levels markedly affect the progression and outcome of Streptococcus pneumoniae infections. A reduction in dietary zinc intake results in diminished phagocyte zinc stores, leading to a compromised capacity for Streptococcus pneumoniae elimination due to hindered zinc-dependent antimicrobial mechanisms [Citation139]. Furthermore, zinc is vital for maintaining the structural integrity of the respiratory epithelium, a natural barrier against infectious agents [Citation141,Citation142]. Zinc deficiency in alveolar macrophages and lung epithelia contributes to respiratory distress syndrome by weakening lung barrier defenses [Citation87,Citation143,Citation144]. This, in turn, renders individuals, especially those with COPD, more prone to bacterial invasion. Zinc supplementation has been observed to mitigate infection risks [Citation145], suggesting its potential as a therapeutic adjunct in managing infection susceptibility among COPD patients. Therefore, ensuring adequate zinc intake may represent a viable strategy to bolster host defenses against respiratory pathogens, particularly in populations at increased risk due to underlying pulmonary conditions.
Overall, the complex interplay between biometals and respiratory pathogens represents an emerging area of interest. A deeper mechanistic understanding of how biometal dyshomeostasis influences infections in COPD would open new therapeutic avenues, such as selective metal chelation or repletion to restrict pathogen growth while minimizing inflammation.
The intricate interplay of metal ions
A significant revelation in the field of biological trace element research is the intricate relationships that bind iron, copper, and zinc. Instead of operating in solitary isolation, these essential metals emerge as interwoven threads within a complex network of pathways [Citation146–148]. This discovery not only underscores the depth of their connectivity but also illuminates the interplay that exists between metal homeostasis and COPD progression.
A notable illustration of this intricate interdependence can be found in the multifaceted actions of the versatile multicopper oxidase ceruloplasmin. This ‘moonlighting’ protein, beyond its primary role in copper transportation within the bloodstream, extends its influence into the realm of iron homeostasis [Citation149–151].
Ceruloplasmin’s copper-laden molecular structure empowers it to catalyze crucial oxidation reactions [Citation152]. A key biochemical process it facilitates is the oxidation of ferrous iron to ferric iron, an essential reaction for iron mobilization. Once exported from cells via ferroportin, ferrous iron undergoes oxidation by ceruloplasmin, enabling its binding to transferrin for effective transport [Citation153–155]. Thus, the ceruloplasmin-ferroportin system constitutes the primary mechanism for cellular iron efflux and plays a pivotal role in the regulation of cellular iron homeostasis [Citation156]. Consequently, disruptions in ceruloplasmin function can profoundly impact iron metabolism. Meanwhile, zinc emerges as a pivotal player in modulating the equilibrium of both iron and copper. Notably, zinc triggers the uptake of iron and its transcellular transport through the induction of DMT1 and ferroportin [Citation157]. Furthermore, zinc exerts inhibitory effects on copper uptake mediated by copper transporter 1 (CTR1) [Citation158]. The profound interconnectedness among these essential metal ions underscores the necessity of addressing COPD-associated metal dyshomeostasis as a collective entity, rather than as isolated anomalies.
Monitoring and measuring metal fluxes in COPD
The advancement and implementation of metal-targeted therapies in clinical settings would greatly benefit from techniques that allow for the longitudinal measurement of metal levels in both the airspaces and systemic circulation of COPD patients. Such capabilities would help elucidate the mechanistic links between systemic metal homeostasis and the dynamic metal fluxes within the airspaces. While systemic metal dyshomeostasis can impact pulmonary metal levels through circulating metal-protein complexes, the lung possesses intricate regulatory mechanisms that maintain metal equilibrium locally [Citation159,Citation160]. Disentangling the crosstalk between systemic and pulmonary metal handling holds the key to unlocking the mysteries of metal dyshomeostasis in COPD progression. Developing techniques to longitudinally track both systemic and airspace metal fluxes in patients represents a vital step toward this goal.
One such technique is the use of inductively coupled plasma mass spectrometry (ICP-MS) to analyze bronchoalveolar lavage (BAL) fluid, sputum, and lung tissue, providing quantitative assessments of total metal concentrations, including iron, copper, and zinc [Citation25,Citation161]. Another technique, X-ray fluorescence microscopy, allows for the quantification and spatial mapping of metals within lung tissues [Citation162].
While these approaches provide valuable snapshots of metal distribution, they lack the ability to provide dynamic longitudinal information. Recently, emerging in vivo imaging methods such as laser ablation-ICP-MS and X-ray fluorescence tomography have shown promise in enabling three-dimensional spatial mapping of metal pools, albeit primarily in preclinical models [Citation163–165]. Additionally, positron emission tomography utilizing radio-labeled tracers like 64-copper (64Cu) has shown potential in measuring labile copper pools, thus paving the way for translational prospects [Citation166].
The development of minimally-invasive biomarkers that correlate with alterations in lung metal pools would also allow for longitudinal monitoring. For instance, the level of serum ceruloplasmin has been found to modestly reflect soluble copper content [Citation167]. Furthermore, zinc availability can be assessed through oral zinc tolerance testing [Citation100]. Yet, amidst these findings, a pressing need lingers for robust biomarkers that can specifically indicate metal dyshomeostasis within the pulmonary airspaces. The identification of such biomarkers, along with the development of platforms that enable noninvasive tracking of lung metal content, remains a crucial future objective in order to facilitate responsive therapies.
Moreover, despite the well-established circadian fluctuations in plasma metals in healthy individuals [Citation168,Citation169], these dynamic patterns remain unexplored in patients with COPD. To bridge this knowledge gap, dedicated longitudinal studies are recommended to track blood metal levels in COPD patients over time under standardized conditions. Frequent serial sampling coupled with circadian mapping would illuminate patterns and help discern the stability versus variability of these potential biomarkers. Elucidating the intricacies of trace metal homeostasis and its perturbations in COPD will provide key insights into disease pathogenesis and progression. In addition, quantifying the magnitude and time scale of fluctuations in blood metal levels may unveil new prognostic biomarkers and help inform the design of future metal-modifying interventions.
Conclusion
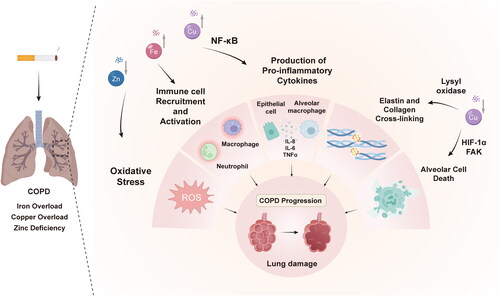
In conclusion, the extensive body of evidence now unequivocally demonstrates that imbalances in transition metal ions play a fundamental role in driving the characteristic inflammation, oxidative stress, and lung damage observed in COPD progression (). Our understanding has evolved from mere associative observations to a refined mechanistic comprehension of the intricate interplay between iron, copper and zinc. This knowledge underscores the restoration of metal homeostasis as a promising yet underutilized therapeutic opportunity. By improving clinical biomarkers that monitor metal fluxes and conducting well-designed trials focusing on chelators, supplements, and combination therapies, we can ultimately determine the optimal role of metal-targeted approaches within the COPD treatment arsenal. Although much work lies ahead, further exploration of this underexplored field holds tremendous potential for attenuating the relentless cycle that underlies this significant cause of global morbidity and mortality.
Figure 3. The roles of metal dyshomeostasis in COPD progression.
Disclosure statement
The authors report there are no competing interests to declare.
Additional information
Funding
References
- Chen S, Kuhn M, Prettner K, et al. The global economic burden of chronic obstructive pulmonary disease for 204 countries and territories in 2020-50: a health-augmented macroeconomic modelling study. Lancet Glob Health. 2023;11(8):1–13. doi: 10.1016/S2214-109X(23)00217-6.
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. doi: 10.1371/journal.pmed.0030442.
- Wang C, Hao X, Chen S. Calling for improved pulmonary and critical care medicine in China and beyond. Chinese Med J Pulm Crit Care Med. 2023;1(1):1–2. doi: 10.1016/j.pccm.2023.03.005.
- Yadav AK, Gu W, Zhang T, et al. Current perspectives on biological therapy for COPD. COPD. 2023;20(1):197–209. doi: 10.1080/15412555.2023.2187210.
- Chen D, Curtis JL, Chen Y. Twenty years of changes in the definition of early chronic obstructive pulmonary disease. Chinese Med J Pulm Crit Care Med. 2023;1(2):84–93. doi: 10.1016/j.pccm.2023.03.004.
- DeVries R, Kriebel D, Sama S. Outdoor air pollution and COPD-related emergency department visits, hospital admissions, and mortality: a meta-analysis. COPD. 2017;14(1):113–121. doi: 10.1080/15412555.2016.1216956.
- Hansel NN, McCormack MC, Kim V. The effects of air pollution and temperature on COPD. COPD. 2016;13(3):372–379. doi: 10.3109/15412555.2015.1089846.
- Tajbakhsh A, Gheibihayat SM, Mortazavi D, et al. The effect of cigarette smoke exposure on efferocytosis in chronic obstructive pulmonary disease; molecular mechanisms and treatment opportunities. COPD. 2021;18(6):723–736. doi: 10.1080/15412555.2021.1978419.
- Terzikhan N, Verhamme KM, Hofman A, et al. Prevalence and incidence of COPD in smokers and non-smokers: the Rotterdam study. Eur J Epidemiol. 2016;31(8):785–792. doi: 10.1007/s10654-016-0132-z.
- Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi: 10.1016/j.jaci.2016.05.011.
- Barnes PJ. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020;33:101544. doi: 10.1016/j.redox.2020.101544.
- Choudhury G, MacNee W. Role of inflammation and oxidative stress in the pathology of ageing in COPD: potential therapeutic interventions. COPD. 2017;14(1):122–135. doi: 10.1080/15412555.2016.1214948.
- Barnes PJ. Oxidative stress in chronic obstructive pulmonary disease. Antioxidants. 2022;11(5):965. doi: 10.3390/antiox11050965.
- Lin JL, Thomas PS. Current perspectives of oxidative stress and its measurement in chronic obstructive pulmonary disease. COPD. 2010;7(4):291–306. doi: 10.3109/15412555.2010.496818.
- Oberley-Deegan RE, Regan EA, Kinnula VL, et al. Extracellular superoxide dismutase and risk of COPD. COPD. 2009;6(4):307–312. doi: 10.1080/15412550903085193.
- Moustakas M. The role of metal ions in biology, biochemistry and medicine. Materials (Basel). 2021;14(3):549. doi: 10.3390/ma14030549.
- Jomova K, Makova M, Alomar SY, et al. Essential metals in health and disease. Chem Biol Interact. 2022;367:110173. doi: 10.1016/j.cbi.2022.110173.
- Lu Y. Metal ions as matchmakers for proteins. Proc Natl Acad Sci U S A. 2010;107(5):1811–1812. doi: 10.1073/pnas.0914008107.
- Salgado EN, Ambroggio XI, Brodin JD, et al. Metal templated design of protein interfaces. Proc Natl Acad Sci U S A. 2010;107(5):1827–1832. doi: 10.1073/pnas.0906852107.
- MacDonald RS. The role of zinc in growth and cell proliferation. J Nutr. 2000;130(5S Suppl):1500S–1508S. doi: 10.1093/jn/130.5.1500S.
- Prasad AS. Zinc: an antioxidant and anti-inflammatory agent: role of zinc in degenerative disorders of aging. J Trace Elem Med Biol. 2014;28(4):364–371. doi: 10.1016/j.jtemb.2014.07.019.
- Hirota K. An intimate crosstalk between iron homeostasis and oxygen metabolism regulated by the hypoxia-inducible factors (HIFs). Free Radic Biol Med. 2019;133:118–129. doi: 10.1016/j.freeradbiomed.2018.07.018.
- Bleackley MR, Macgillivray RT. Transition metal homeostasis: from yeast to human disease. Biometals. 2011;24(5):785–809. doi: 10.1007/s10534-011-9451-4.
- Kunutsor SK, Voutilainen A, Laukkanen JA. Serum copper-to-zinc ratio and risk of chronic obstructive pulmonary disease: a cohort study. Lung. 2023;201(1):79–84. doi: 10.1007/s00408-022-00591-6.
- Fei Q, Weng X, Liu K, et al. The relationship between metal exposure and chronic obstructive pulmonary disease in the general US population: NHANES 2015-2016. Int J Environ Res Public Health. 2022;19(4):2085. doi: 10.3390/ijerph19042085.
- Ghio AJ, Pavlisko EN, Roggli VL, et al. Cigarette smoke particle-induced lung injury and iron homeostasis. Int J Chron Obstruct Pulmon Dis. 2022;17:117–140. doi: 10.2147/COPD.S337354.
- Healy C, Munoz-Wolf N, Strydom J, et al. Nutritional immunity: the impact of metals on lung immune cells and the airway microbiome during chronic respiratory disease. Respir Res. 2021;22(1):133. doi: 10.1186/s12931-021-01722-y.
- Szabo R, Bodolea C, Mocan T. Iron, copper, and zinc homeostasis: physiology, physiopathology, and nanomediated applications. Nanomaterials. 2021;11(11):2958. doi: 10.3390/nano11112958.
- Cotroneo E, Ashek A, Wang L, et al. Iron homeostasis and pulmonary hypertension: iron deficiency leads to pulmonary vascular remodeling in the rat. Circ Res. 2015;116(10):1680–1690. doi: 10.1161/CIRCRESAHA.116.305265.
- Lu J, Liu X, Li X, et al. Copper regulates the host innate immune response against bacterial infection via activation of ALPK1 kinase. Proc Natl Acad Sci U S A. 2024;121(4):e2311630121. doi: 10.1073/pnas.2311630121.
- Luan R, Ding D, Xue Q, et al. Protective role of zinc in the pathogenesis of respiratory diseases. Eur J Clin Nutr. 2023;77(4):427–435. doi: 10.1038/s41430-022-01191-6.
- Chang CJ, Brady DC. Capturing copper to inhibit inflammation. Nat Chem Biol. 2023;19(8):937–939. doi: 10.1038/s41589-023-01383-6.
- Maywald M, Wessels I, Rink L. Zinc signals and immunity. Int J Mol Sci. 2017;18(10):2222. doi: 10.3390/ijms18102222.
- Walter R, Gottlieb DJ, O’Connor GT. Environmental and genetic risk factors and gene-environment interactions in the pathogenesis of chronic obstructive lung disease. Environ Health Perspect. 2000;108 (Suppl 4):733–742. doi: 10.1289/ehp.00108s4733.
- Eisner MD, Anthonisen N, Coultas D, et al. An official American Thoracic Society public policy statement: novel risk factors and the global burden of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182(5):693–718. doi: 10.1164/rccm.200811-1757ST.
- Shen Y, Chen L, Chen J, et al. Mitochondrial damage-associated molecular patterns in chronic obstructive pulmonary disease: pathogenetic mechanism and therapeutic target. J Transl Int Med. 2023;11(4):330–340. doi: 10.2478/jtim-2022-0019.
- Pappas RS. Toxic elements in tobacco and in cigarette smoke: inflammation and sensitization. Metallomics. 2011;3(11):1181–1198. doi: 10.1039/c1mt00066g.
- Bernhard D, Rossmann A, Wick G. Metals in cigarette smoke. IUBMB Life. 2005;57(12):805–809. doi: 10.1080/15216540500459667.
- Caliri AW, Tommasi S, Besaratinia A. Relationships among smoking, oxidative stress, inflammation, macromolecular damage, and cancer. Mutat Res Rev Mutat Res. 2021;787:108365. doi: 10.1016/j.mrrev.2021.108365.
- Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10(11):822. doi: 10.1038/s41419-019-2064-5.
- Zhou S, Du X, Xie J, et al. Interleukin-6 regulates iron-related proteins through c-Jun N-terminal kinase activation in BV2 microglial cell lines. PLoS One. 2017;12(7):e0180464. doi: 10.1371/journal.pone.0180464.
- Hansen JB, Tonnesen MF, Madsen AN, et al. Divalent metal transporter 1 regulates iron-mediated ROS and pancreatic beta cell fate in response to cytokines. Cell Metab. 2012;16(4):449–461. doi: 10.1016/j.cmet.2012.09.001.
- Lin YS, Caffrey JL, Chang MH, et al. Cigarette smoking, cadmium exposure, and zinc intake on obstructive lung disorder. Respir Res. 2010;11(1):53. doi: 10.1186/1465-9921-11-53.
- Galy B, Conrad M, Muckenthaler M. Mechanisms controlling cellular and systemic iron homeostasis. Nat Rev Mol Cell Biol. 2024;25(2):133–155. doi: 10.1038/s41580-023-00648-1.
- Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10(1):9–17. doi: 10.1038/nchembio.1416.
- Kawabata T. Iron-Induced oxidative stress in human diseases. Cells. 2022;11(14):2152. doi: 10.3390/cells11142152.
- Galaris D, Barbouti A, Pantopoulos K. Iron homeostasis and oxidative stress: an intimate relationship. Biochim Biophys Acta Mol Cell Res. 2019;1866(12):118535. doi: 10.1016/j.bbamcr.2019.118535.
- Vogt AS, Arsiwala T, Mohsen M, et al. On iron metabolism and its regulation. Int J Mol Sci. 2021;22(9):4591. doi: 10.3390/ijms22094591.
- Recalcati S, Cairo G. Macrophages and iron: a special relationship. Biomedicines. 2021;9(11):1585. doi: 10.3390/biomedicines9111585.
- Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD. 2004;1(1):59–70. doi: 10.1081/COPD-120028701.
- DeRosa A, Leftin A. The iron curtain: macrophages at the interface of systemic and microenvironmental iron metabolism and immune response in cancer. Front Immunol. 2021;12:614294. doi: 10.3389/fimmu.2021.614294.
- Soares MP, Hamza I. Macrophages and iron metabolism. Immunity. 2016;44(3):492–504. doi: 10.1016/j.immuni.2016.02.016.
- Anderson GJ, Frazer DM. Current understanding of iron homeostasis. Am J Clin Nutr. 2017;106(Suppl 6):1559S–1566S. doi: 10.3945/ajcn.117.155804.
- Mizumura K, Gon Y. Iron-regulated reactive oxygen species production and programmed cell death in chronic obstructive pulmonary disease. Antioxidants. 2021;10(10):1569. doi: 10.3390/antiox10101569.
- Mumby S, Saito J, Adcock IM, et al. Decreased breath excretion of redox active iron in COPD: a protective failure? Eur Respir J. 2016;47(4):1267–1270. doi: 10.1183/13993003.01710-2015.
- Cloonan SM, Mumby S, Adcock IM, et al. The "iron"-y of iron overload and iron deficiency in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;196(9):1103–1112. doi: 10.1164/rccm.201702-0311PP.
- Zhang WZ, Oromendia C, Kikkers SA, et al. Increased airway iron parameters and risk for exacerbation in COPD: an analysis from SPIROMICS. Sci Rep. 2020;10(1):10562. doi: 10.1038/s41598-020-67047-w.
- Cloonan SM, Glass K, Laucho-Contreras ME, et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. 2016;22(2):163–174. doi: 10.1038/nm.4021.
- Masilamani V, AlZahrani K, Devanesan S, et al. Smoking induced hemolysis: spectral and microscopic investigations. Sci Rep. 2016;6(1):21095. doi: 10.1038/srep21095.
- Lloyd RV, Hanna PM, Mason RP. The origin of the hydroxyl radical oxygen in the fenton reaction. Free Radic Biol Med. 1997;22(5):885–888. doi: 10.1016/s0891-5849(96)00432-7.
- Halliwell B, Adhikary A, Dingfelder M, et al. Hydroxyl radical is a significant player in oxidative DNA damage in vivo. Chem Soc Rev. 2021;50(15):8355–8360. doi: 10.1039/d1cs00044f.
- Graille M, Wild P, Sauvain JJ, et al. Urinary 8-isoprostane as a biomarker for oxidative stress. A systematic review and meta-analysis. Toxicol Lett. 2020;328:19–27. doi: 10.1016/j.toxlet.2020.04.006.
- Sindrilaru A, Peters T, Wieschalka S, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. 2011;121(3):985–997. doi: 10.1172/JCI44490.
- Ni S, Yuan Y, Kuang Y, et al. Iron metabolism and immune regulation. Front Immunol. 2022;13:816282. doi: 10.3389/fimmu.2022.816282.
- Tacchini L, Gammella E, De Ponti C, et al. Role of HIF-1 and NF-kappaB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J Biol Chem. 2008;283(30):20674–20686. doi: 10.1074/jbc.M800365200.
- Wessling-Resnick M. Iron homeostasis and the inflammatory response. Annu Rev Nutr. 2010;30(1):105–122. doi: 10.1146/annurev.nutr.012809.104804.
- Renassia C, Peyssonnaux C. New insights into the links between hypoxia and iron homeostasis. Curr Opin Hematol. 2019;26(3):125–130. doi: 10.1097/MOH.0000000000000494.
- Lee JW, Ko J, Ju C, et al. Hypoxia signaling in human diseases and therapeutic targets. Exp Mol Med. 2019;51(6):1–13. doi: 10.1038/s12276-019-0235-1.
- Frost JN, Wideman SK, Preston AE, et al. Plasma iron controls neutrophil production and function. Sci Adv. 2022;8(40):eabq5384. doi: 10.1126/sciadv.abq5384.
- Haschka D, Hoffmann A, Weiss G. Iron in immune cell function and host defense. Semin Cell Dev Biol. 2021;115:27–36. doi: 10.1016/j.semcdb.2020.12.005.
- Kim K, Zhang WZ, Kikkers SA, et al. Use of the iron chelator deferiprone to restore function in BAL fluid macrophages in smoking and chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2023;68(4):458–462. doi: 10.1165/rcmb.2022-0372LE.
- Ho T, Nichols M, Nair G, et al. Iron in airway macrophages and infective exacerbations of chronic obstructive pulmonary disease. Respir Res. 2022;23(1):8. doi: 10.1186/s12931-022-01929-7.
- Mohan S, Ho T, Kjarsgaard M, et al. Hemosiderin in sputum macrophages may predict infective exacerbations of chronic obstructive pulmonary disease: a retrospective observational study. BMC Pulm Med. 2017;17(1):60. doi: 10.1186/s12890-017-0408-4.
- Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7(1):378. doi: 10.1038/s41392-022-01229-y.
- Janssen R, Wouters EF, Janssens W, et al. Copper-heparin inhalation therapy to repair emphysema: a scientific rationale. Int J Chron Obstruct Pulmon Dis. 2019;14:2587–2602. doi: 10.2147/COPD.S228411.
- Gaun S, Ali SA, Singh P, et al. Melatonin ameliorates chronic copper-induced lung injury. Environ Sci Pollut Res Int. 2023;30(10):24949–24962. doi: 10.1007/s11356-022-19930-4.
- Isik B, Isik RS, Ceylan A, et al. Trace elements and oxidative stress in chronic obstructive pulmonary disease. Saudi Med J. 2005;26(12):1882–1885.
- Tanrikulu AC, Abakay A, Evliyaoglu O, et al. Coenzyme Q10, copper, zinc, and lipid peroxidation levels in serum of patients with chronic obstructive pulmonary disease. Biol Trace Elem Res. 2011;143(2):659–667. doi: 10.1007/s12011-010-8897-5.
- Jiang C, Wu B, Xue M, et al. Inflammation accelerates copper-mediated cytotoxicity through induction of six-transmembrane epithelial antigens of prostate 4 expression. Immunol Cell Biol. 2021;99(4):392–402. doi: 10.1111/imcb.12427.
- Karadag F, Cildag O, Altinisik M, et al. Trace elements as a component of oxidative stress in COPD. Respirology. 2004;9(1):33–37. doi: 10.1111/j.1440-1843.2003.00534.x.
- Ko JW, Park JW, Shin NR, et al. Copper oxide nanoparticle induces inflammatory response and mucus production via MAPK signaling in human bronchial epithelial cells. Environ Toxicol Pharmacol. 2016;43:21–26. doi: 10.1016/j.etap.2016.02.008.
- Persichini T, Percario Z, Mazzon E, et al. Copper activates the NF-kappaB pathway in vivo. Antioxid Redox Signal. 2006;8(9-10):1897–1904. doi: 10.1089/ars.2006.8.1897.
- Pham AN, Xing G, Miller CJ, et al. Fenton-like copper redox chemistry revisited: hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production. J Catal. 2013;301:54–64. doi: 10.1016/j.jcat.2013.01.025.
- Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28(1):219–242. doi: 10.1183/09031936.06.00053805.
- Mizuno S, Yasuo M, Bogaard HJ, et al. Copper deficiency induced emphysema is associated with focal adhesion kinase inactivation. PLoS One. 2012;7(1):e30678. doi: 10.1371/journal.pone.0030678.
- Hamon R, Homan CC, Tran HB, et al. Zinc and zinc transporters in macrophages and their roles in efferocytosis in COPD. PLoS One. 2014;9(10):e110056. doi: 10.1371/journal.pone.0110056.
- Liu X, Ali MK, Dua K, et al. The role of zinc in the pathogenesis of lung disease. Nutrients. 2022;14(10): 2115. doi: 10.3390/nu14102115.
- Nedić O, Šunderić M, Robajac D, et al. Major trace elements and their binding proteins in the early phase of covid-19 infection. J Biol Inorg Chem. 2022;27(2):261–269. doi: 10.1007/s00775-022-01931-w.
- Faghfouri AH, Zarezadeh M, Aghapour B, et al. Clinical efficacy of zinc supplementation in improving antioxidant defense system: a comprehensive systematic review and time-response meta-analysis of controlled clinical trials. Eur J Pharmacol. 2021;907:174243. doi: 10.1016/j.ejphar.2021.174243.
- Cheng Y, Chen H. Aberrance of zinc metalloenzymes-induced human diseases and its potential mechanisms. Nutrients. 2021;13(12):4456. doi: 10.3390/nu13124456.
- Wu CY, Steffen J, Eide DJ. Cytosolic superoxide dismutase (SOD1) is critical for tolerating the oxidative stress of zinc deficiency in yeast. PLoS One. 2009;4(9):e7061. doi: 10.1371/journal.pone.0007061.
- Marreiro DD, Cruz KJ, Morais JB, et al. Zinc and oxidative stress: current mechanisms. Antioxidants. 2017;6(2):24. doi: 10.3390/antiox6020024.
- Liu M-J, Bao S, Gálvez-Peralta M, et al. ZIP8 regulates host defense through zinc-mediated inhibition of NF-kappaB. Cell Rep. 2013;3(2):386–400. doi: 10.1016/j.celrep.2013.01.009.
- Uzzo RG, Leavis P, Hatch W, et al. Zinc inhibits nuclear factor-kappa B activation and sensitizes prostate cancer cells to cytotoxic agents. Clin Cancer Res. 2002;8(11):3579–3583.
- Li C, Guo S, Gao J, et al. Maternal high-zinc diet attenuates intestinal inflammation by reducing DNA methylation and elevating H3K9 acetylation in the A20 promoter of offspring chicks. J Nutr Biochem. 2015;26(2):173–183. doi: 10.1016/j.jnutbio.2014.10.005.
- Kuźmicka W, Manda-Handzlik A, Cieloch A, et al. Zinc supplementation modulates NETs release and neutrophils’ degranulation. Nutrients. 2020;13(1):51. doi: 10.3390/nu13010051.
- Chamardani TM, Amiritavassoli S. Inhibition of NETosis for treatment purposes: friend or foe? Mol Cell Biochem. 2022;477(3):673–688. doi: 10.1007/s11010-021-04315-x.
- Mousavi SM, Djafarian K, Mojtahed A, et al. The effect of zinc supplementation on plasma C-reactive protein concentrations: a systematic review and meta-analysis of randomized controlled trials. Eur J Pharmacol. 2018;834:10–16. doi: 10.1016/j.ejphar.2018.07.019.
- Kırkıl G, Hamdi Muz M, Seçkin D, et al. Antioxidant effect of zinc picolinate in patients with chronic obstructive pulmonary disease. Respir Med. 2008;102(6):840–844. doi: 10.1016/j.rmed.2008.01.010.
- Atalay Y, Arcasoy A, Kürkçüoğlu M. Oral plasma zinc tolerance test in patients with protein energy malnutrition. Arch Dis Child. 1989;64(11):1608–1611. doi: 10.1136/adc.64.11.1608.
- Agarwal AR, Kadam S, Brahme A, et al. Systemic immuno-metabolic alterations in chronic obstructive pulmonary disease (COPD). Respir Res. 2019;20(1):171. doi: 10.1186/s12931-019-1139-2.
- Bruzzaniti S, Bocchino M, Santopaolo M, et al. An immunometabolic pathomechanism for chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2019;116(31):15625–15634. doi: 10.1073/pnas.1906303116.
- Liu Z, Villareal L, Goodla L, et al. Iron promotes glycolysis to drive colon tumorigenesis. Biochim Biophys Acta Mol Basis Dis. 2023;1869(8):166846. doi: 10.1016/j.bbadis.2023.166846.
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809.
- Kao CC, Hsu JW, Bandi V, et al. Glucose and pyruvate metabolism in severe chronic obstructive pulmonary disease. J Appl Physiol. 2012;112(1):42–47. doi: 10.1152/japplphysiol.00599.2011.
- Zhao H, Dennery PA, Yao H. Metabolic reprogramming in the pathogenesis of chronic lung diseases, including BPD, COPD, and pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2018;314(4):L544–L554. doi: 10.1152/ajplung.00521.2017.
- Soto-Heredero G, Gomez de Las Heras MM, Gabande-Rodriguez E, et al. Glycolysis - a key player in the inflammatory response. FEBS J. 2020;287(16):3350–3369. doi: 10.1111/febs.15327.
- Ishida S, Andreux P, Poitry-Yamate C, et al. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc Natl Acad Sci U S A. 2013;110(48):19507–19512. doi: 10.1073/pnas.1318431110.
- Ramchandani D, Berisa M, Tavarez DA, et al. Copper depletion modulates mitochondrial oxidative phosphorylation to impair triple negative breast cancer metastasis. Nat Commun. 2021;12(1):7311. doi: 10.1038/s41467-021-27559-z.
- Zeng H, Saari JT, Johnson WT. Copper deficiency decreases complex IV but not complex I, II, III, or V in the mitochondrial respiratory chain in rat heart. J Nutr. 2007;137(1):14–18. doi: 10.1093/jn/137.1.14.
- Ruiz LM, Libedinsky A, Elorza AA. Role of copper on mitochondrial function and metabolism. Front Mol Biosci. 2021;8:711227. doi: 10.3389/fmolb.2021.711227.
- Lukaski HC. Low dietary zinc decreases erythrocyte carbonic anhydrase activities and impairs cardiorespiratory function in men during exercise. Am J Clin Nutr. 2005;81(5):1045–1051. doi: 10.1093/ajcn/81.5.1045.
- Kim JK, Lee C, Lim SW, et al. Elucidating the role of metal ions in carbonic anhydrase catalysis. Nat Commun. 2020;11(1):4557. doi: 10.1038/s41467-020-18425-5.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7(2):168–181. doi: 10.1038/nrd2467.
- Shen H, MacDonald R, Bruemmer D, et al. Zinc deficiency alters lipid metabolism in LDL receptor deficient mice treated with rosiglitazone. J Nutr. 2007;137(11):2339–2345. doi: 10.1093/jn/137.11.2339.
- Shi Y, Zou Y, Shen Z, et al. Trace elements, PPARs, and metabolic syndrome. Int J Mol Sci. 2020;21(7):2612. doi: 10.3390/ijms21072612.
- Allison ME, Fearon DT. Enhanced immunogenicity of aldehyde-bearing antigens: a possible link between innate and adaptive immunity. Eur J Immunol. 2000;30(10):2881–2887. doi: 10.1002/1521-4141(200010)30:10.
- Feghali-Bostwick CA, Gadgil AS, Otterbein LE, et al. Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(2):156–163. doi: 10.1164/rccm.200701-014OC.
- Packard TA, Li QZ, Cosgrove GP, et al. COPD is associated with production of autoantibodies to a broad spectrum of self-antigens, correlative with disease phenotype. Immunol Res. 2013;55(1-3):48–57. doi: 10.1007/s12026-012-8347-x.
- Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173(10):1114–1121. doi: 10.1164/rccm.200506-859OC.
- Sethi S. Infection as a comorbidity of COPD. Eur Respir J. 2010;35(6):1209–1215. doi: 10.1183/09031936.00081409.
- Han Y, Hua J, He B, et al. Inhaled antibiotics and airway bacterial decolonization for patients with chronic obstructive pulmonary disease: the rationale and future. J Transl Int Med. 2022;10(3):181–184. doi: 10.2478/jtim-2022-0005.
- Sethi S, Murphy TF. Bacterial infection in chronic obstructive pulmonary disease in 2000: a state-of-the-art review. Clin Microbiol Rev. 2001;14(2):336–363. doi: 10.1128/CMR.14.2.336-363.2001.
- Nairz M, Weiss G. Iron in infection and immunity. Mol Aspects Med. 2020;75:100864. doi: 10.1016/j.mam.2020.100864.
- Kosman DJ. Energy metabolism, oxygen flux, and iron in bacteria: the Mossbauer report. J Biol Chem. 2019;294(1):63–64. doi: 10.1074/jbc.H118.006703.
- Qiu DH, Huang ZL, Zhou T, et al. In vitro inhibition of bacterial growth by iron chelators. FEMS Microbiol Lett. 2011;314(2):107–111. doi: 10.1111/j.1574-6968.2010.02153.x.
- Cross JH, Bradbury RS, Fulford AJ, et al. Oral iron acutely elevates bacterial growth in human serum. Sci Rep. 2015;5(1):16670. doi: 10.1038/srep16670.
- Kao JK, Wang SC, Ho LW, et al. Chronic iron overload results in impaired bacterial killing of THP-1 derived macrophage through the inhibition of lysosomal acidification. PLoS One. 2016;11(5):e0156713. doi: 10.1371/journal.pone.0156713.
- Ridge PG, Zhang Y, Gladyshev VN. Comparative genomic analyses of copper transporters and cuproproteomes reveal evolutionary dynamics of copper utilization and its link to oxygen. PLoS One. 2008;3(1):e1378. doi: 10.1371/journal.pone.0001378.
- Giachino A, Waldron KJ. Copper tolerance in bacteria requires the activation of multiple accessory pathways. Mol Microbiol. 2020;114(3):377–390. doi: 10.1111/mmi.14522.
- Fu Y, Chang FM, Giedroc DP. Copper transport and trafficking at the host-bacterial pathogen interface. Acc Chem Res. 2014;47(12):3605–3613. doi: 10.1021/ar500300n.
- Li C, Li Y, Ding C. The role of copper homeostasis at the host-pathogen axis: from bacteria to fungi. Int J Mol Sci. 2019;20(1):175. doi: 10.3390/ijms20010175.
- Prohaska JR, Lukasewycz OA. Copper deficiency suppresses the immune response of mice. Science. 1981;213(4507):559–561. doi: 10.1126/science.7244654.
- Babu U, Failla ML. Respiratory burst and candidacidal activity of peritoneal macrophages are impaired in copper-deficient rats. J Nutr. 1990;120(12):1692–1699. doi: 10.1093/jn/120.12.1692.
- Samanovic MI, Ding C, Thiele DJ, et al. Copper in microbial pathogenesis: meddling with the metal. Cell Host Microbe. 2012;11(2):106–115. doi: 10.1016/j.chom.2012.01.009.
- Babu U, Failla ML. Copper status and function of neutrophils are reversibly depressed in marginally and severely copper-deficient rats. J Nutr. 1990;120(12):1700–1709. doi: 10.1093/jn/120.12.1700.
- Ramani PK, Parayil Sankaran B. Menkes disease. In: statPearls. Treasure Island (FL): StatPearls Publishing; 2024.
- Wong SM, Gawronski J, Akerley BJ. Copper efflux system required in murine lung infection by Haemophilus influenzae composed of a canonical ATPase gene and tandem chaperone gene copies. Infect Immun. 2023;91(5):e0009123. doi: 10.1128/iai.00091-23.
- Eijkelkamp BA, Morey JR, Neville SL, et al. Dietary zinc and the control of Streptococcus pneumoniae infection. PLoS Pathog. 2019;15(8):e1007957. doi: 10.1371/journal.ppat.1007957.
- McDevitt CA, Ogunniyi AD, Valkov E, et al. A molecular mechanism for bacterial susceptibility to zinc. PLoS Pathog. 2011;7(11):e1002357. doi: 10.1371/journal.ppat.1002357.
- Brazel EB, Tan A, Neville SL, et al. Dysregulation of Streptococcus pneumoniae zinc homeostasis breaks ampicillin resistance in a pneumonia infection model. Cell Rep. 2022;38(2):110202. doi: 10.1016/j.celrep.2021.110202.
- Zalewski PD, Truong-Tran AQ, Grosser D, et al. Zinc metabolism in airway epithelium and airway inflammation: basic mechanisms and clinical targets. A review. Pharmacol Ther. 2005;105(2):127–149. doi: 10.1016/j.pharmthera.2004.09.004.
- Joshi PC, Mehta A, Jabber WS, et al. Zinc deficiency mediates alcohol-induced alveolar epithelial and macrophage dysfunction in rats. Am J Respir Cell Mol Biol. 2009;41(2):207–216. doi: 10.1165/rcmb.2008-0209OC.
- Skalny AV, Skalnaya MG, Grabeklis AR, et al. Zinc deficiency as a mediator of toxic effects of alcohol abuse. Eur J Nutr. 2018;57(7):2313–2322. doi: 10.1007/s00394-017-1584-y.
- Lassi ZS, Moin A, Bhutta ZA. Zinc supplementation for the prevention of pneumonia in children aged 2 months to 59 months. Cochrane Database Syst Rev. 2016;12(12):CD005978. doi: 10.1002/14651858.CD005978.pub3.
- Xu Z, Wang P, Wang H, et al. Zinc excess increases cellular demand for iron and decreases tolerance to copper in Escherichia coli. J Biol Chem. 2019;294(45):16978–16991. doi: 10.1074/jbc.RA119.010023.
- Kambe T, Weaver BP, Andrews GK. The genetics of essential metal homeostasis during development. Genesis. 2008;46(4):214–228. doi: 10.1002/dvg.20382.
- Li Y, Du Y, Zhou Y, et al. Iron and copper: critical executioners of ferroptosis, cuproptosis and other forms of cell death. Cell Commun Signal. 2023;21(1):327. doi: 10.1186/s12964-023-01267-1.
- Chen Z, Jiang R, Chen M, et al. Multi-copper ferroxidase deficiency leads to iron accumulation and oxidative damage in astrocytes and oligodendrocytes. Sci Rep. 2019;9(1):9437. doi: 10.1038/s41598-019-46019-9.
- Bielli P, Calabrese L. Structure to function relationships in ceruloplasmin: a ‘moonlighting’ protein. Cell Mol Life Sci. 2002;59(9):1413–1427. doi: 10.1007/s00018-002-8519-2.
- Doguer C, Ha JH, Collins JF. Intersection of iron and copper metabolism in the mammalian intestine and liver. Compr Physiol. 2018;8(4):1433–1461. doi: 10.1002/cphy.c170045.
- Bento I, Peixoto C, Zaitsev VN, et al. Ceruloplasmin revisited: structural and functional roles of various metal cation-binding sites. Acta Crystallogr D Biol Crystallogr. 2007;63(Pt 2):240–248. doi: 10.1107/S090744490604947X.
- Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr. 2002;22(1):439–458. doi: 10.1146/annurev.nutr.22.012502.114457.
- Ward DM, Kaplan J. Ferroportin-mediated iron transport: expression and regulation. Biochim Biophys Acta. 2012;1823(9):1426–1433. doi: 10.1016/j.bbamcr.2012.03.004.
- Vasilyev VB. Looking for a partner: ceruloplasmin in protein-protein interactions. Biometals. 2019;32(2):195–210. doi: 10.1007/s10534-019-00189-1.
- Musci G, Polticelli F, Bonaccorsi di Patti MC. Ceruloplasmin-ferroportin system of iron traffic in vertebrates. World J Biol Chem. 2014;5(2):204–215. doi: 10.4331/wjbc.v5.i2.204.
- Kondaiah P, Yaduvanshi PS, Sharp PA, et al. Iron and zinc homeostasis and interactions: does enteric zinc excretion cross-talk with intestinal iron absorption? Nutrients. 2019;11(8):1885. doi: 10.3390/nu11081885.
- Lee J, Peña MMO, Nose Y, et al. Biochemical characterization of the human copper transporter Ctr1. J Biol Chem. 2002;277(6):4380–4387. doi: 10.1074/jbc.M104728200.
- Kim J, Wessling-Resnick M. The role of iron metabolism in lung inflammation and injury. J Allergy Ther. 2012;3(Suppl 4):004. doi: 10.4172/2155-6121.S4-004.
- Neves J, Haider T, Gassmann M, et al. Iron homeostasis in the lungs-a balance between health and disease. Pharmaceuticals. 2019;12(1):5. doi: 10.3390/ph12010005.
- Clases D, Gonzalez de Vega R. Facets of ICP-MS and their potential in the medical sciences-part 1: fundamentals, stand-alone and hyphenated techniques. Anal Bioanal Chem. 2022;414(25):7337–7361. doi: 10.1007/s00216-022-04259-1.
- Zhang R, Li L, Sultanbawa Y, et al. X-ray fluorescence imaging of metals and metalloids in biological systems. Am J Nucl Med Mol Imaging. 2018;8:169–188.
- Doble PA, de Vega RG, Bishop DP, et al. Laser ablation-inductively coupled plasma-mass spectrometry imaging in biology. Chem Rev. 2021;121(19):11769–11822. doi: 10.1021/acs.chemrev.0c01219.
- Stewart TJ. Across the spectrum: integrating multidimensional metal analytics for in situ metallomic imaging. Metallomics. 2019;11(1):29–49. doi: 10.1039/c8mt00235e.
- Hare DJ, New EJ, de Jonge MD, et al. Imaging metals in biology: balancing sensitivity, selectivity and spatial resolution. Chem Soc Rev. 2015;44(17):5941–5958. doi: 10.1039/c5cs00055f.
- Emilie Munk D, Teicher Kirk F, Vendelbo M, et al. Positron emission tomography using 64-copper as a tracer for the study of copper-related disorders. J Vis Exp. 2023;(194). doi: 10.3791/65109.
- Danzeisen R, Araya M, Harrison B, et al. How reliable and robust are current biomarkers for copper status? Br J Nutr. 2007;98(4):676–683. doi: 10.1017/S0007114507798951.
- Scales WE, Vander AJ, Brown MB, et al. Human circadian rhythms in temperature, trace metals, and blood variables. J Appl Physiol. 1988;65(4):1840–1846. doi: 10.1152/japl.1988.65.4.1840.
- Parmalee NL, Aschner M. Metals and circadian rhythms. Adv Neurotoxicol. 2017;1:119–130. doi: 10.1016/bs.ant.2017.07.003.