
Abstract
Accumulating evidence has highlighted the importance of immune cells in the pathogenesis of chronic obstructive pulmonary disease (COPD). However, the understanding of the causal association between immunity and COPD remains incomplete due to the existence of confounding variables. In this study, we employed a two-sample Mendelian randomization (MR) analysis, utilizing the genome-wide association study database, to investigate the causal association between 731 immune-cell signatures and the susceptibility to COPD from a host genetics perspective. To validate the consistency of our findings, we utilized MR analysis results of lung function data to assess directional concordance. Furthermore, we employed MR-Egger intercept tests, Cochrane’s Q test, MR-PRESSO global test, and "leave-one-out" sensitivity analyses to evaluate the presence of horizontal pleiotropy, heterogeneity, and stability, respectively. Inverse variance weighting results showed that seven immune phenotypes were associated with the risk of COPD. Analyses of heterogeneity and pleiotropy analysis confirmed the reliability of MR results. These results highlight the interactions between the immune system and the lungs. Further investigations into their mechanisms are necessary and will contribute to inform targeted prevention strategies for COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive lung inflammatory disease, with pathological changes involving both large and small airways [Citation1, Citation2]. It has a significant global prevalence and leads to high mortality rates [Citation3]. The disease is characterized by persistent airflow restriction and chronic airway inflammation, which are responsible for decreased lung function and altered airway structure [Citation4]. Despite significant advances in understanding its pathophysiology, there remain several unresolved issues, particularly regarding precise control of the risk factors.
Immune dysregulation is one of the causes of COPD, which is thought to have a complicated pathogenesis [Citation5]. Innate and adaptive immune cell infiltration is related to the abnormal immune response in the lower respiratory tract in COPD [Citation6–8]. It appears that COPD develops and progresses primarily as a result of altered innate immune cell function [Citation9, Citation10]. The airway of COPD patients is invaded by more monocytes, macrophages, and neutrophils than usual. These invasive leukocytes control airway inflammation, bacterial colonization, and airway remodeling, which results in progressive airflow obstruction as COPD progresses. In acute exacerbation patients, the dysregulation of the innate immune system in COPD is considerably more serious [Citation11]. Furthermore, evidence highlights the relationship between adaptive immune cells and COPD. During the progression of COPD, the adaptive immune system is activated, which is characterized by the activation of CD8+ T cells, T helper (Th) type 17 CD4+ T cells, and B cells, as well as the inhibition of regulatory T cells (Tregs) [Citation12]. These results imply that immune cells may contribute to the development and progression of COPD, potentially through interactions with lung immunity. Although there is direct, indirect, and circumstantial evidence that immunity plays a role in COPD patients, no cause-and-effect relationship between immunity and COPD has been demonstrated.
The link between immune cells and COPD in observational studies was susceptible to confounding variables such as smoking. Chronic exposure to harmful gas leads to enhanced immune responses and persistent changes in airway structure and function. In addition, low body weight, respiratory infections, depression, and underlying antibody deficiency syndrome are predisposing factors associated with an abnormal immune response that may increase susceptibility to COPD and cause more frequent exacerbations [Citation13, Citation14]. Studies have demonstrated that genetic factors may influence airway immunity [Citation15]. Therefore, using genetic variants for causal inference is an effective approach. Mendelian randomization (MR) is a method that can overcome problems of unmeasured confounding and reverse causation typical of conventional observational epidemiology [Citation16]. MR allows causal inference through the use of genetic variants as proxies for modifiable risk factors or health outcomes [Citation17].
In this study, we performed two-sample MR analysis to explore the potential causal relationship between immune cells and COPD. These immune cell signatures may serve as potential biomarkers for the prevention of COPD and shed light on the treatment of COPD.
Methods
Study design
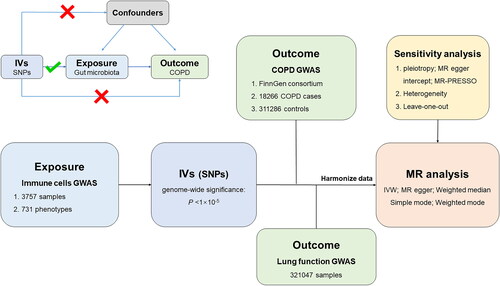
In this investigation, each immune phenotype was classified as a single exposure. The causal associations between immune phenotypes and COPD were investigated using a two-sample MR analysis. The flowchart of the MR investigation between immune cell signatures and COPD is shown in . Furthermore, in order to achieve trustworthy results, MR analysis had to meet the following three assumptions [Citation17]: (1) the instrumental variables (IVs) that were subsequently used have to be strongly connected to immune cell signatures; (2) the included IVs and confounder were independent of each other; and (3) IVs only affected COPD via the immune cell signatures pathway. In the meantime, we reported our findings in accordance with MRSTROBE guidance [Citation18].
Figure 1. Overview of MR analyses process and major assumptions.
Data sources
Subjects with persistent airflow limitation, respiratory symptoms, and a post-bronchodilator forced expiratory volume in one second (FEV1)/forced vital capacity (FVC) ratio of 70% are considered to have COPD [Citation19]. The latest genome-wide relationship studies (GWASs) summary statistics for COPD were obtained from a dataset in the FinnGen biobank analysis round 9 (https://r9.finngen.fi/), comprising 18,266 COPD cases and 311,286 controls. FinnGen biobank focuses on specific genotypes and phenotypes through genomic research. The COPD phenotype was defined based on ICD codes, including J10_EMPHYSEMA and J10_COPDNAS. The lung function GWASs summary statistics were obtained from the GWAS Catalog (https://www.ebi.ac.uk/gwas/) (ebi-a-GCST007431). This GWAS was a pooled analysis of the refined UK Biobank spirometry and the expanded SpiroMeta consortium data set, comprising 321047 European individuals [Citation20]. During the analysis, the GWAS summary statistics were adjusted for sex, age, first ten main components, and genotyping batch [Citation21].
Summary statistics containing 731 immune cell signatures were obtained from the GWAS Catalog (accession numbers from GCST0001391 to GCST0002121) [Citation22], including median fluorescence intensities (MFI) morphological parameters [MP] (n = 32), absolute cell (AC) counts (n = 118), relative cell (RC) counts (n = 192), and reflecting surface antigen levels (n = 389). This GWAS analysis on immune cell signatures was conducted based on 3,757 European samples. Imputing with a Sardinian sequence-based reference panel [Citation23], approximately 22 million single nucleotide polymorphisms (SNPs) genotyped with high-density arrays were imputed after adjusting for covariates.
Instrumental variable (IV)
To ensure the robustness of the data and the precision of the results, the SNPs were evaluated for quality to acquire compliant IVs: (1) at the locus-wide significance threshold (p < 5 × 10−5) [Citation24], SNPs linked to immune phenotypes were selected as potential IVs; (2) to guarantee the independence of the chosen IV, the Linkage Disequilibrium (LD) test was conducted using PLINK (v 1.9), and LD r2<0.1 within a window of 500 kb was applied; (3) palindromic SNPs were deleted to keep alleles from influencing the outcome of causality between immune phenotypes and COPD.
MR analysis
In this study, multiple methods including inverse variance weighting (IVW), MR-Egger, weighted median, weighted mode, and simple mode were used to examine whether there was a causal association between immune cell signatures and COPD. The IVW method combines a meta-analysis approach with Wald estimates for each SNP to generate a comprehensive assessment of immune cell signature’s impact on COPD [Citation25]. It was the main technique for computing causal impact values in the lack of horizontal pleiotropy in order to get unbiased estimates. The MR-Egger regression allows the SNPs for which the MR model is performed to be pleiotropic, and the pleiotropic effects of the SNPs in the model are reflected by intercept term. When pleiotropic SNPs are present in the model, MR-Egger can still give unbiased causal estimates [Citation26]. The weighted median (WM) method ranked the estimated proportion of each SNP from the lowest to the highest, and then weighted average by the median. When up to 50% of IVs are invalid, WM provides for the correct calculation of causal connection [Citation27]. The MR-PRESSO analysis identifies horizontal pleiotropy and eliminates it by removing significant outliers. The MR-PRESSO analysis first identified whether there was pleiotropy of the IVs included in the study by global test. When there was no pleiotropy (p > 0.05), all SNPs included in the model were considered to be valid IVs, and the result of MR-PRESSO was the final causal association estimation result. When the global test showed outliers (p < 0.05), it indicated that the overall IVs of the included studies had pleiotropic effects [Citation28]. In addition, we harmonized the summary statistics and deleted palindromic SNPs to ensure that each IV was connected with the same effect allele.
Cochrane’s Q statistics were applied to quantify and evaluate the potential heterogeneity of IVs, while the MR-Egger intercept test was used to estimate horizontal pleiotropy. In addition, to identify any potential outliers that independently influence the observed causal relationship, leave-one-out analysis was conducted. The MR-PRESSO test was also utilized to find outliers and adjust for heterogeneity for significant MR estimates. If there was heterogeneity in the IVs, the outliers were deleted and the MR analysis was repeated. The strength of IVs was assessed by calculating the F-statistic. If the corresponding F-statistic was more than 10, it was considered that there was no significant weak instrumental bias. In addition, to obtain more robust results, we validated the MR results of the COPD data in the lung function data, that is, the immune cell signatures associated with COPD risk also needed to show consistent directionality in the MR results of the lung function, otherwise excluded. All statistical analyses were performed using R version 4.3.1. MR analyses were performed using the Two-sample MR, MR-PRESSO.
Results
An overview of IVs in immune cell signatures
Each of 731 immune cell signatures received numerous SNPs as proxies after screening the genome-wide significance level (p < 1 × 10−5), harmonizing, MR-PRESSO test and verifying F statistics. All SNPs that the MR-PRESSO global test determined to be outliers were excluded (p < 0.05). The F statistics of all remaining SNPs were greater than 10, showing a strong association between IVs and the relevant phenotypes. Supplementary Table S1 displayed the final list of retained SNPs as well as pertinent statistical data.
Associations of genetically proxied immune cell signatures with COPD
We tested the potential causality from COPD to all available immune cell signatures and all of the results are displayed in Supplementary Table S2. The primary analysis IVW method revealed that the relative abundance of the 27 genetically predicted immune cell signatures was causally related with COPD after MR analysis (). In addition, we further excluded immune cell signatures with a different directionality from the lung function MR results, retaining seven immune cell signatures ().
Figure 2. MR results of immune cell signatures with a causal relationship to COPD. From the inner to outer circles, they represent the p-value of weighted mode, simple mode, weighted median, MR-Egger, and inverse-variance weighted methods, respectively. The shades of color reflect the magnitude of the pvalue.
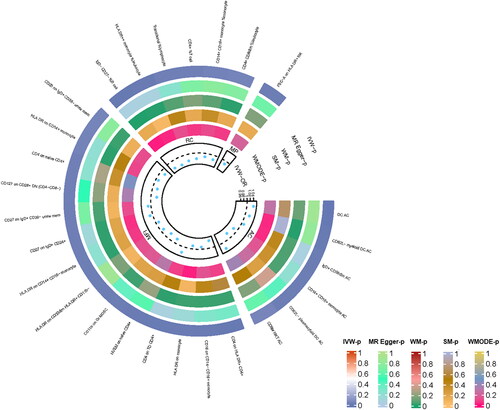
Figure 3. Forrest plot for summary causal effects of seven immune cell signatures on COPD risk.
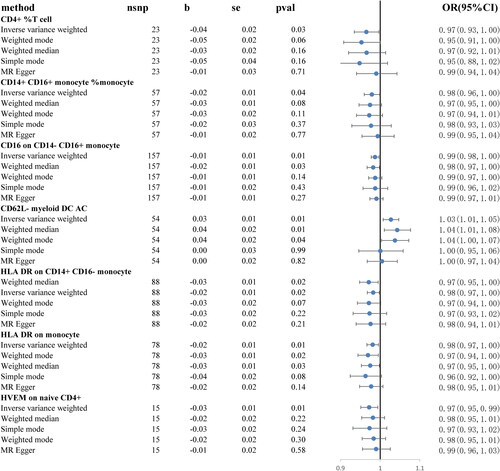
Specifically, it was discovered that seven immune cell signatures were causally linked to the risk of COPD. These included CD4+% T cell, CD14 + CD16+ monocyte% monocyte, CD16 on CD14-CD16+ monocyte, HLA DR on CD14 + CD16- monocyte, HLA DR on monocyte, HVEM on naive CD4+ and CD62L- myeloid DC AC. CD4+% T cell (IVW odds ratio [OR] = 0.97, confidence interval [CI] = 0.93–1.00, p = 0.03), CD14 + CD16+ monocyte% monocyte (OR = 98, CI = 0.96–1.00, p = 0.04), CD16 on CD14-CD16+ monocyte (OR = 0.98, CI = 0.98–1.00, p = 0.01), HLA DR on CD14 + CD16- monocyte (OR = 0.97, CI = 0.95–1.00, p = 0.02), HLA DR on monocyte (OR = 0.98, CI = 0.97–1.00, p = 0.01) and HVEM on naive CD4+ (OR = 0.97, CI = 0.95–0.99, p = 0.01) demonstrated a protective effect on COPD, whereas CD62L- myeloid DC AC may be related with an increased risk of COPD (OR = 1.03, CI = 1.01–1.05, p = 0.01). For the causal effects of immune cell signatures on COPD risk with specific SNPs, scatter plots () and forest plots (supplemental Figure S1) were used to show the above results. In addition, the functions of these seven immune cells () and their actions on the airways were summarized ().
Figure 4. Scatter plots of the MR analyses for the association of immune cell signatures and the risk of COPD.
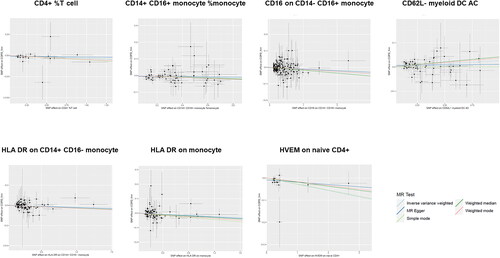
Figure 5. Schematic diagram illustrating how immune cells act on the airways.
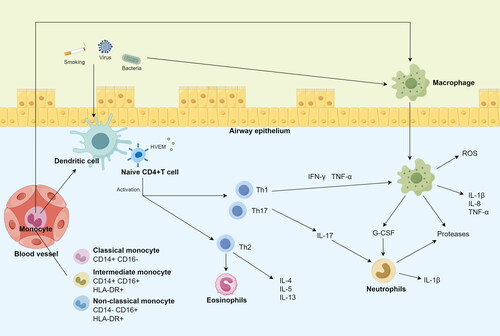
Table 1. The functions of these seven immune cells.
Sensitivity analyses and detection of potential pleiotropy
The results of the Cochrane’s Q test demonstrated that the SNPs exhibited no significant heterogeneity (p > 0.05, supplementary Table S3). Also, the funnel plots indicating no heterogeneity in the results (Supplementary Figure S2). MR analysis revealed no pleiotropy based on the results of the MR Egger test (p > 0.05, supplementary Table S4). The leave-one-out method was analyzed by eliminating each SNP step by step to find out whether the remaining SNP IVW results were similar to the original results. As can be seen from supplementary Figure S3, the removal of any SNP did not fundamentally affect the results, indicating that the MR results were robust. However, the additional MR-PRESSO global test showed no significant outliers (p > 0.05). Furthermore, MR-PRESSO revealed that our MR analysis results exhibited no horizontal pleiotropy (p > 0.05, supplementary Table S5).
Discussion
The lung is a major lymphoid organ with a sophisticated functional structure. A normal adult human lung has 30 × 109 lymphocytes, or 7% of the body’s total lymphocyte population [Citation29, Citation30]. However, confounding factors such as age and smoking have hampered the progress of the immune-lung interaction study, making it difficult to cross-sectionally analyze the relationship between immune cells and COPD. In this study, we evaluated the potential causal link between immunophenotypes and COPD from the standpoint of the host genetics. To our knowledge, this is the first MR analysis to investigate the causal link between multiple immune cell signatures and COPD.
Inflammation is the core feature of COPD, which causes the activation and changes of airway and lung structural cells, as well as the release and recruitment of infiltrating inflammatory cells [Citation31–33]. These changes eventually lead to structural changes and airway blockage, resulting in emphysema, tissue damage, and mucus hypersecretion [Citation34,Citation35]. In general, immunosuppressive network induction is intimately linked to chronic inflammation and weakened resistance to respiratory infections, which may contribute to the pathogenesis of COPD. COPD patients have higher levels of macrophages, neutrophils, T and B lymphocytes, and dendritic cells, which are markers of lower airway and lung inflammation [Citation36]. However, it has been suggested that the inflammatory response seen in the small airways of COPD patients is an amplification of the inflammatory response to stimuli (such as smoking) in smokers with normal lung function [Citation37]. Therefore, it is necessary to exclude the interference of confounding factors to explore the causal relationship between immune cells and COPD.
Our research employed extensive GWAS data to confirm the causal effect of immune cell on COPD. We explored causal relationship between 731 immune cell signatures and COPD. The results suggested that seven immune cell signatures may have a causal impact on COPD. Furthermore, the directionality of these seven immune cells features in the lung function MR results was validated, although they did not have a significant causal relationship with lung function. These signatures deserved further attention as potential indicators for assessing COPD risk and their effects on lung function. Interestingly, six out of these immune cells (CD4+% T cell, CD14 + CD16+ monocyte % monocyte, CD16 on CD14-CD16+ monocyte, HLA DR on CD14 + CD16- monocyte, HLA DR on monocyte and HVEM on naive CD4+) were discovered possibly decreasing the risk of COPD, which has implications for clinical translational applications.
In this study, CD4+ T was identified as an important immune cell signature associated with COPD risk. In the presence of co-stimulatory signals, CD4+ T-helper lymphocytes can be activated by major histocompatibility complexes (MHCs) and generate proinflammatory cytokines that coordinate the inflammatory cell networks [Citation38]. In COPD patients, CD4+ T cells primarily assist pulmonary B cells and CD8+ T cells, amplifying and prolonging the inflammatory response in the lung microenvironment. Studies have reported that CD4 is associated with lung function, emphysema, frequent exacerbations of COPD, and disease progression [Citation39–41]. However, studies on the link between CD4+ T and COPD have thus far produced inconsistent findings. Kim et al. [Citation42] reported that there was no significant difference in peripheral blood CD4+ and other T cell subsets between COPD patients and healthy people, whereas Shirai et al. [Citation43] reported that the ratio of CD4+ T cells was increased compared with the control group. Casio et al. found that the number of CD4+ T cells in the trachea and lung tissues of COPD patients increased [Citation44, Citation45]. This may be the result of confounding variables that are outside the scope of the existing investigations. In this study, we found that higher CD4+ T was related with a reduced risk of COPD. Interestingly, higher expression of protective trait HVEM on naive CD4+ cell subtypes were also found. In recent years, the tumor necrosis factor receptor superfamily member known as herpes virus entry mediator (HVEM) has been identified as a novel immunological checkpoint [Citation46]. The B- and T-lymphocyte attenuator on other immune cells is activated by HVEM, which is predominantly expressed in immune cells [Citation47]. However, there are no studies on its role in COPD, but given its role in immune regulation and inflammation, HVEM may serve as a potential therapeutic target for COPD. The regulation of HVEM signaling pathway by targeted therapy or biological agents may provide new therapeutic strategies for COPD patients to control inflammation, reduce acute exacerbations, and improve clinical outcomes.
In addition, several monocyte subsets are highlighted in this study. The heterogeneity and plasticity of circulating monocytes have been demonstrated in recent studies [Citation48, Citation49]. Different monocyte subgroup patterns were connected to disease development or prognosis [Citation50–52]. Our study found that four monocyte phenotypes were associated with the risk of COPD, including CD14+ CD16+ monocyte, CD16 on CD14- CD16+ monocyte, HLA-DR on CD14+ CD16- monocyte and HLA DR on monocyte. Monocytes can be categorized into three main subgroups: classical (CD14 + CD16-), intermediate (CD14 + CD16+), and non-classical (CD14-CD16+) [Citation53]. The various monocyte subgroups are thought to have diverse roles. Classical monocytes are recruited to places of inflammation during bacterial infection, where they identify and phagocytose pathogens, release a variety of proinflammatory cytokines, and draw in more immune cells to control the inflammatory response [Citation54–56]. In contrast to intermediate monocytes, which are recruited at a later stage of inflammation and are linked to the release of cytokines and chemokines, non-classical monocytes have a unique capacity to protect the vascular endothelium under both homeostatic and inflammatory conditions [Citation57]. Studies have shown that peripheral blood non-classical monocytes may have potential as diagnostic and prognostic biomarkers for acute exacerbation of COPD, as the percentage of non-classical monocytes correlates with length of hospital stay (remission time) and disease duration [Citation58]. In addition, the human leukocyte antigen DR (HLA-DR) is widely expressed in human monocytes and macrophages. It is an MHC class II antigen that is responsible for antigen presentation to CD4-positive T cells. HLA-DR expression modulates cell-mediated immunity in the lung [Citation59]. The expression of HLA-DR by non-classical was lower than classical in COPD patients [Citation58]. This is consistent with our study, which found a causal relationship between HLA-DR on classical monocyte and the risk of COPD. Low expression of HLA-DR leads to frequent exacerbations of COPD [Citation41]. In addition, patients with higher expression of HLA-DR on immune cells at baseline may show better responses to anti-inflammatory or immunomodulatory therapies [Citation60]. Monitoring the changes in HLA-DR expression during COPD treatment may help to evaluate treatment effects and guide treatment decisions.
Although a serious of new immunomodulatory medications have been developed in recent years to help in the repair or immunological rebuilding of injured airways, there is currently no strategy for reversing airway remodeling and the ensuing irreversible restricted airflow. This study identified six immune cells signatures that were negatively connected with the risk of developing COPD. These immune cells signatures may be employed as a therapeutic target for COPD in the future and may play a part in controlling the immunological dysregulation in the lung of COPD patients.
Additionally, our study revealed that higher CD62L- myeloid DC AC levels were associated with an increased risk of COPD. CD62L is a type of adhesion molecule, also known as L-selectin (SELL), that is involved in T cell homing and T cell quiescence [Citation61]. CD62L is highly expressed in mature neutrophils [Citation62]. It can recruit leukocytes to sites of inflammation [Citation63], and this process is critical for the pathogenesis of emphysema [Citation64, Citation65]. A deficiency of CD62L prevents leukocyte rolling and migration [Citation66]. It was reported that sputum neutrophils CD62L expression was reduced in COPD patients [Citation67]. Reduced CD62L expression suggests blood neutrophils have undergone priming in COPD, which may be the result of systemic inflammation [Citation68]. To date, a variety of different selectin inhibitors have been identified and validated in animal models. It has been established that selectin inhibitors reduce inflammatory responses, but these results have not been translated into clinical practice. Treatment of chronic airway inflammation with selectin inhibitors seems promising. Our result revealed that CD62L deficient in myeloid DC AC was associated with COPD risk. However, the mechanism is still unclear, which needs to be further studied in the future.
However, there are some limitations worth noting. Firstly, because all GWAS data included in this investigation were acquired from European participants only, additional validation is required to evaluate whether these findings may be generalized to non-European populations. Secondly, we were unable to carry out a further stratified analysis of the population due to the lack of personal data of the participants. Thirdly, we were incapable to investigate potential stratification effects or nonlinear correlations due to the nature of IVs obtained from GWAS meta-analysis data sources used in this study. Despite these potential limitations, a range of sensitivity analysis showed that the estimates of causality in this investigation were powerful, indicating that this study precisely illustrated the substantial link between immune cell signatures and COPD risk.
Conclusion
In conclusion, we have established a causal link between COPD and specific immune cell signatures. Immunomodulation targeting the airway remodeling process in COPD may be a promising treatment option for delaying or even reversing disease progression. The development of a potentially beneficial immunotherapy is of great significance for the prevention or treatment of COPD.
Ethics statement
The summary-level data used in this study are de-identified public data and are available for download. Each GWAS involved in this study was ethically approved by the respective institutions.
Author contributions
Qian Zhang and Xinru Xiao designed the study and wrote the manuscript. Xinru Xiao, Ziqi Ding, and Yujia Shi analyzed data and drew the figures. All authors read and approved the final manuscript.
Supplemental Material
Download Zip (11 MB)Data availability
The data that supports the findings of this study are available upon reasonable request from the corresponding author.
Conflicts of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflicts with the subject matter or materials discussed in the manuscript apart from those disclosed.
Additional information
Funding
References
- Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):1–10. doi: 10.1056/NEJMoa032158.
- Holtzman MJ, Byers DE, Alexander-Brett J, et al. The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat Rev Immunol. 2014;14(10):686–698. doi: 10.1038/nri3739.
- Sin DD, Anthonisen N, Soriano J, et al. Mortality in COPD: role of comorbidities. Eur Respir J. 2006;28(6):1245–1257. doi: 10.1183/09031936.00133805.
- Jeffery PK. Structural and inflammatory changes in COPD: a comparison with asthma. Thorax. 1998;53(2):129–136. doi: 10.1136/thx.53.2.129.
- Wang C, Zhou J, Wang J, et al. Progress in the mechanism and targeted drug therapy for COPD. Sig Transduct Target Ther. 2020;5(1):248. doi: 10.1038/s41392-020-00345-x.
- Meng H, Long Q, Wang R, et al. Identification of the key immune-related genes in chronic obstructive pulmonary disease based on immune infiltration analysis. Int J Chron Obstruct Pulmon Dis. 2022;17:13–24. doi: 10.2147/COPD.S333251.
- Zhang Y, Xia R, Lv M, et al. Machine-learning algorithm-based prediction of diagnostic gene biomarkers related to immune infiltration in patients with chronic obstructive pulmonary disease. Front Immunol. 2022;13:740513. doi: 10.3389/fimmu.2022.740513.
- Yang Y-C, Zhang M-Y, Liu J-Y, et al. Identification of ferroptosis-related hub genes and their association with immune infiltration in chronic obstructive pulmonary disease by bioinformatics analysis. Int J Chron Obstruct Pulmon Dis. 2022;17:1219–1236. doi: 10.2147/COPD.S348569.
- Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378(9795):1015–1026. doi: 10.1016/S0140-6736(11)60988-4.
- Holloway RA, Donnelly LE. Immunopathogenesis of chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2013;19(2):95–102. doi: 10.1097/MCP.0b013e32835cfff5.
- Mackay AJ, Hurst JR. COPD exacerbations: causes, prevention, and treatment. Immunol Allergy Clin North Am. 2012;33(1):95–115. doi: 10.1016/j.iac.2012.10.006.
- Kheradmand F, Zhang Y, Corry DB. Contribution of adaptive immunity to human COPD and experimental models of emphysema. Physiol Rev. 2023;103(2):1059–1093. doi: 10.1152/physrev.00036.2021.
- Montes de Oca M, Pérez-Padilla R. Global initiative for chronic obstructive lung disease (GOLD)-2017: the alat perspective. Arch Bronconeumol. 2017;53(3):87–88. doi: 10.1016/j.arbres.2017.01.002.
- McCullagh BN, Comellas AP, Ballas ZK, et al. Antibody deficiency in patients with frequent exacerbations of chronic obstructive pulmonary disease (COPD). PLoS One. 2017;12(2):e0172437. doi: 10.1371/journal.pone.0172437.
- Hoonhorst SJ, ten Hacken NH, Lo Tam Loi AT, et al. Lower corticosteroid skin blanching response is associated with severe COPD. PLoS One. 2014;9(3):e91788. doi: 10.1371/journal.pone.0091788.
- Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22. doi: 10.1093/ije/dyg070.
- Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–R98. doi: 10.1093/hmg/ddu328.
- Skrivankova VW, Richmond RC, Woolf BA, et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomization: the STROBE-MR statement. JAMA. 2021;326(16):1614–1621. doi: 10.1001/jama.2021.18236.
- Singh D, Agusti A, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease: the GOLD science committee report 2019. Eur Respir J. 2019;53(5):1900164. doi: 10.1183/13993003.00164-2019.
- Shrine N, Guyatt AL, Erzurumluoglu AM, et al. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet. 2019;51(3):481–493. doi: 10.1038/s41588-018-0321-7.
- Kurki MI, Karjalainen J, Palta P, et al. FinnGen: unique genetic insights from combining isolated population and national health register data. MedRxiv. 2022. doi: 10.1101/2022.03.03.22271360.
- Orrù V, Steri M, Sidore C, et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat Genet. 2020;52(10):1036–1045. doi: 10.1038/s41588-020-0684-4.
- Sidore C, Busonero F, Maschio A, et al. Genome sequencing elucidates sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat Genet. 2015;47(11):1272–1281. doi: 10.1038/ng.3368.
- Wang C, Zhu D, Zhang D, et al. Causal role of immune cells in schizophrenia: mendelian randomization (MR) study. BMC Psychiatry. 2023;23(1):590. doi: 10.1186/s12888-023-05081-4.
- Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in mendelian randomization: comparison of allele score and summarized data methods. Stat Med. 2016;35(11):1880–1906. doi: 10.1002/sim.6835.
- Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi: 10.1093/ije/dyv080.
- Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985–1998. doi: 10.1093/ije/dyx102.
- Verbanck M, Chen C-Y, Neale B, et al. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–698. doi: 10.1038/s41588-018-0099-7.
- Blum KS, Pabst R. Lymphocyte numbers and subsets in the human blood: do they mirror the situation in all organs? Immunol Lett. 2007;108(1):45–51. doi: 10.1016/j.imlet.2006.10.009.
- Ganusov VV, De Boer RJ. Do most lymphocytes in humans really reside in the gut? Trends Immunol. 2007;28(12):514–518. doi: 10.1016/j.it.2007.08.009.
- Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):71–86. doi: 10.1016/j.ccm.2013.10.004.
- Postma DS, Reddel HK, ten Hacken NH, et al. Asthma and chronic obstructive pulmonary disease: similarities and differences. Clin Chest Med. 2014;35(1):143–156. doi: 10.1016/j.ccm.2013.09.010.
- Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4(1):435–459. doi: 10.1146/annurev.pathol.4.110807.092145.
- Barnes PJ, Shapiro SD, Pauwels R. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–688. doi: 10.1183/09031936.03.00040703.
- Stockley RA, Mannino D, Barnes PJ. Burden and pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6(6):524–526. doi: 10.1513/pats.200904-016DS.
- Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet. 2004;364(9435):709–721. doi: 10.1016/S0140-6736(04)16900-6.
- Barnes PJ. Small airways in COPD. N Engl J Med. 2004;350(26):2635–2637. doi: 10.1056/NEJMp048102.
- Smyth LJ, Starkey C, Vestbo J, et al. CD4-regulatory cells in COPD patients. Chest. 2007;132(1):156–163. doi: 10.1378/chest.07-0083.
- Glader P, von Wachenfeldt K, Löfdahl C-G. Systemic CD4+ T-cell activation is correlated with FEV1 in smokers. Respir Med. 2006;100(6):1088–1093. doi: 10.1016/j.rmed.2005.09.025.
- Sullivan AK, Simonian PL, Falta MT, et al. Oligoclonal CD4+ T cells in the lungs of patients with severe emphysema. Am J Respir Crit Care Med. 2005;172(5):590–596. doi: 10.1164/rccm.200410-1332OC.
- Geerdink JX, Simons SO, Pike R, et al. Differences in systemic adaptive immunity contribute to the ‘frequent exacerbator’COPD phenotype. Respir Res. 2016;17(1):140. doi: 10.1186/s12931-016-0456-y.
- Kim W-D, Kim W-S, Koh Y, et al. Abnormal peripheral blood T-lymphocyte subsets in a subgroup of patients with COPD. Chest. 2002;122(2):437–444. doi: 10.1378/chest.122.2.437.
- Shirai T, Suda T, Inui N, et al. Correlation between peripheral blood T-cell profiles and clinical and inflammatory parameters in stable COPD. Allergol Int. 2010;59(1):75–82. doi: 10.2332/allergolint.09-OA-0126.
- Cosio MG, Majo J, Cosio MG. Inflammation of the airways and lung parenchyma in COPD: role of T cells. Chest. 2002;121(5 Suppl):160S–165S. doi: 10.1378/chest.121.5_suppl.160s.
- Turato G, Zuin R, Miniati M, et al. Airway inflammation in severe chronic obstructive pulmonary disease: relationship with lung function and radiologic emphysema. Am J Respir Crit Care Med. 2002;166(1):105–110. doi: 10.1164/rccm.2111084.
- Aubert N, Brunel S, Olive D, et al. Blockade of HVEM for prostate cancer immunotherapy in humanized mice. Cancers (Basel). 2021;13(12):3009. doi: 10.3390/cancers13123009.
- Deng Z, Zheng Y, Cai P, et al. The role of B and T lymphocyte attenuator in respiratory system diseases. Front Immunol. 2021;12:635623. doi: 10.3389/fimmu.2021.635623.
- Ziegler-Heitbrock H. Definition of human blood monocytes. J Leukoc Biol. 2000;67(5):603–606. doi: 10.1002/jlb.67.5.603.
- Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14(6):392–404. doi: 10.1038/nri3671.
- Han J, Wang B, Han N, et al. CD14highCD16+ rather than CD14lowCD16+ monocytes correlate with disease progression in chronic HIV-infected patients. J Acquir Immune Defic Syndr. 2009;52(5):553–559. doi: 10.1097/qai.0b013e3181c1d4fe.
- Valcour VG, Shiramizu BT, Shikuma CM. HIV DNA in circulating monocytes as a mechanism to dementia and other HIV complications. J Leukoc Biol. 2010;87(4):621–626. doi: 10.1189/jlb.0809571.
- Heine G, Ulrich C, Seibert E, et al. CD14++ CD16+ monocytes but not total monocyte numbers predict cardiovascular events in dialysis patients. Kidney Int. 2008;73(5):622–629. doi: 10.1038/sj.ki.5002744.
- Kapellos TS, Bonaguro L, Gemünd I, et al. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front Immunol. 2019;10:2035. doi: 10.3389/fimmu.2019.02035.
- Serbina NV, Jia T, Hohl TM, et al. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26(1):421–452. doi: 10.1146/annurev.immunol.26.021607.090326.
- Tolouei Semnani R, Moore V, Bennuru S, et al. Human monocyte subsets at homeostasis and their perturbation in numbers and function in filarial infection. Infect Immun. 2014;82(11):4438–4446. doi: 10.1128/IAI.01973-14.
- Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. 2017;17(6):349–362. doi: 10.1038/nri.2017.28.
- Stolk J, Aggarwal N, Hochnadel I, et al. Blood monocyte profiles in COPD patients with PiMM and PiZZ α1-antitrypsin. Respir Med. 2019;148:60–62. doi: 10.1016/j.rmed.2019.02.001.
- Yang J, Qiao M, Li Y, et al. Expansion of a population of large monocytes (atypical monocytes) in peripheral blood of patients with acute exacerbations of chronic obstructive pulmonary diseases. Mediators Inflamm. 2018;2018:9031452–9031413. doi: 10.1155/2018/9031452.
- Bühling F, Ittenson A, Kaiser D, et al. MRP8/MRP14, CD11b and HLA-DR expression of alveolar macrophages in pneumonia. Immunol Lett. 2000;71(3):185–190. doi: 10.1016/s0165-2478(00)00164-4.
- Monneret G, Lepape A, Voirin N, et al. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med. 2006;32(8):1175–1183. doi: 10.1007/s00134-006-0204-8.
- Ivetic A, Hoskins Green HL, Hart SJ. L-selectin: a major regulator of leukocyte adhesion, migration and signaling. Front Immunol. 2019;10:1068. doi: 10.3389/fimmu.2019.01068.
- Tak T, Wijten P, Heeres M, et al. Human CD62Ldim neutrophils identified as a separate subset by proteome profiling and in vivo pulse-chase labeling. Blood. 2017;129(26):3476–3485. doi: 10.1182/blood-2016-07-727669.
- Tedder TF, Steeber DA, Pizcueta P. L-selectin-deficient mice have impaired leukocyte recruitment into inflammatory sites. J Exp Med. 1995;181(6):2259–2264. doi: 10.1084/jem.181.6.2259.
- Noguera A, Sala E, Pons AR, et al. Expression of adhesion molecules during apoptosis of circulating neutrophils in COPD. Chest. 2004;125(5):1837–1842. doi: 10.1378/chest.125.5.1837.
- Romano SJ. Selectin antagonists: therapeutic potential in asthma and COPD. Treat Respir Med. 2005;4(2):85–94. doi: 10.2165/00151829-200504020-00002.
- Arbonés ML, Ord DC, Ley K, et al. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity. 1994;1(4):247–260. doi: 10.1016/1074-7613(94)90076-0.
- Mallia P, Contoli M, Gray KK, et al. Neutrophil adhesion molecules in experimental rhinovirus infection in COPD. Respir Res. 2013;14(1):72. doi: 10.1186/1465-9921-14-72.
- Lokwani R, Wark PA, Baines KJ, et al. Blood neutrophils in COPD but not asthma exhibit a primed phenotype with downregulated CD62L expression. Int J Chron Obstruct Pulmon Dis. 2019;14:2517–2525. doi: 10.2147/COPD.S222486.