
ABSTRACT
Translation fidelity relies on accurate aminoacylation of transfer RNAs (tRNAs) by aminoacyl-tRNA synthetases (AARSs). AARSs specific for alanine (Ala), leucine (Leu), serine, and pyrrolysine do not recognize the anticodon bases. Single nucleotide anticodon variants in their cognate tRNAs can lead to mistranslation. Human genomes include both rare and more common mistranslating tRNA variants. We investigated three rare human tRNALeu variants that mis-incorporate Leu at phenylalanine or tryptophan codons. Expression of each tRNALeu anticodon variant in neuroblastoma cells caused defects in fluorescent protein production without significantly increased cytotoxicity under normal conditions or in the context of proteasome inhibition. Using tRNA sequencing and mass spectrometry we confirmed that each tRNALeu variant was expressed and generated mistranslation with Leu. To probe the flexibility of the entire genetic code towards Leu mis-incorporation, we created 64 yeast strains to express all possible tRNALeu anticodon variants in a doxycycline-inducible system. While some variants showed mild or no growth defects, many anticodon variants, enriched with G/C at positions 35 and 36, including those replacing Leu for proline, arginine, alanine, or glycine, caused dramatic reductions in growth. Differential phenotypic defects were observed for tRNALeu mutants with synonymous anticodons and for different tRNALeu isoacceptors with the same anticodon. A comparison to tRNAAla anticodon variants demonstrates that Ala mis-incorporation is more tolerable than Leu at nearly every codon. The data show that the nature of the amino acid substitution, the tRNA gene, and the anticodon are each important factors that influence the ability of cells to tolerate mistranslating tRNAs.
Introduction
In all cells, accurate aminoacylation of each tRNA is required for high fidelity translation of the genetic code. Two independently evolved families [Citation1] of aminoacyl-tRNA synthetases (class I and II AARSs) are responsible for catalysing the ligation of a specific amino acid to its cognate tRNA in an ATP-dependent reaction [Citation2]. Each tRNA includes a set of identity element nucleotides that are essential for accurate and efficient aminoacylation [Citation3]. For many of the tRNAs, the anticodon or a portion of the anticodon sequence are critical identity elements, thus physically linking the amino acid with the anticodon to define codon assignments. For alanyl- [Citation4], seryl- [Citation5], pyrrolysyl- [Citation6,Citation7], and leucyl-tRNA synthetases (LeuRS) [Citation8,Citation9], the anticodon is not recognized. In the case of LeuRS, the major identity elements in human tRNALeu include the discriminator base (A73), base pairs in the acceptor stem (A4:U69, G5:C68), and the extra-long variable arm () [Citation10].
Figure 1. Schematic of natural and synthetic tRNALeu variants.
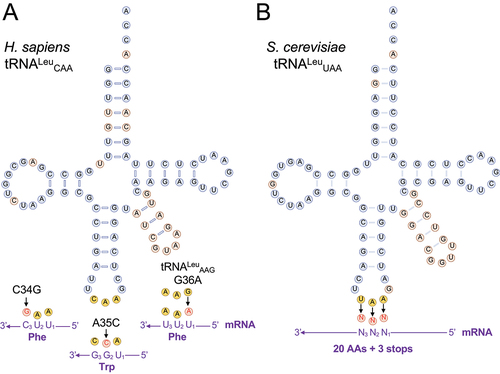
Table 1. Naturally occurring human tRNALeu variants characterized in this study.
Protein synthesis is normally a high-fidelity process, with error rates indicating that 1 in every 1,000–10,000 codons are mis-read [Citation11]. Specific measurements with sensitive luminescent reporters suggest that background error rates could be as low as 1 mistake in every 106 to 108 codons [Citation12–14]. Despite these low basal error rates, cells representing the diversity of life including bacterial [Citation15,Citation16], yeast [Citation17–19], and mammalian cells [Citation20–23] are able to tolerate significantly elevated mistranslation levels of 1–10% per codon, in some cases without substantial growth defects [Citation17,Citation21,Citation22]. There are several distinct mechanisms that cause errors in protein synthesis by affecting the accuracy of aminoacylation or decoding on the ribosome. While AARSs show significant selectivity for their cognate amino acids, some AARSs require editing domains that function in cis [Citation24,Citation25] or in trans [Citation26,Citation27] to discriminate between similar amino acids and hydrolyse mis-aminoacylated tRNAs. Editing-defective AARSs are known to cause mistranslation and some mutants are associated with neurodegenerative disease [Citation28].
Mistranslating tRNAs were first identified in selections for suppressors of nonsense or missense mutations in essential genes, such as tryptophan synthase [Citation29]. Recently, we characterized mistranslation resulting from both synthetic and naturally occurring tRNA variants in yeast [Citation17–19] and mammalian cells [Citation20–23]. Analysing sequence data from the 1000 Genomes and TOPMED projects, we identified many distinct tRNA variants in humans that cause mistranslation [Citation30]. In a targeted sequencing study, we observed that individuals carry 60–70 single nucleotide polymorphisms (SNPs) in their tRNA genes with ~10 rare alleles per person [Citation31]. For example, a tRNASerAGA G35A variant occurs in 2% of the population [Citation30–32] and causes mis-incorporation of serine (Ser) at phenylalanine (Phe) codons in murine and human cells [Citation20]. The resulting mistranslation leads to toxicity and defects in protein production and degradation. Furthermore, the mutant tRNA showed super-additive or synthetic toxicity with a causative allele for amyotrophic lateral sclerosis [Citation23] and inhibited the ability of cells to degrade poly-glutamine aggregates that cause Huntington’s disease [Citation20]. Conversely, we investigated alanine (Ala) and glycine (Gly) tRNA variants that occur in 2–8% of the population [Citation21]. These enabled substantial Ala mis-incorporation at Gly codons, yet the conservative Ala for Gly substitution was well tolerated by mammalian cells [Citation21].
Based on the demonstrated potential of natural human tRNA variants to generate mistranslation and cause negative genetic interactions with disease-causing alleles, we characterized mistranslation, protein production, and cytotoxicity from rare human tRNALeu variants that decode Phe or Trp codons in neuroblastoma cells (). Because each variant caused significant yet relatively minor defects in protein production and no cytotoxicity despite ample evidence of mistranslation from mass spectrometry, we investigated the complete catalogue of tRNALeu anticodon variants using an inducible system in yeast. Together the data reveal a large library of tRNALeu missense suppressor variants that show the cell’s ability to tolerate Leu mis-incorporation depends on the nature of the tRNA and the amino acid substitution.
Material and methods
DNA constructs
Genomic DNA from HEK 293T cells (ATCC #CRL-3216) was used as a template to PCR amplify human tRNALeu genes (Leu-CAA-3-1 and Leu-AAG-3-1). Anticodon mutants were generated using overlap extension PCR as before [Citation20]. Forward and reverse primers (all DNA primers are listed in Table S1) captured the tRNA gene and 300 base pairs of surrounding tRNA sequence, which are sufficient for regulated tRNA expression in mammalian cells [Citation23]. The tRNALeu anticodon mutants were generated by an overlap extension PCR protocol as before [Citation20]. PciI sites in each primer flank the tRNA allele and enable subsequent digestion and ligation into a permissive site in the WT-PAN plasmid, which was a gift from Tao Pan (Addgene plasmid # 99638) [Citation37]. WT-PAN is a pCDNA derived vector expressing a GFP-mCherry gene that we use to identify transfected cells and monitor protein production levels. All plasmids were verified by DNA sequencing (Azenta Life Sciences, Plainfield, NJ, USA).
Yeast tRNALeu (UAA; YNCB0012W) including approximately 300 base pairs of up and downstream-flanking sequence was amplified by PCR using genomic DNA as template and primers UL0676 and YI6283A (Table S1). The tRNA was cloned into the LEU2 centromeric plasmid YCplac111 with a tetracycline inducible promoter positioned such that its transcription would run into the 3’ side of the tRNA [Citation38]. The plasmid pCB4727 was obtained through a triple ligation with the PCR tRNALeu fragment digested EcoRI (5’ end) - NotII (3’ end), the TetO promoter from pCB4695 [Citation38] digested BamHI-NotI and YCplac111 digested EcoRI-BamHI. tRNALeu (UAA) variants were obtained as a GeneString fragment (Life Technologies, ThermoFisher Scientific, Burlington, ON, Canada) containing a random mix of each nucleotide at anticodon positions 34, 35 and 36 flanked by up- and downstream sequence as above (Fig. S1). Unique anticodons were identified by sequencing with primer AD0128. As the synthetic fragment contained a G26A mutation in the tRNA, plasmids were converted to wild-type G26 by two-step PCR mutagenesis using primer pairs AB9159/YI6283 and AB9160/UL0676 in the first round and UL0676-YI6283 in the second and ligating the EcoRI-NotI digested product into pCB4727. For variants that were not selected out of the randomized anticodon pool, constructs expressing these tRNALeu variants were engineered via two-step PCR using the common 5’ primer AB9160 paired with Ul0676 and the specific mutagenic primer (Table S2) with YI6283 in the first round and UL0676-YI6283 in the second. An additional set was made by pairing of different specific forward and reverse primers (AA8950-AB8955) in the two-step reaction and identifying clones by sequencing. This set required conversion of A26 to G as above. The UGG variant used the mutagenic primers UL1165 and UL1166 in the two-step PCR reaction using CB4727 as the initial template. Plasmid numbers and the method for constructing the 64 tRNALeuUAA variants is listed (Table S2).
S. cerevisiae tRNALeu (UAG; YNCJ0029W) and tRNA (CAA; YNCG0040C) were amplified by PCR from genomic DNA using primers AH2795/AH2796 and AH2797/2798, respectively. These were substituted into pCB4727 as EcoRI-NotI fragments to generate pCB5212 and pCB5197. GCG (Arg), AAA (Phe), CGU (Thr) and AAC (Val) anticodon variants of YNCJ0029W were made by two-step PCR mutagenesis using primer pairs AI3080/AH2795 and a specific primer (Table S2)/AH2796 in the first round and AH2795/AH2796 in the second and ligating the EcoRI-NotI digested product into pCB4727. Similarly, mutagenesis of tRNALeu (CAA; YNCG0040C) used AI2977/AH2798 and a specific primer (Table S2)/AH2797 in the first round and AH2798/AH2797 in the second before ligating EcoRI-NotI into pCB4727. YNCJ0029W and YNCG0040C variants were confirmed by DNA sequencing at the London Regional Genomics Centre (London, ON, Canada) with primers AI2975 and AI2976, respectively. The plasmid numbers for these constructs are listed in Table S3. For ochre suppression assays, tRNALeuUAA or tRNALeuUUA were expressed in a URA3 centromeric plasmid by subcloning EcoRI-BamHI fragments from pCB4718 and pCB5165 into YCplac33 to give pCB5223 and pCB5224, respectively.
Mammalian cell culture and fluorescence microscopy
Mammalian cell culture experiments were performed in murine neuroblastoma Neuro2a (N2a; ATCC #CCL-131) cells derived from a male mouse of the C-1300 line. Cells were grown and maintained at 37°C, with 5% CO2 and humidity. Dulbecco’s modified Eagle medium (DMEM 4.5 g/L; Gibco) with 10% fetal bovine serum (FBS; Gibco, ThermoFisher Scientific) and 1% penicillin-streptomycin (P: 100 IU/mL; S: 100 µg/mL; Wisent Bioproducts, St. Bruno, Quebec, Canada) was used for culturing the cells. Lipofectamine 3000 (Invitrogen, ThermoFisher Scientific) was used to transfect cells with WT-PAN plasmids containing wild-type or mutant tRNA alleles according to the manufacturer’s instructions for the appropriate plate size. All fluorescent images were captured using the BioTek Cytation C10 confocal imaging reader (Agilent, Santa Clara, CA, USA). eGFP fluorescence was visualized using the GFP channel (excitation 469 ± 37 nm; emission 525 ± 39 nm) and the TexasRed channel (excitation 586 ± 15 nm; emission 647 ± 57 nm). All images were captured with the Olympus 10× plan fluorite phase-contrast objective (working distance 10 mm, numerical aperture 0.03). After plating and transfection, cells were incubated at 37°C, with 5% CO2 and humidity. Images were captured 24-h post-transfection and the level of eGFP and mCherry fluorescence was quantified in individual transfected cells using the BioTek Gen5 software (Agilent).
Mass spectrometry
GFP-mCherry was purified from N2a cells and analysed by liquid-chromatography tandem mass spectrometry (MS/MS) as before [Citation21]. Briefly, at 24-h post-transfection, N2a cells were harvested and stored in −80°C prior to use. Cells were lysed and GFP-mCherry was purified from cell lysates using RFP-Trap Agarose kit (Chromotek, Munich, Germany) following manufacturer’s instructions. Purified GFP-mCherry was diluted with 2 × sodium dodecyl sulphate (SDS)-running buffer and separated using SDS-polyacrylamide gel electrophoresis (PAGE) with 10% acrylamide. Bands corresponding to GFP-mCherry were excised from the Coomassie stained gel. Excised bands were stored in 5% acetic acid and submitted for LC-MS/MS analysis at the Biological Mass Spectrometry Laboratory (The University of Western Ontario, London, Canada). The protein samples were digested with trypsin and an Orbitrap Elite Velos Pro mass spectrometer (ThermoFisher) was used in FT/IT/CID configuration to identify amino acids mis-incorporated in GFP-mCherry.
Strain CY8652 containing pCB4718 (UAA Leu), pCB5164 (AAA Phe) or pCB5181 (GAA Phe) were grown in minimal medium lacking leucine and uracil to stationary phase at 25°C. Cells were diluted 1:50 for pCB4718, 1:40 for pCB5164 and 1:10 for pCB5181 into minimal medium lacking leucine containing 1.0 ug/mL doxycycline and grown for 17 h at 30°C. Cells were pelleted by centrifugation, washed twice with lysis buffer (50 mM HEPES pH 7.5, 100 mM NaCl) with 5 mM EDTA, then washed 2 times with lysis buffer lacking EDTA. Protein extracts were prepared by cell lysis with glass beads in lysis buffer lacking EDTA. Additional details of mass spectrometry are in the Supplementary Methods.
Western blotting
Following harvest and lysis of cells, 20 μg of protein from each lysate was separated by SDS-PAGE (12% acrylamide, 120 volts for 3 h). Proteins were transferred from gel to methanol-activated polyvinylidene difluoride (PVDF) membranes using a Trans-Blot Turbo transfer system (25 V, 1.3 A, 14 min; BioRad, Hercules, CA, USA). All blocking and wash solutions were made using tris-buffered saline (TBS; 50 mM Tris-HCl, pH 7.5, 150 mM NaCl). Following transfer, membranes were incubated in blocking solution (5% bovine serum albumin, 0.1% Tween-20, 1 × TBS) for 1 h at room temperature. Membranes were initially incubated overnight at 4°C in anti-mCherry primary antibody (ab213511, AbCam, Cambridge, UK) at a dilution of 1:1,000. The following day, membranes were washed in 1% BSA TBS-Tween 3 × 10 min, before a 2-h incubation at room temperature in IRDye 800CW donkey anti-rabbit (mCherry 926–32213, Li-cor Biosciences, Lincoln, NB, USA) fluorescent secondary at a dilution of 1:10,000. Membranes were then washed 3 × 10 min in 1 × TBS-Tween, and once in 1 × TBS for 10 min before imaging. Membranes were imaged using fluorescence on a ChemiDoc MP imager (BioRad). After imaging of the mCherry antibody, the same membranes were incubated overnight with anti-GAPDH primary antibody (MAB374m, Sigma-Aldrich, Oakville, Ontario, Canada) at a dilution of 1:5,000. Following the second overnight incubation, the same washing steps were performed, and an IRDye 680RD goat anti-mouse (GAPDH 926–68070, Li-cor Biosciences) fluorescent secondary antibody was diluted 1:10,000 for visualizing GAPDH on the membranes.
Protein synthesis assay
Protein synthesis was measured using the SUrface SEnsing of Translation (SUnSET) method [Citation39] which measures the incorporation of puromycin into nascent peptide chains. At 24 h after transfection, N2a cells were incubated in puromycin treatment media (1 μg/mL puromycin in DMEM (10% FBS, 1% P/S)). After a 30-min incubation at 37°C, puromycin treatment media was removed and cells were incubated with DMEM (10% FBS, 1% P/S) for 15 min. Cells were then washed with PBS two times and harvested for cell lysis. 20 μg of protein was separated on a 10% SDS-PAGE gel. Proteins were transferred to PVDF membrane using a Trans-Blot Turbo Transfer System (25 V, 1.3 A constant for 20 min; BioRad) and subsequently incubated in TBST buffer containing 5% BSA. Membranes were then incubated overnight with a monoclonal puromycin antibody (clone 12D10, EMD Millipore, Temecula, CA, United States, diluted 1:5000 in TBST and 5% BSA w/v), then washed three times with TBST and 1% BSA for 10 min each. Membranes were incubated for 2 h in TBST containing 3% skim milk plus a secondary mouse antibody (Sigma-Aldrich, 1:5000) then washed three times with TBST for 15 min each. Membranes were visualized on BioRad ChemiDoc imaging system. The total lane density was analysed using Image J [Citation40].
Ordered two-template relay sequencing for tRnas (OTTR-seq for tRNAs)
From N2a cells expressing the indicated tRNA variant, total RNA was purified from TRIzol (Thermo Fisher 15,596,026) treated cells and then incubated with T4 Polynucleotide Kinase (NEB, M0201L) to resolve 2’,3’-cyclic phosphate ends as described previously [Citation41], followed by deacylation at pH 9.0 for 30 min at 45°C using 100 mM Na2B4O7 [Citation42]. Treated RNA was recovered using the RNA Clean and Concentrator-5 Kit (Zymo Research, R1013). OTTR-seq libraries were prepared according to previously described methods [Citation43] (OTTR reagents and protocol kindly provided by Kathleen Collins’s Lab, UC Berkeley), with modifications for tRNA sequencing. Briefly, 40 ng of total RNA was used as input. RNA was labelled for 2 h at 30°C using only dideoxyATP (ddATP). cDNA synthesis was conducted using the RNA-DNA primer duplex with + 1T overhang and 7-nt unique molecular identifiers (UMI). cDNA was recovered using the MinElute reaction cleanup kit (Qiagen 28,204), then size selected for 110–200 bp on a 1 × Tris-borate-EDTA (TBE, 89 mM Tris, 89 mM boric acid, 2 mM EDTA, pH 8.3) buffer, 8 M urea, and 9% polyacrylamide gel to remove adapter dimers. cDNA was recovered by diffusion at 70°C for 1 h with shaking in a solution of 10 mM Tris-HCl pH 8, 300 mM NaCl, 1 mM EDTA, and 0.25% SDS, followed by precipitation in 66% ethanol with 0.6 M ammonium acetate and 1 μg/mL GlycoBlue (Thermo Fisher, AM9515). cDNA was amplified with Q5 polymerase (NEB, M0493L) for 13 cycles using the NEBNext Multiplex Oligos for Illumina (adaptors and index primers) (NEB, E6609) then size selected for 160–250 bp on a 1 × TBE, 6% polyacrylamide gel to remove remaining adapter dimer. Libraries were extracted from the gel by diffusion at 70°C for 1 h with shaking in a solution of 10 mM Tris-HCl pH 8, 300 mM NaCl, and 1 mM EDTA, followed by precipitation in 70% ethanol with 80 mM sodium acetate and 1 μg/mL GlycoBlue (Thermo Fisher, AM9515). The recovered libraries were quantified using the Qubit dsDNA High-Sensitivity Kit (Invitrogen, Q32854), Agilent Bioanalyzer with High-Sensitivity DNA Kit (Aglient, 5067–4626), and finally by qPCR, which was conducted using PrimeTime Gene Expression Master Mix (IDT 1,055,772) according to a previously published protocol [Citation44]. Libraries were sequenced on the Illumina NextSeq 2000 platform to generate 100-bp single-end reads with a minimum of 10 million reads per sample.
tRNA sequencing data analysis
Adaptors/primers in raw sequencing reads generated using OTTR-seq were trimmed using Cutadapt [Citation45] with options -j 8 -m 15 -a AGATCGGAAGAGCACACGTC. Only reads with a minimum of 15 nts were retained. Reads were further processed to extract the UMI using UMI-tools [Citation46] with options – extract-method=string – bc-pattern=NNNNNNN. Trimmed reads were processed through the tRNA Analysis of eXpression pipeline (tRAX) [Citation47], using a reference database that contained the genomic sequence of mouse GRCm39/mm39 assembly and the annotated tRNAs obtained from the Genomic tRNA Database (GtRNAdb) [Citation33]. In brief, the tRAX pipeline aligned sequence reads to mature tRNA sequences and the reference genome, computed read counts for annotated tRNAs and other small noncoding RNAs, performed normalization and differential expression analysis between samples, and identified nucleotide mismatches at specific tRNA positions that may represent mutations in tRNA variants, RNA modifications, or RNA editing events. Because OTTR-seq is capable of detecting both tRNA-derived RNAs and mature tRNAs, we extracted read alignments that were above 60 bp and reprocessed them through tRAX to only include reads corresponding with mature tRNAs. Misincorporations identified in the tRNA variants were determined by using the base counts piled up at each position along the tRNAs provided in the tRAX outputs. tRNA variant abundance was determined by counting the expected misincorporations (e.g. G36A for tRNALeuAAA). On the other hand, RNA modifications were identified by the presence of base mismatches at positions previously reported as modification sites in Modomics [Citation48]. A-to-I editing rate was computed with G34 mismatches versus the A34 reference allele for sequencing reads aligned to tRNALeuAAA and tRNALeuAAG.
Proteasome inhibition and cytotoxicity assays
Cytotoxicity and proteasome inhibition assays were conducted with N = 3 biological replicates for each condition. At 24-h post-transfection, cells were washed, and media was replaced with media containing either dimethyl sulphoxide (DMSO, BioBasic, New York, USA), or varying concentrations of proteasome inhibitor (0.1, 1.0, or 10 μM; MG132, Sigma-Aldrich). Cells were incubated at 37°C for 4 h before images were captured using the BioTek Cytation C10 imaging reader (Agilent). Cytotoxicity was measured with the CytotoxGlo assay (Promega, Fitchburg, WI, USA). Following the manufacturer’s instructions, 50 µL of assay reagent were added to each well and an initial luminescence reading was taken using a Biotek Cytation C10 imaging reader. Next, 50 μL of assay buffer including digitonin was added to each well and a final luminescence reading was taken. The initial luminescence reading quantifies the population of dead cells in each well, and the addition of digitonin allowed a determination of the total number of cells present.
Yeast strains and growth
Wild-type haploid yeast strains are derivatives of BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) [Citation49]. The haploid strain CY8652 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 tTA*-URA3) containing the tet ‘off’ activator (tTA*) marked with URA3 was derived from R1158 [Citation50] after crossing with BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) [Citation49] and sporulation. Yeast strains were grown at 30°C in yeast peptone medium or in synthetic medium supplemented with nitrogenous bases and amino acids containing 2% glucose. Transformations of YCplac111 derivative plasmids into CY8652 were performed using the lithium acetate method [Citation51] with growth at room temperature for up to 6 days. Transformants containing tRNALeu plasmids were grown at room temperature to reduce toxicity of mistranslation, in synthetic media lacking uracil and leucine. Growth curves were generated by diluting the resulting cultures to an OD600 ~0.1 in synthetic medium lacking uracil and leucine with 0.01 mM, 1.0 mM, or without doxycycline (Sigma-Aldrich) as indicated and incubating at 30°C. A600 was measured every 15 min for 24 h in a BioTek Epoch 2 microplate spectrophotometer. Doubling time was calculated using the R package ‘growthcurver’ [Citation52]. For ochre suppression assays, the strain CY6874 (MATα his3Δ1 leu2Δ0 met5Δ0 ura3Δ0 tti2Δ-met5Δ::Tn10luk YCplac111-DED1- tti2Q276TAA) [Citation53] was transformed with plasmids bearing either wild-type or ochre suppressor tRNALeuUUA variants. Individual transformants were grown in synthetic complete medium lacking uracil to stationary phase. Cells from each were 10-fold serially diluted, spotted onto minimal plates lacking uracil and grown two days at 30°C or 37°C. The same cells were also grown on minimal plates lacking uracil and with the addition of 6 μg/mL calcofluor white (CW) at 30°C for 4 days.
Heatshock reporter assay
Yeast strain BY4741 containing the heatshock promoter element (HSE)-GFP reporter [Citation54] and pCB4718 (tRNALeuUAA) or pCB5164 (tRNALeuAAA(Phe)) were grown to saturation in medium lacking leucine and uracil, diluted 1:500 in the same medium and grown for 18 h at 30°C. Prior to measuring fluorescence (ex. 485 nm, em. 528 nm), all fluorescence measurements were normalized by optical density (A600). Fluorescence was measured with a BioTek Synergy H1 microplate reader and analysed using Gen5 2.08 software. A BY4741 strain lacking HSE-GFP was used to subtract background fluorescence from the cells. The average GFP fluorescence intensity was calculated across five technical replicates from three biological replicates for each strain.
Results
Natural human tRNALeu missense suppressors cause Leu mis-incorporation
Anticodon variants in human tRNALeu genes are rare, yet they have been identified in independent sequencing studies. The human tRNALeu anticodon variants, which are catalogued in the Genomic tRNA Database [Citation33] and in the single nucleotide polymorphism database (dbSNP) [Citation55], occur with allele frequencies ranging from 0.0004% to 0.03% in different sequencing projects (). Two variants derived from the same tRNA gene (Leu-CAA-3-1) include a Trp-decoding variant A35C (tRNALeuCCA) and a Phe-decoding variant C34G (tRNALeuGAA). Given their rare, but persistent occurrence in the population, we cloned each variant, characterized their ability to cause mistranslation, and assessed impacts both on protein levels and cytotoxicity in N2a cells. N2a cells transfect to high efficiency and show characteristic reductions in protein production in response to mistranslation [Citation20,Citation21,Citation23].
Following our previous approach to characterize mistranslating human tRNAs [Citation20], we co-expressed a GFP-mCherry marker with either a wild-type tRNALeu or an anticodon variant derived from the same tRNA gene. In combination with epigenetic chromatin state, expression of tRNAs in eukaryotic cells is regulated by internal A and B box RNA polymerase III promoter sequences and transcription factor-binding sites in the upstream region, as well as the downstream tail region which is important for transcription termination and tRNA processing [Citation56]. Thus, capturing 300 base pairs up and downstream of a native tRNA allele is sufficient to enable regulated expression from a plasmid construct [Citation20,Citation22,Citation23]. We previously used tRNA sequencing to show that this approach enables expression of a tRNA allele at physiological levels in mammalian cells [Citation23].
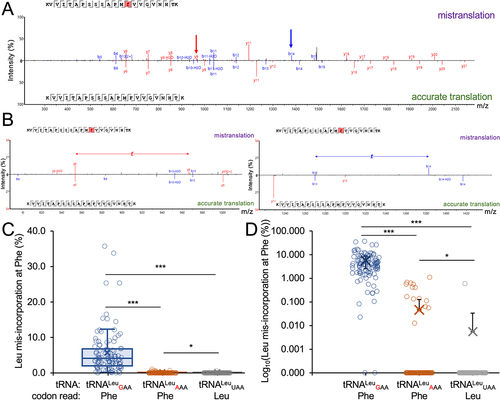
We purified the GFP-mCherry protein produced in cells expressing wild-type or mutant tRNA and used MS/MS analysis to identify peptides containing Leu mis-incorporation (, ). Cells expressing the mutant tRNA variants showed ample evidence of mistranslation supported by multiple peptide hits identified at individual and across multiple sites (). In cells expressing the wild-type tRNA, only a single peptide () at a single location was identified that may represent endogenous mistranslation of a Phe codon with Leu or a misidentification of the peptide. In contrast, cells expressing the Trp- (); A35C) or Phe-decoding (); C34G) tRNALeu variants derived from tRNALeuCAA both displayed multiple, high quality (, −10logP) peptide hits. Our MS/MS data show that the Trp-decoding mutant mis-incorporates Leu at Trp codons, which we did not observe in cells expressing wild-type tRNALeu. Based on spectral counts relative to the number of properly translated peptides observed at the same sites, the Trp-decoding mutant caused mistranslation at an apparently lower level than the Phe-decoding tRNALeu. The Trp-decoding mutant is also 5 to 15 times more prevalent in the population (). We note that the Trp codon usage is low in human and mouse genomes (UGG 12/1000 codons) and in our GFP protein reporter relative to Phe codons (36/1000 = UUU 17/1000, UUC 19/1000).
Figure 2. Identification of Leu mis-incorporation in mammalian cells by tandem mass spectrometry.
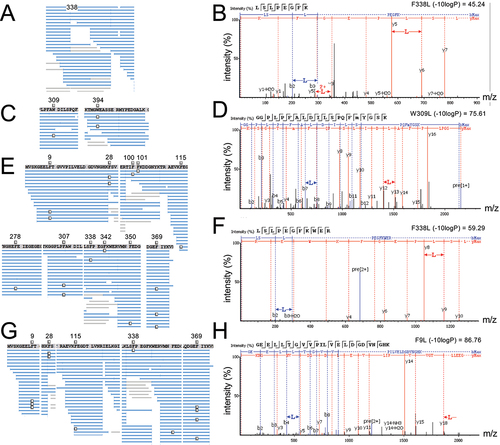
Table 2. Summary of Leu mis-incorporation identified by MS/MS in mammalian cells.
We next identified multiple and generally high-quality matches of peptide to spectra that demonstrate Leu mis-incorporation in cells expressing the tRNALeuAAG-derived Phe-decoding variant (G36A) (). Based on spectral counts (), the tRNALeuAAA variant leads to a reduced level of mis-incorporation compared to the Phe-decoding tRNALeuGAA mutant. Our GFP-mCherry construct contains only UUC Phe codons. Adenosine deaminase acting on transfer RNA (ADATs) catalyse the conversion the A34 base to inosine [Citation57], which can read codons ending in U, C, and A [Citation58]. We observed before that a tRNASer with an AAA anticodon decodes UUC Phe codons with Ser [Citation20], thus it is not surprising that tRNALeuAAA is able to decode UUC. Indeed, human tRNALeuAAG is well established as an ADAT substrate [Citation59]. Together the observations suggest the A34 is converted to I in the tRNALeu mutant. In addition to Phe UUU codons, the tRNALeuAAA may also decode the A-ending codon (UUA), which is a Leu codon and would not contribute to translation error.
tRNA-seq quantifies tRNALeu anticodon variant levels in mammalian cells
To confirm expression and measure the levels of each human tRNALeu anticodon variant in murine N2a cells (, S2, Supplementary Data File 1), we applied a generalized sequencing method called OTTR-seq [Citation43] for sequencing tRNAs in combination with tRNA Analysis of eXpression (tRAX) [Citation47], an analysis pipeline for the tRNA-seq data. To gauge mutant tRNA expression, we compared the levels of each tRNA variant to its parent tRNALeu isodecoder pool (). The data confirmed expression of the anticipated anticodon variants only in cells where the mutant tRNA genes were expressed, and we found no evidence of the mutant tRNAs in cells transfected with either wild-type tRNALeu. For cells expressing the Trp or Phe-decoding tRNALeu species, the A35C or C34G variants comprised ~5% of the tRNALeuCAA pool. In contrast, in cells transfected with the tRNALeuAAA mutant, the G35A mutant was 17% of the tRNALeuAAG pool.
Figure 3. OTTR-seq analysis of N2a cells expressing wild-type human tRNA or anticodon variants.
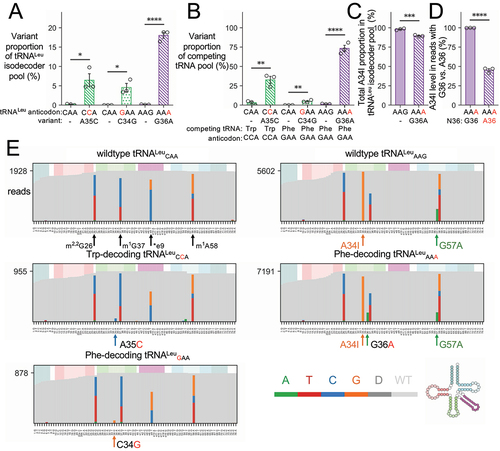
We then compared the level of each tRNALeu anticodon mutant with the level of the Trp- or Phe-decoding tRNAs that compete with the anticodon mutants for decoding on the ribosome (). The Trp-decoding tRNALeu accounts for ~25% of the total pool of Trp-decoding tRNAs in the cell. The tRNALeuGAA represents ~5% of Phe-decoding tRNAs, while tRNALeuAAA is much more abundant and represents 70% of the Phe-decoding tRNA pool. The read data indicate the tRNALeuAAG is expressed at a greater level relative to tRNAPhe or tRNALeuCAA (, Supplementary Data File 1). Thus, even though the tRNALeuAAA mutant is a smaller fraction of the total tRNALeu isoacceptor pool, the variant represents a major proportion of Phe-decoders in the cell.
Mis-incorporation of G at position 34 in the OTTR-seq reads confirms A-to-I editing [Citation60] at this position in both the wild-type tRNALeuAAG and the mutant tRNALeuAAA. We observed a small but significant reduction in the total level of I34 in the tRNALeu isodecoder pool in cells expressing the mutant tRNALeuAAA (). In cells transfected with tRNALeuAAA, we then compared the level of A34I in reads from endogenous tRNAs with G36 to reads from the mutant tRNA with A36. Strikingly, while nearly all the A34 bases are converted to I34 in wild-type tRNALeuAAG, the stoichiometry of I34 in tRNALeuAAA is ~45% (). These data validate our observations with MS/MS that showed tRNALeuAAA decodes UUC codons as expected for an I34-bearing tRNA. In addition, and even though we observed greater abundance of tRNALeuAAA compared to tRNALeuGAA relative to the pool of Phe-decoders (), less than half of the tRNALeuAAA pool is competent to decode both UUU and UUC Phe codons.
We also identified misincorporation of bases at three positions in all tRNALeu variants. The modified bases N2,N2-dimethylguanosine (m2,2G26), 1-methylguanosine (m1G37), and 1-methyladenosine (m1A58) were previously confirmed [Citation61] (tdbR00000269) to occur at positions where we observed consistent mis-incorporation of other bases, suggestive of methylation [Citation60]. The transfected human tRNALeuCAA and anticodon variants have C at position e9, while the mouse isodecoders contain C, A, or U at this position. Thus, some of the Ce9 observed comes from the transfected human gene, while the observed G and T in the OTTR-seq reads are from the mouse background () and were also observed at similar levels in cells transfected with the tRNALeuAAG variants. Since Ge9 is not found in the mouse genome, this may represent an unknown modification or a natural variant in N2a cells. In the human tRNALeuAAG variants, a single base differentiates the human (A57) from the mouse (G57) sequence (). Thus, the abundance of the G57A variant at this position measures the level of our expressed human tRNALeuAAG and tRNALeuAAA variants. Indeed, we observe a similar level of A57 and A36, which are both unique to the human tRNALeuAAA mutant ().
OTTR-seq also provides the relative abundance level of each tRNA in the cell. Overall, we recorded minor changes across the tRNome (Fig. S2). All statistically significant (p-value <0.05) and differentially expressed tRNAs showed a change of at least 1.5-fold. Two of the most significant changes were in cells expressing the Phe- or Trp-decoding anticodon variants of tRNALeuCAA, which showed a 1.7 to 2-fold decrease in abundance of the tRNA-Gly-CCC-2 and tRNA-Leu-CAA-2 compared to wild-type cells. Cells expressing the Trp-decoding tRNALeuCCA (Fig. S2A) had several tRNAs enriched in abundance by 1.5 to 2-fold relative to cells expressing wild-type tRNA (tRNA-His-GTG-2, tRNA-iMet-CAT-1, tRNA-Met-CAT-2, tRNA-Arg-TCG-2, tRNA-Arg-CCT-4). Relative to cells expressing the Phe-decoding tRNALeuGAA, several tRNAs were enriched by 1.5 to 2.4-fold in cells transfected with wild-type tRNALeu (tRNA-Arg-ACG-1,3, tRNA-Gly-GCC-1,2,3,4, tRNA-Cys-GCA-21, tRNA-Trp-CCA-3,4, tRNA-Leu-CAA-3). Cells expressing the other Phe-decoder (tRNALeuAAA) had the least perturbed tRNAome, where we found only one tRNA (tRNA-iMet-CAT-3) that was significantly increased by 1.8-fold (Fig S2C).
Human tRNALeu anticodon variants cause defects in protein production
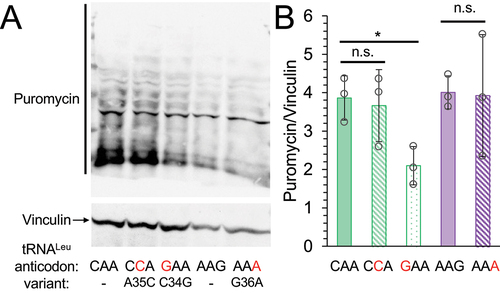
In mammalian cells, depending on the kind and level of amino acid mis-incorporation, inhibition of protein synthesis [Citation21–23,Citation62,Citation63] and defects in protein degradation [Citation20] are hallmarks of mistranslation. To determine if the tRNA mutants showed a global impact on protein synthesis, we used a puromycylation assay to measure the rate of protein synthesis in N2a cells expressing either wild-type or mutant tRNAs ()). In cells expressing the Phe-decoding tRNALeuGAA mutant we observed a significant 2-fold reduction in global protein synthesis relative to cells expressing wild-type tRNA. Cells expressing the Phe-decoding tRNALeuAAA or Trp-decoding tRNALeuCCA variant did not significantly reduce global protein synthesis.
Figure 4. Global protein synthesis levels in cells expressing tRNA variants.
Previously, we [Citation21,Citation23] and others [Citation62] have measured fluorescent proteins as markers for the impact of mistranslation on protein production in cells. Here, we used a GFP-mCherry protein as a proxy of potential defects in protein production and initially assessed total GFP-mCherry levels by western blotting (Fig. S3). In cells expressing tRNALeuCCA (Trp) or tRNALeuGAA (Phe) variants, analysis of the western blots indicated minor reductions in GFP-mCherry levels that were not significantly different than those from cells expressing wild-type tRNALeuCAA (Fig. S3A, S3C). We observed a significant 1.4-fold reduction of GFP-mCherry levels in cells expressing the tRNALeuAAA variant compared to cells expressing the wild-type tRNALeuAAG control.
Since western blotting is only semi-quantitative and to provide a more sensitive measure of GFP-mCherry protein levels, we used fluorescence microscopy (, S4, S5) to determine the level of GFP and mCherry fluorescence in individual cells, following our established protocols [Citation23]. In comparison to blotting, this approach also has the advantage of selectively quantifying protein production in transfected cells. Under normal conditions, there were significant 1.7-fold reductions in both GFP and mCherry fluorescence in cells expressing the Phe-decoding tRNALeuGAA (Fig. S4A, S4B) in comparison to cells expressing wild-type tRNALeuCAA. In cells expressing the Trp-decoding mutant (Fig. S4A, S4B) mCherry fluorescence was significantly reduced by 1.3-fold, but the reduction in GFP fluorescence per cell was not significant. Conversely, cells expressing the Phe-decoding tRNALeuAAA variants showed a significant 1.5-fold reduction in GFP fluorescence and a reduction in mCherry fluorescence that was not significant.
Figure 5. eGFP fluorescence and cytotoxicity in normal and mistranslating cells under proteasome inhibition.
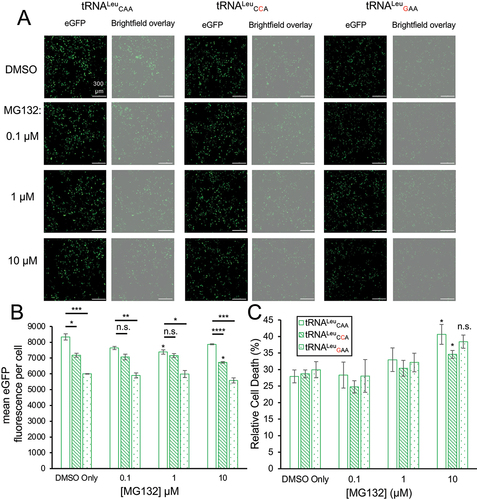
Because two of the variants showed a somewhat greater effect on one fluorescent protein versus the other, we considered that the fluorescent properties of GFP or mCherry might be impaired by mistranslation. To determine if mistranslation altered eGFP relative to mCherry fluorescence in individual cells, we plotted the ratio of green to red fluorescence per cell (Fig. S4E). In normal and mistranslating cells, the ratio was not significantly different except for cells expressing the Trp-decoding tRNALeuCCA, where we recorded a small but significant 8% increase in eGFP relative to mCherry fluorescence. Overall, the data suggest that the reduced GFP and/or mCherry fluorescence in the cells containing the tRNA variants is indicative of defective protein synthesis.
Mistranslation with Leu without toxicity to N2a cells
In previous studies, we measured cytotoxicity in mammalian cells expressing tRNAs that mis-incorporate Ser at Phe codons [Citation20,Citation23], Ser at Pro codons [Citation20], Ala at Pro codons [Citation22], and Ala at Gly codons [Citation21]. While the tRNA that directs mistranslation of Phe codons with Ser showed significant cytotoxicity on its own and synthetic toxicity in combination with proteasome inhibition [Citation20], the other mistranslating tRNAs showed no significant cytotoxicity. For the tRNALeu variants, we measured cytotoxicity under growth conditions without or with increasing concentrations of the proteasome inhibitor, MG132 (, S5C). In the absence of MG132, none of the mutant tRNAs significantly increased cytotoxicity. At 10 µM MG132, we observed a minor but significant increase in cell death in cells expressing wild-type tRNALeuCAA and the Trp-decoding tRNALeuCCA variant compared to cells with the same tRNAs and no MG132. At any given MG132 concentration, there was no significant difference in the toxicity of the Trp or Phe-decoding tRNALeu variants as compared to the wild-type tRNA (, S5C). Our data suggest that the mistranslation generated by each of the tRNALeu variants was not sufficiently disruptive to cause toxicity even in the context of proteasome inhibition.
Defective protein production in normal and mistranslating cells under proteasome inhibition
We found that each mutant tRNA caused a significant reduction of GFP fluorescence per cell in the DMSO control relative to cells expressing wild-type tRNA (, S5B). With increasing concentration of the proteasome inhibitor MG132, we observed a more significant reduction in GFP fluorescence in each mistranslating cell line (, S5B). Cells expressing tRNALeu with a Phe-decoding anticodon (GAA) showed a greater (1.4-fold) reduction in GFP fluorescence per cell compared to the 1.2-fold reduction in cells expressing the Trp-decoding tRNALeuCAA variant (). The greater defect in GFP fluorescence evident in cells expressing the Phe-decoding compared to Trp-decoding tRNALeu agrees with our MS/MS findings (). We also compared GFP fluorescence per cell in N2a cells expressing wild-type tRNALeuAAG or the Phe-decoding variant, tRNALeuAAA (Fig. S5A, S5B). There was also a significant 1.3-fold reduction in GFP fluorescence per cell in cells expressing the mutant tRNALeuAAA relative to wild-type tRNA in the DMSO control (Fig. S5).
Consistent with previous data [Citation64], in N2a cells expressing wild-type tRNA, inhibiting the proteasome with MG132 leads to a minor defect in protein production, i.e. ~1.1-fold reduced at each MG132 concentration compared to the DMSO control (). For cells expressing wild-type tRNA, the defect in protein production was only statistically significant at 1 µM MG132. In N2a cells expressing the Trp-decoding tRNALeu, we observed a statistically significant reduction in GFP levels compared to the DMSO control only at 10 µM MG132. Compared to cells expressing wild-type tRNA, GFP fluorescence in cells expressing the Trp-decoding tRNALeu was significantly reduced by 1.2-fold in conditions with DMSO (p < 0.05) and more significantly reduced at 10 µM MG132 (p < 0.0001) (). At intermediate concentrations of MG132 (0.1 and 1 µM), the defect in GFP production for cells mis-reading Trp codons with Leu was not significantly different from the reduced level observed in wild-type cells under proteasome inhibition.
In cells expressing the Phe-decoding tRNALeuGAA variant, we recorded a consistent and statistically significant defect in protein production as evidenced by a 1.4-fold reduction in GFP fluorescence per cell compared to cells expressing wild-type tRNALeuCAA at each MG132 concentration ()). In N2a cells expressing the other Phe-decoding tRNALeuAAA variant, we observed significant 1.2 to 1.3-fold reductions in GFP fluorescence per cell compared to cells expressing the wild-type tRNALeuAAG. At increasing MG132 concentrations, the defect in GFP fluorescence became increasingly significant (DMSO p < 0.05; at 1 µM MG132 p < 0.01; at 10 µM MG132 p < 0.001) in cells expressing tRNALeuAAA (Fig. S5A, S5B). The fact that MG132 did not increase the magnitude of the defect in GFP production caused by each tRNA mutant agrees with our findings above showing that proteasome inhibition was not synthetic toxic with any of the tRNALeu variants.
The anticodon is a critical determinant of phenotypic impact from tRNALeu variants
To allow a comprehensive evaluation of all tRNALeu anticodon variants, we engineered centromeric plasmids to express each one of the 64 possible tRNALeu anticodon derivatives (Table S2) and analysed their impact on yeast growth. Since the expression of some variants might fully inhibit growth, expression of the tRNA was regulated by a tetracycline inducible system whereby in the absence of the tetracycline analog doxycycline, tRNA expression is repressed [Citation38]. Addition of doxycycline to the growth media allows tRNA variant expression in a titratable manner. The tRNALeu locus used in this analysis was tL(UAA)B2 (Saccharomyces Genome Database: YNCB0012W; Genomic tRNA Database: Leu-TAA-1-2) including approximately 300 base pairs of up and downstream native flanking sequence (Fig. S1).
Plasmids containing one of 64 tRNALeu anticodon variants were transformed into yeast strain CY8652 that constitutively expresses Tet-VP16 (34). Individual transformants were colony purified and their growth characterized in liquid minimal media or in minimal media containing 0.01 or 1.0 µg/mL doxycycline (Supplementary Data File 2). Increased doxycycline concentrations will increase tRNA variant expression levels, and as seen previously [Citation38,Citation65], were expected to decrease growth rate. Growth was measured with four biological replicates of each strain at 30°C for 24 h. Representative growth curves for strains containing the wild-type control tRNALeuUAA(Leu) and anticodon variants tRNALeuCAU(Met) and tRNALeuUCA(Stop) show a range of impact on cell growth (). Less severe growth defects were seen at lower doxycycline concentrations for the Met-decoding tRNALeu variant that significantly slows cell growth.
Figure 6. Impact of tRNALeu anticodon variants on yeast growth.
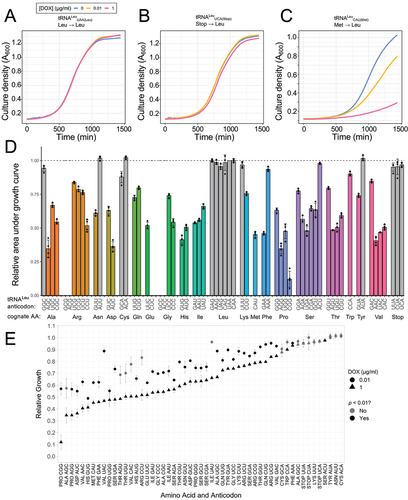
In medium containing 1.0 µg/mL doxycycline (), relative growth data of strains with each of the 64 possible tRNALeu variants show that of the 58 variants with non-leucine anticodons, 45 significantly decreased growth (p-value <0.01; Benjamini–Hochberg correction applied to t-test) relative to the wild-type tRNALeu. Five of the variants with anticodons GCG (Arg), ACG (Arg), CUC (Glu), GCC (Gly), and ACC (Gly) could not be transformed into yeast. Each is represented as 0% relative growth. These tRNAs are likely too toxic to yeast as the result of a low level of leaky expression. In future work, we can engineer in secondary mutations (as in [Citation54] and Fig. S8) that weaken tRNA activity that would facilitate transformation of these variants into yeast.
At least one anticodon variant for each of the 19 non-leucine amino acids significantly reduced growth. The data agree with reports that no position of the anticodon serves as a major identity element for LeuRS [Citation8,Citation9]. We observed a range of relative growth rates in strains containing different anticodon variants that mis-read codons for the same amino acid. For example, of the four Ala-decoding anticodons, Leu mis-incorporation reduced growth significantly () with three variants to 35% (AGC), 54% (CGC), and 67% (UGC) of wild-type growth. The Ala-decoding GGC anticodon variant did not show a significant growth defect. Similarly, the Phe-decoding tRNALeu variants showed markedly different growth defects. The tRNALeuGAA variant reduced growth rate to 45% of the control in medium containing 1.0 μg/mL Dox (), whereas cells expressing tRNALeuAAA grew at 93% of the control rate at 1.0 μg/mL Dox. Interestingly, although the growth defect caused by tRNALeuAAA was mild, it was sufficient to significantly increase heat shock protein expression as measured using an eGFP fluorescent reporter containing Hsf1-activated promoter elements (Fig. S6).
We measured the level of mis-incorporation in each of these strains using a label-free LC-MS/MS approach. Both Phe-decoding tRNALeu mutants generated mistranslation of Leu at Phe codons. Compared to the control tRNALeu, the tRNALeuGAA mutant showed ~10 to 100-fold greater abundance of mistranslated relative to properly translated peptides with mistranslation levels of 0.02–35% (, Supplementary Data File 3). The tRNALeuAAA mutant generated less mistranslation (0.1–1%) than tRNALeuGAA, but still significantly more than the control cells expressing wild-type tRNA.
Figure 7. Identification of Leu mis-incorporation in yeast by tandem mass spectrometry.
Each of the stop codon suppressing tRNALeu variants showed no statistically significant growth defect (), consistent with previous findings [Citation66–68]. This may arise because of the limited number of stop codons in the genome and/or the ability of many proteins to tolerate a C-terminal extension rather than the inability of the tRNA mutant to mistranslate. Therefore, we used a nonsense suppression assay to evaluate the ability of the ochre decoding tRNALeuUUA to readthrough the UAA stop codon. The assay takes advantage of our previous finding that an ochre codon at amino acid position 276 in the gene encoding the protein chaperone Tti2 (tti2Q276TAA) results in slow growth with a more severe phenotype in conditions of stress [Citation53]. Tti2 is essential but because low levels of the protein support viability, endogenous readthrough of tti2Q276TAA is sufficient to support some growth [Citation53]. To examine if tRNALeuUUA suppresses the nonsense ochre codon, we compared the growth of yeast strain CY6874 (tti2Q276TAA) transformed with a centromeric plasmid expressing either wild-type tRNALeuUAA or tRNALeuUUA at 30°C or 37°C in serial dilutions on synthetic minimal medium (Fig. S7). Consistent with tRNALeuUUA mistranslating the UAA stop codon, the strain expressing this plasmid showed increased growth at both temperatures. As a further indication of nonsense suppression under stress conditions, we grew the cells on synthetic minimal medium plates with 6 μg/mL Calcofluor white (CW). CW binds to chitin in the cell wall and inhibits growth. The strain with the wild-type tRNA showed essentially no growth, while the nonsense suppressor tRNALeu enabled normal growth under stress (Fig. S7). Together the data confirm that tRNALeuUAA robustly suppresses the UAA codon in the tti2Q276TAA transcript and provides a greater selective advantage under stress.
To determine how expression level affects the impact of the different tRNA variants on growth, we compared the relative growth of strains containing the tRNALeu variants in medium containing 0.01 or 1.0 µg/mL doxycycline. The impact of each variant on growth at the two doxycycline concentrations depends on the anticodon sequence (). For several of the variants, the reduction in relative growth at 0.01 μg/mL doxycycline was approximately 50% of that seen for 1.0 μg/mL doxycycline; however, for some variants, most notably tRNALeuUAU (Ile; 66% of wild-type growth at 1.0 μg/mL to 96% at 0.01 μg/mL) and tRNALeuUAC (Val; 47% at 1.0 μg/mL to 89% at 0.01 μg/mL), impact on growth was minimal at the lower doxycycline concentration and significantly reduced compared to wild-type at the higher doxycycline concentration.
Our previous work has shown that secondary mutations in the body of a mistranslating tRNA can reduce its phenotypic impact while still enabling mistranslation [Citation54]. We compared the growth of yeast strains expressing tRNALeuUGG(Pro) variants in the context of the wild-type tRNA and with the backbone including a G26A mutation. G26 is modified to N2,N2-dimethylguanosine [Citation69], as we observed for the human tRNAs (). In the absence of the modification, the tRNA is subject to degradation by the rapid tRNA decay pathway [Citation54,Citation69,Citation70]. Plasmids expressing tRNALeuUAA (wild-type) or tRNALeuUGG with or without the G26A mutation were introduced into yeast strain CY8652, and growth was measured in minimal medium at 30°C for 24 h. The relative growth rates were calculated from the area under the growth curve (Fig. S8). The G26A mutation significantly reduced the phenotypic impact of the defect caused by the tRNA that mis-incorporates Leu at Pro codons as compared to the variant with the UGG anticodon mutation and an otherwise wild-type tRNA body.
Factors determining the impact of the anticodon on growth
The ‘error minimization theory’ of genetic code evolution suggests that to minimize the deleterious effect of mutations, related amino acids are decoded by triplets of the most similar sequence [Citation71,Citation72]. To determine if a relationship exists between the similarity of the anticodon to those for Leu, we separated the anticodons into two groups depending on whether they contain one or two base changes from being complementary to a Leu codon (UUR or CUN). The plots of these two groups are largely overlapping with each group containing anticodons with a range of growth defects (Fig. S9). Each group also contains multiple variants that could not be introduced into yeast as well as multiple variants that did not impact growth. Comparison of the median growth defect between the two groups shows that there is no significant difference (Mann Whitney p = 0.2) between tRNALeu variants that differ by 1 or 2 bases from wild-type tRNALeu. The analysis suggests that similarity of the anticodon to those for Leu is not a primary determinant of impact on growth.
To visualize trends in phenotypic defects across the genetic code, we constructed a heat map of the relative growth superimposed on a circular representation of the genetic code based on the codon-anticodon duplex binding energy between anticodon positions 35 and 36 with the first two positions of the codon (). ‘Weak anticodons’, based on the codon-anticodon binding energy, are A/U rich. These A/U rich anticodon variants tended to show more moderate growth defects. In contrast, tRNALeu variants with a ‘strong’ or G/C rich anticodon tended to cause a greater growth defect. Indeed, among the most impactful variants overall were those with G/C rich anticodons for Arg, Gly, and Pro. To further address how the anticodon sequence affects growth, we plotted the relative growth of each variant versus the Turner binding energy of the base pairing between the codon and anticodon at positions 35 and 36. We observed a significant correlation between the Turner binding energetics and the reduction in relative growth (R = 0.42; p = 0.00055) (Fig. S10).
Figure 8. Relative growth of yeast strains with tRNALeu anticodon variants.
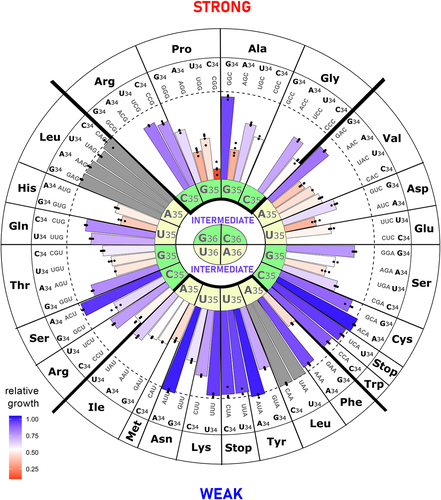
Because of the important role of base 34 in wobble decoding and translation fidelity, we next evaluated the contribution of tRNALeu variants at base 34 towards growth. For the 4-box anticodons including those for Arg and Ser, in 6 of 7 cases, the anticodon with A34 is either the most impactful or approximately equally impactful to a second variant (Fig. S11). For native four-box tRNAs, if present, A34 is normally modified to inosine [Citation73] and thus has the potential to decode three of the four codons, those ending with A, U, or C [Citation58]. In contrast, for two-box tRNAs in 5 of 6 cases, with a purine at position 34, the G34 variant is more impactful. It is possible that either the A34 in tRNA variants that mis-read two-box codons are either not modified to inosine in yeast, or perhaps the G at position 34 has increased impact by contributing a greater G/C content.
Of the 57 possible anticodons not encoding leucine or a stop, 18 are non-native to yeast. In 5 of 6 cases for the 2-box tRNAs with one anticodon native and the other non-native for the same amino acid, the non-native anticodon causes the greatest phenotypic defect (Fig. S11). In contrast, for the 4-box anticodons, including those for Arg and Ser, in 6 of 7 cases, a non-native anticodon was least impactful, the exception being GGG for proline, which reduces growth to ~10% of wild-type.
Finally, we examined the possibility that codon usage may impact efficiency of the suppressor tRNA variant. Assuming normal wobble GU pairs and the fact that A34I reads U, C and A, we summed the frequencies of codons read (per 1000 in the yeast genome) by each tRNA variant and plotted the values as a function of impact on growth of the relevant tRNA mutant (Fig. S12). We observed no overall correlation between codon usage and impact on growth. When looking at amino acids encoded by three or more codons, only Arg and Gly showed a correlation between increased impact on growth and increased number of codons read.
Impact of the amino acid substitution on relative growth with tRNALeu variants
Although the nature of the anticodon clearly impacts growth defects associated with mistranslating tRNALeu variants, we also considered the extent to which properties of the amino acid replaced by Leu contribute to such growth defects. We compared the impact on relative growth to the amino acid substitution scores for Leu in the BLOSUM62 matrix [Citation74] (Fig. S13A). The BLOSUM matrix was developed from blocks of aligned amino acids sequences in homologous proteins. The matrix records whether a Leu substitution for each of the other 19 amino acids is under-represented (negative scores), equally likely (0), or enriched (positive) in the substitutions observed in the natural evolution of proteins. We observed a positive (r = 0.43) and significant (p = 0.0006) correlation of increased growth rate with increased similarity of the replaced amino acid to Leu as defined by the BLOSUM matrix.
We then considered another metric that correlates well with error minimization theories of the genetic code [Citation75]. The polar requirement (PR) is a measure of amino acid migration on chromatography paper in a solution of pyridine [Citation76]. We observed a significant (p = 0.012) and moderately negative (r = −0.31) correlation between relative growth and similarity of the amino acids according to the PR metric (Fig. S13B). To identify other features of the amino acid replacement that may be important in determining impact on growth, we plotted the relative growth of strains containing tRNALeu variants versus amino acid volume, electron-ion-interaction potential (eiip), hydrophobicity, α-helix propensity, and π-helix propensity. These five factors were identified using repeated binary separation to attempt to best distinguish the 20 amino acids from each other [Citation77]. We only observed a significant correlation with amino acid replacements for Leu in their propensity to form π-helical structure (r = −0.35; p = 0.021) (Fig. S14). π-helices are comparatively rare yet found in 15% of proteins, often with functional significance [Citation78,Citation79]. The basis for the correlation is unclear since Leu generally supports π-helical structure [Citation79]. The correlation with each of the other parameters tested showed no statistical significance (Fig. S14), indicating that none of these amino acid properties individually explain the variance in our growth data.
Comparison of anticodon variants in different tRNALeu isoacceptors
In S. cerevisiae four isoacceptors decode Leu codons with three encoded by multiple copies: tRNALeuUAA (7 genes), tRNALeuCAA (10 genes), tRNALeuUAG (3 genes), and tRNALeuGAG (1 gene). To determine if anticodon variants in different tRNALeu genes give rise to similar growth defects, we engineered variants of the tRNALeuCAA and tRNALeuUAG isoacceptors to compare with those of tRNALeuUAA. Based on alignment of the tRNA sequences (Fig. S15), excluding the anticodon, there are 31 of 79 identical bases in the three tRNAs with the tRNALeuUAA isoacceptor having an additional base pair in the variable arm. The A box (consensus TRGYNNARNNG) and B box (consensus RGTTCRANTCC) promoter elements required for RNA polymerase III transcription (Fig. S15) are well conserved in each tRNALeu. The tRNALeuUAG strictly adheres to the A and B box consensus sequences, while the UAA and CAA isoacceptors differ at a single base in the B box. We note that the genomic copy of the tRNALeuUAG used contains an intron that is removed during tRNA maturation [Citation80].
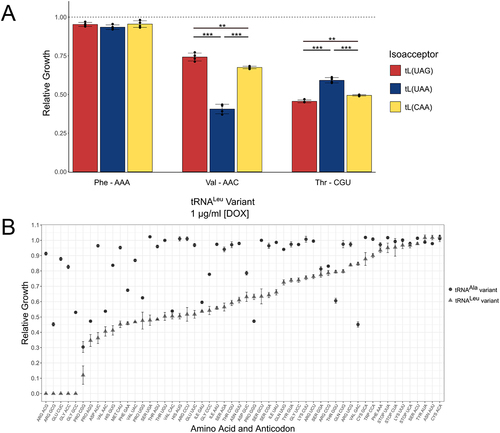
Genes for the tRNALeuCAA (YNCG0040C; tRNA-Leu-CAA-1-6) and tRNALeuUAG (YNCJ0029W; tRNA-Leu-TAG-1-1) isoacceptors were introduced into the doxycycline-inducible expression plasmid as above, with each including their native flanking sequences of ~300 base pairs. Four different anticodons were analysed in the three different tRNALeu genes: GCG (Arg), AAA (Phe), CGU (Thr), and AAC (Val). As was the case with the tRNALeuUAA isoacceptor, no transformants were obtained when the GCG (Arg) anticodon was substituted into the tRNALeuCAA or tRNALeuUAG isoacceptors. Transformants were obtained for the AAA (Phe), CGU (Thr), and AAC (Val) anticodons. We measured the impact of each tRNA allele on growth when cells were grown in minimal media containing 1.0 μg/mL doxycycline (). Consistent with our data above in yeast and mammalian cells, the AAA (Phe) anticodon had minimal impact on relative growth in each of the tRNALeu isoacceptors. AAC (Val) and CGU (Thr) anticodons reduced growth in each tRNALeu isoacceptor, but to a different extent. The ACA (Val) anticodon caused a greater growth defect than the CGU (Thr) anticodon when located in the tRNALeuUAA isoacceptor. Conversely, in the UAG and CAG isoacceptors, the Thr-decoding tRNA displayed a greater growth defect. Thus, the anticodon sequence and the nature isoacceptor both contribute to phenotypic defects associated with a tRNA anticodon mutant.
Figure 9. Analysis of anticodon variants in different contexts.
Comparison of tRNALeu and tRNAAla variants
Similar to tRNALeu, aminoacylation of tRNAAla is independent of its anticodon [Citation4,Citation10]. Having recently [Citation65] examined growth of yeast strains with all possible tRNAAla anticodon variants, we compared their impact to that of the tRNALeu variants characterized here. We observed that more tRNALeu variants reduced growth than tRNAAla variants, and in general, the individual tRNALeu variants have greater impact than the tRNAAla variants (). There are, however, exceptions to this trend. For example, the mistranslating tRNAAla variant with a GAC (Val) anticodon grew at a level of ~45% compared to cells with wild-type tRNA, while the tRNALeu variant with the same anticodon led to a more moderate growth defect, which was ~75% that of wild-type growth. These results are again consistent with the conclusion that the nature of the amino acid substitution and of the tRNA together determine the phenotypic impact of mistranslation in cells.
Discussion
Leu mis-incorporation from human tRNA anticodon variants in mammalian cells and yeast
We found three rare examples of tRNALeu anticodon mutants in human genomes. Expression of each allele in mammalian cells led to mistranslation, the greatest being for the Phe-decoding GAA variant, which also showed a significant defect in global protein synthesis. In the conditions tested, we found no significant cytotoxicity associated with any of the variants, but each of the cell lines expressing a tRNALeu anticodon variant showed defects in GFP protein production per cell with increasingly significant defects seen in cells under proteasome inhibition. Interestingly, these defects in protein homoeostasis were much less severe than the synthetic toxicity with proteosome inhibition that we observed in cells expressing a naturally occurring human Phe-decoding tRNASer variant [Citation20]. The greater defect in protein homoeostasis observed in cells mis-incorporating Ser at Phe codons compared to Leu at Phe codons likely results from multiple factors. Physiochemically and according to conservation in proteins [Citation74], Leu is more similar to Phe than Ser. The less conservative substitution of Ser at Phe codons may lead to greater disruption in protein folding and activity.
Human tRNALeu anticodon variants exist in our population, but the known variants are rare and represent only a few different anticodon sequences. This suggests that other tRNALeu anticodon variants might be significantly more toxic than the Phe- and Trp-decoding variants we characterized. To test our hypothesis, we used an inducible system in yeast to express all possible tRNALeu anticodon variants at low and high levels of induction. In agreement with our studies in mammalian cells, the Trp- and Phe (AAA)-decoding tRNALeu variants had minimal phenotypic impact. In contrast, the Phe (GAA) decoding variant showed a significant (50%) reduction in yeast growth. Although tRNALeuGAA was not cytotoxic in murine cells, the finding in yeast is consistent with our mass spectrometry analysis in yeast and mammalian cells, which showed that tRNALeu with the GAA (Phe) anticodon displays more evidence of mistranslation than the Phe-decoding tRNALeuAAA in N2a cells. Despite a reduced level of mistranslation, we did record elevated heat shock response in yeast cells expressing tRNALeuAAA, indicating the accumulation of misfolded proteins, which is a hallmark of mistranslation [Citation20,Citation62,Citation81,Citation82]. In yeast, missense tRNAAla suppressors [Citation33] behaved similarly to the tRNALeu variants, with tRNAAlaAAA and Trp-decoding variants showing little or no growth defect, while a significant growth reduction was identified in cells expressing tRNAAlaGAA.
In future work, it will be interesting to examine how particular pathways or proteins, perhaps based on the frequency of mistranslated codons contained in their open reading frames, may be differentially impacted by amino acid mis-incorporation. Using mass spectrometry data for such an analysis should be met with some caution, since mistranslated peptides may not be as readily detected. Indeed, mistranslation that generates insoluble or aggregated protein may be especially challenging to detect. Further work, including more detailed proteomic analysis and validation studies, will likely identify changes to the composition of the proteome that reveal cellular responses to tRNA-dependent mistranslation.
Factors affecting the impact of tRNALeu variants on growth
The facile nature of the yeast genetic system enabled us to generate yeast strains containing tRNALeu with all possible anticodons. We identified a range of growth defects and found the nature of the tRNA anticodon as well as the tRNA gene in which the variant was placed have strong impacts on the magnitude of the observed phenotypic defect in yeast. Together the data show that the impact of the tRNALeu missense suppressors ranges from well tolerated to strongly inhibitory of growth. While we were unable to identify a single property that individually could predict impact, substitutions with amino acids more distinct from Leu, according to the BLOSUM matrix, showed greater growth defects on average. Despite this trend, we found exceptions, such as the variants that decode the highly similar isoleucine (Ile) reduced growth by ~40%. Although one might assume the substitution of one hydrophobic amino acid with another of similar size would be minimally disruptive, nature has evolved important editing activities in several tRNA synthetases so that chemically similar amino acids do not contaminate the proteome. For example, IleRS has editing activities towards mis-charged Val-tRNAIle, while LeuRS de-acylates Val-tRNALeu and Ile-tRNALeu (reviewed in [Citation83]).
Analysis of our data on the complete catalogue of tRNALeu variants in yeast suggests that specific features of the tRNA anticodon influence toxicity of mistranslating tRNALeu variants. tRNALeu variants with more GC-rich anticodon:codon duplexes tended to produce greater and more significant growth defects. In addition, of the 15 missense suppressor tRNALeu mutants that include an A34 nucleotide, approximately half showed the strongest growth defect of all anticodons with the same cognate amino acid. The data suggest that, at least in some of these cases as we showed for tRNALeuAAA, the A34 is modified to inosine, which can efficiently decode codons that end in U, C, or A [Citation58]. Additional studies will be needed to assess the decoding efficiency on different codons for the tRNALeu variants characterized here.
The nature of the tRNA gene also affects the level of missense suppression and impact on growth. Although only 32 tRNAs are required to decode the 61 sense codons, yeast have multiple copies of each tRNA and encode 275 tRNA genes. Placing the same nonsynonymous anticodon in different tRNALeu isoacceptors also led to differential growth defects. The isoacceptors have differences in the tRNA body sequence, with two of the alleles we tested having SNPs in the B box portion of the promoter, and their flanking sequences are distinct. The nature of the amino acid was also a complicating factor. For example, among tRNA mutants derived from tRNALeuUAA, the Val-decoding variant showed the greatest phenotypic impact, while the Thr-decoding tRNA derived from the tRNALeuCAA isoacceptor was more toxic in yeast than the Val or Phe-decoding variants derived from the same tRNALeu. The data suggest that there are both optimal and less ideal pairings of anticodon, tRNA isoacceptor sequence and replaced amino acid that can tune the phenotypic impact of tRNA-dependent mistranslation.
There are other factors that contribute to the missense suppression levels and phenotypic defects caused by the tRNA variants. Our future work will focus on further defining mechanisms that tune the missense and nonsense suppression levels of tRNA variants. Previous studies identified multiple mechanisms that impact amino acid mis-incorporation, including tRNA expression level and copy number [Citation84]. For example, E. coli and yeast ribosome profiling has shown that across the transcriptome, increased tRNA levels correlate with decreased decoding times on the ribosome [Citation85]. Decoding efficiency of the tRNA on the ribosome [Citation86,Citation87], tRNA modifications [Citation88], aminoacylation level [Citation89], and the interaction of the mis-aminoacylated tRNA with elongation factor [Citation90] are additional factors contributing to tRNA activity. Some of our results were likely impacted by different aminoacylation levels of the tRNALeu variants. Although the anticodon is not a major identity element, previous studies in yeast found that tRNALeuUAG anticodon variants A35G (UGG, Pro) and G36U (UAU, Ile) were leucylated in vivo in yeast cells, but at a level that was 1.5-fold reduced compared to wild-type tRNA [Citation8].
Some of the tRNALeu variants could in principle be aminoacylated with the aaRS that is ‘cognate’ to the anticodon, which would moderate mistranslation. Although it is possible that these interactions could occur, the anticodon is not the only essential identity element for any of the aaRSs. For example, the eukaryotic PheRS requires A35 and A36 in the anticodon along with G20, G34, A37, and A73. Eukaryotic TrpRS requires the complete anticodon (C34, C35, A36) and G1–C72, U5–G68, and A73. The tRNALeu with a Phe-anticodon lacks G20 and A37 [Citation10] needed for PheRS, while tRNALeu with a Trp-anticodon lacks the U5-G68 pair for mammalian and human TrpRS [Citation91,Citation92]. The unusually large extra variable arm in tRNALeu acts as an anti-determinant towards non-cognate aaRSs, even those such as tRNASer that also contain a long variable arm [Citation93,Citation94].
Expression levels of tRNA variants, competition between suppressor tRNAs and the pool of cognate tRNAs that decode the same codon [Citation21,Citation23] or competition with release factor [Citation95] in the case of nonsense suppressors, may all contribute to the level of translation error. To begin to understand the role of expression levels, we used OTTR-seq to determine the levels of the tRNALeu anticodon variants in mammalian cells. We confirmed expression of each mutant tRNA and quantified the tRNA anticodon mutant expression levels relative to parent and competing tRNAs. The approach revealed differential base modification in tRNALeuAAA that showed 45% conversion of A34 to I. Thus, only half of the tRNALeuAAA pool can decode both UUU/C Phe codons, while all the tRNALeuGAA, which is expressed at a lower level, efficiently decodes both Phe codons. Fascinatingly, relative to tRNALeuAAA, tRNALeuGAA had the higher mistranslation level according to MS/MS analysis and the greatest impact on protein homoeostasis in mammalian cells and on growth in yeast. The data indicate that tRNALeuGAA more effectively competes against tRNAPhe for insertion of Leu at Phe codons. In addition, we found that cells expressing tRNALeuGAA had the most perturbed tRNAome overall, with 11 other tRNAs showing small but significantly reduced abundance in mistranslating cells. In future studies, we will determine if the changes in the tRNAome contribute the greater phenotypic defect in cells expressing tRNALeuGAA.
Codon usage may also impact the efficiency of missense or nonsense suppression as a result of different competing pools of aminoacyl-tRNAs. In our studies of natural human tRNALeu variants, we also observed a greater level of mis-incorporation at the more common Phe relative to Trp codons in mammalian cells. In considering our anticodon catalogue in yeast, we found no overall correlation between codon usage and impact on growth (Fig. S12). In the replacement of Leu for some individual amino acids (Arg and Gly) there is a correlation between the frequency of codons read by the mutant tRNA with increasing growth defect, while other amino acids showed no correlation. While all these factors play important roles, our data show that mistranslation and associated phenotypic defects depend on the kind of amino acid replacement and the nature of the tRNA, including the anticodon sequence and the tRNA gene in which the mistranslating variant occurs. These are, however, only some of the complex factors that affect mis-incorporation levels and the magnitude of fitness defects.
Applications of missense and nonsense suppressor tRNAs in synthetic biology and therapeutics
Expansion of the genetic code with additional amino acids relies on the natural or synthetic evolution of additional tRNA and AARS pairs that do not cross react with endogenous tRNAs and AARSs and that possess selectivity for an unnatural or non-canonical amino acid (ncAA). These orthogonal tRNA and tRNA synthetase pairs (o-pairs) are based of several different AARSs [Citation96]. For example, an o-pair derived from LeuRS and tRNALeu was established for site-directed incorporation of a photo-caged selenocysteine in proteins in mammalian cells to generate light inducible selenoprotein activity [Citation97]. These o-pairs are desirable because of their ability to incorporate many kinds of ncAAs into recombinant proteins, and the LeuRS systems are mutually orthogonal with genetic code expansion systems based on pyrrolysyl-tRNA synthetase. Indeed, these systems were used together for dual labelling applications of proteins [Citation98]. While most of the genetic code expansion field is focused on reassignment of stop codons, sense codon recoding or reassignment via missense suppression is another approach that can be used for the site-specific incorporation of selenocysteine [Citation99] or diverse ncAAs into proteins [Citation100]. Our findings suggest that nonsense or missense suppression in these systems may be improved by choosing an effective combination of tRNA genes and anticodons.
Beyond applications in synthetic biology, a new era of tRNA therapeutics has arrived with major breakthroughs that already use wild-type [Citation101], nonsense [Citation102] or missense [Citation103] suppressor tRNAs to correct disease-causing alleles in cells and mice. Premature termination codon (PTC) mutations account for 11% of human inherited genetic diseases [Citation104,Citation105]. Interestingly, 3% of the disease-causing PTCs are of a Leu codon (UUG or UUA) to a stop codon [Citation104]. Nonsense suppressor tRNALeu variants may enable readthrough of these disease-causing alleles to produce at least some portion of the full length and wild-type protein. While these approaches might not be applicable to PTCs in genes that require high levels of protein production, many proteins are produced at greater levels than needed [Citation17,Citation106,Citation107].
Established nonsense suppression approaches to therapeutics are based on aminoglycosides (e.g. gentamicin, G418) or small molecules, like Ataluren [Citation106], that promote ribosomal readthrough of stop codons (UGA, UAA, UAG) with different amino acids at levels of 10–30% [Citation106,Citation108,Citation109]. Aminoglycosides are least effective at UAA suppression [Citation110]. Furthermore, suppressor tRNAs are more programmable and selective than aminoglycosides as they will deliver a single kind of amino acid to one of the three stop codons. Suppressor tRNAs will readthrough all instances of a specific stop codon [Citation111], yet off-target effects would be less than Ataluren, an approved treatment of a PTC that causes Duchenne’s Muscular Dystrophy [Citation106]. In a breakthrough, a nonsense suppressor tRNA was recently delivered using an adeno-associated viral vector to effectively treat a mouse model of mucopolysaccharidosis type I, a disease caused by a PTC [Citation112]. In another example, nonsense suppressor tRNAs resolved disease via readthrough of a PTC in cystic fibrosis transmembrane conductance regulator in Xenopus oocytes and mouse models of the disease [Citation107]. In both cases, the tRNA-dependent stop codon readthrough was sufficient to suppress the disease causing-allele without causing toxicity to the animals.
Since many inherited genetic diseases are caused by missense mutations in coding genes [Citation76,Citation104,Citation105], missense suppressors that produce a sufficient level of a wild-type protein from a disease-causing allele will have broad applications for tRNA-based therapies [Citation113]. Although some mutant tRNAs cause toxicity in cells, many missense suppressor tRNAs are well-tolerated in yeast [Citation17], flies [Citation114], human, and murine cells [Citation21,Citation103,Citation115]. Thus, tRNA suppressors can be employed as therapeutic agents to correct genetic defects. For example, in our earlier studies in yeast, we identified a missense suppressor tRNAPro mutant that generated mistranslation of Ala at Pro codons, without significant phenotypic defects [Citation17]. In fact, the mutant tRNA pre-exists in the cell population to spontaneously suppress a stress sensitive missense allele of the co-chaperone, Tti2. A level of 6% mistranslation was sufficient to rescue growth under stress. Here, we identified many tRNALeu anticodon variants that are non-toxic to mammalian cells or yeast cells, at the appropriate induction level. Our data suggest that several different kinds of missense suppressor tRNALeu variants are viable routes to tRNA therapeutics. Overall, our data show that by selecting the appropriate anticodon, tRNA gene and level of expression, tRNALeu variants will provide useful approaches to engineer nonsense and missense suppressor tRNAs for applications in synthetic biology and medicine.
Conclusion
We found that naturally occurring human tRNALeu anticodon mutants mistranslate Phe and Trp codons with Leu and cause defects in protein production that were not toxic in mammalian cells. Two of the same variants were non-toxic to yeast, while a third Phe-decoding tRNALeuGAA variant showed a growth defect in yeast that correlated with a higher level of Leu mis-incorporation in yeast and mammalian cells. We probed the entire genetic code for Leu mis-incorporation in yeast. The data revealed a complex interplay between several factors, including the level of tRNA induction, the sequence of the tRNA isoacceptor, the nature of the anticodon sequence, and the amino acid substitution that all contribute to modulate phenotypic defects in mistranslating cells. We observed generally stronger growth defects in mutant tRNAs with A34 or with G/C rich codon-anticodon duplexes. This work will have broad impacts in synthetic biology for applications in genetic code expansion to reassign stop and sense codons in eukaryotic cells. Our characterization of effective but non-toxic missense and nonsense tRNALeu suppressors will also have applications in medicine as it leads towards tRNA therapeutics to correct genetic defects that cause human disease.
DataFile1.csv
Download Comma-Separated Values File (341.1 KB)DataFile3.csv
Download Comma-Separated Values File (54.8 KB)DataFile4.csv
Download Comma-Separated Values File (6.8 KB)DataFile2.csv
Download Comma-Separated Values File (30.8 KB)tRNALeu_2023SI_R1.docx
Download MS Word (14.4 MB)Acknowledgments
We are grateful to Josh Isaacson, Matthew Berg, Justin Ye, and Ilka Heinemann for insightful discussion and suggestions. We also thank Paula Pittock (Biological Mass Spectrometry Laboratory, University of Western Ontario) for assistance with identification of mistranslated peptides. In addition, we thank Kathleen Collins (University of California, Berkeley) for providing OTTR-seq reagents and protocol.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data are available in the figures, tables, supplementary information, and Supplementary Data Files 1–4.
All MS/MS data files are available:
https://figshare.com/articles/dataset/Mass_spec_tRNALeu_2023_zip/23284907/1
https://bioinfor.sharefile.com/d-sb232430cb59d423c99b1cb5e3b492e58
OTTR-seq data files are available at NCBI BioProject database
http://www.ncbi.nlm.nih.gov/bioproject/1066329
SubmissionID:SUB14155213
BioProject ID: PRJNA1066329
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15476286.2024.2340297
Additional information
Funding
References
- O’Donoghue P, Luthey-Schulten Z. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol Mol Biol Rev. 2003;67(4):550–573. doi: 10.1128/MMBR.67.4.550-573.2003
- Schimmel PR, Söll D. Aminoacyl-tRNA synthetases: general features and recognition of transfer RNAs. Annu Rev Biochem. 1979;48(1):601–648. doi: 10.1146/annurev.bi.48.070179.003125
- Giege R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26(22):5017–5035. doi: 10.1093/nar/26.22.5017
- Hou YM, Schimmel P. A simple structural feature is a major determinant of the identity of a transfer RNA. Nature. 1988;333(6169):140–145. doi: 10.1038/333140a0
- Lenhard B, Orellana O, Ibba M, et al. tRNA recognition and evolution of determinants in seryl-tRNA synthesis. Nucleic Acids Res. 1999;27(3):721–729. doi: 10.1093/nar/27.3.721
- O’Donoghue P, Prat L, Heinemann IU, et al. Near-cognate suppression of amber, opal and quadruplet codons competes with aminoacyl-tRNAPyl for genetic code expansion. FEBS Lett. 2012;586(21):3931–3937. doi: 10.1016/j.febslet.2012.09.033
- Ambrogelly A, Gundllapalli S, Herring S, et al. Pyrrolysine is not hardwired for cotranslational insertion at UAG codons. Proc Natl Acad Sci, USA. 2007;104(9):3141–3146. doi: 10.1073/pnas.0611634104
- Huang Q, Yao P, Eriani G, et al. In vivo identification of essential nucleotides in tRnaleu to its functions by using a constructed yeast tRNALeu knockout strain. Nucleic Acids Res. 2012;40(20):10463–10477. doi: 10.1093/nar/gks783
- Breitschopf K, Achsel T, Busch K, et al. Identity elements of human tRNA Leu: structural requirements for converting human tRNA Ser into a leucine acceptor in vitro. Nucleic Acids Res. 1995;23(18):3633–3637. doi: 10.1093/nar/23.18.3633
- Giege R, Eriani G. The tRNA identity landscape for aminoacylation and beyond. Nucleic Acids Res. 2023;51(4):1528–1570. doi: 10.1093/nar/gkad007
- Parker J. Errors and alternatives in reading the universal genetic code. Microbiol Rev. 1989;53(3):273–298. doi: 10.1128/mr.53.3.273-298.1989
- Joshi K, Bhatt MJ, Farabaugh PJ. Codon-specific effects of tRNA anticodon loop modifications on translational misreading errors in the yeast Saccharomyces cerevisiae. Nucleic Acids Res. 2018;46(19):10331–10339. doi: 10.1093/nar/gky664
- Manickam N, Nag N, Abbasi A, et al. Studies of translational misreading in vivo show that the ribosome very efficiently discriminates against most potential errors. RNA. 2014;20(1):9–15. doi: 10.1261/rna.039792.113
- Zhang J, Ieong KW, Mellenius H, et al. Proofreading neutralizes potential error hotspots in genetic code translation by transfer RNAs. RNA. 2016;22(6):896–904. doi: 10.1261/rna.055632.115
- Rozik P, Szabla R, Lant JT, et al. A novel fluorescent reporter sensitive to serine mis-incorporation. RNA Biol. 2022;19(1):221–233. doi: 10.1080/15476286.2021.2015173
- Ruan B, Palioura S, Sabina J, et al. Quality control despite mistranslation caused by an ambiguous genetic code. Proc Natl Acad Sci, USA. 2008;105(43):16502–16507. doi: 10.1073/pnas.0809179105
- Hoffman KS, Berg MD, Shilton BH, et al. Genetic selection for mistranslation rescues a defective co-chaperone in yeast. Nucleic Acids Res. 2017;45(6):3407–3421. doi: 10.1093/nar/gkw1021
- Berg MD, Zhu Y, Genereaux J, et al. Modulating mistranslation potential of tRNASer in Saccharomyces cerevisiae. Genetics. 2019;213(3):849–863. doi: 10.1534/genetics.119.302525
- Berg MD, Zhu Y, Ruiz BY, et al. The amino acid substitution affects cellular response to mistranslation. G3 (Bethesda). 2021;11(10):11. doi: 10.1093/g3journal/jkab218
- Lant JT, Kiri R, Duennwald ML, et al. Formation and persistence of polyglutamine aggregates in mistranslating cells. Nucleic Acids Res. 2021;49(20):11883–11899. doi: 10.1093/nar/gkab898
- Hasan F, Lant JT, O’Donoghue P. Perseverance of protein homeostasis despite mistranslation of glycine codons with alanine. Philos Trans R Soc Lond B Biol Sci. 2023;378:20220029. doi: 10.1098/rstb.2022.0029
- Lant JT, Berg MD, Sze DHW, et al. Visualizing tRNA-dependent mistranslation in human cells. RNA Biol. 2018;15(4–5):567–575. doi: 10.1080/15476286.2017.1379645
- Lant JT, Hasan F, Briggs J, et al. Genetic interaction of tRNA-dependent mistranslation with fused in sarcoma protein aggregates. Genes (Basel). 2023;14(2):14. doi: 10.3390/genes14020518
- Wu J, Fan Y, Ling J. Mechanism of oxidant-induced mistranslation by threonyl-tRNA synthetase. Nucleic Acids Res. 2014;42(10):6523–6531. doi: 10.1093/nar/gku271
- Roy H, Ibba M. Phenylalanyl-tRNA synthetase contains a dispensable RNA-binding domain that contributes to the editing of noncognate aminoacyl-tRNA. Biochemistry. 2006;45(30):9156–9162. doi: 10.1021/bi060549w
- Chen M, Kuhle B, Diedrich J, et al. Cross-editing by a tRNA synthetase allows vertebrates to abundantly express mischargeable tRNA without causing mistranslation. Nucleic Acids Res. 2020;48(12):6445–6457. doi: 10.1093/nar/gkaa469
- Ma X, Bakhtina M, Shulgina I, et al. Structural basis of tRNAPro acceptor stem recognition by a bacterial trans-editing domain. Nucleic Acids Res. 2023;51(8):3988–3999. doi: 10.1093/nar/gkad192
- Lee JW, Beebe K, Nangle LA, et al. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443(7107):50–55. doi: 10.1038/nature05096
- Murgola EJ, Yanofsky C. Suppression of glutamic acid codons by mutant glycine transfer ribonucleic acid. J Bacteriol. 1974;117(2):439–443. doi: 10.1128/jb.117.2.439-443.1974
- Lant JT, Berg MD, Heinemann IU, et al. Pathways to disease from natural variations in human cytoplasmic tRNAs. J Biol Chem. 2019;294(14):5294–5308. doi: 10.1074/jbc.REV118.002982
- Berg MD, Giguere DJ, Dron JS, et al. Targeted sequencing reveals expanded genetic diversity of human transfer RNAs. RNA Biol. 2019;16(11):1574–1585. doi: 10.1080/15476286.2019.1646079
- Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature. 2021;590(7845):290–299. doi: 10.1038/s41586-021-03205-y
- Chan PP, Lowe TM. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016;44(D1):D184–9. doi: 10.1093/nar/gkv1309
- Consortium UK, Walter K, Min JL, et al. The UK10K project identifies rare variants in health and disease. Nature. 2015;526(7571):82–90. doi: 10.1038/nature14962
- Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi: 10.1038/s41586-020-2308-7
- Tadaka S, Katsuoka F, Ueki M, et al. 3.5KJPNv2: an allele frequency panel of 3552 Japanese individuals including the X chromosome. Hum Genome Var. 2019;6(1):28. doi: 10.1038/s41439-019-0059-5
- Gomes AC, Kordala AJ, Strack R, et al. A dual fluorescent reporter for the investigation of methionine mistranslation in live cells. RNA. 2016;22(3):467–476. doi: 10.1261/rna.054163.115
- Berg MD, Isaacson JR, Cozma E, et al. Regulating expression of mistranslating tRNAs by readthrough RNA polymerase II transcription. ACS Synth Biol. 2021;10(11):3177–3189. doi: 10.1021/acssynbio.1c00461
- Schmidt EK, Clavarino G, Ceppi M, et al. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6(4):275–277. doi: 10.1038/nmeth.1314
- Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089
- Das U, Shuman S. Mechanism of RNA 2′,3′-cyclic phosphate end healing by T4 polynucleotide kinase–phosphatase. Nucleic Acids Res. 2013;41(1):355–365. doi: 10.1093/nar/gks977
- Watkins CP, Zhang W, Wylder AC, et al. A multiplex platform for small RNA sequencing elucidates multifaceted tRNA stress response and translational regulation. Nat Commun. 2022;13(1):2491. doi: 10.1038/s41467-022-30261-3
- Upton HE, Ferguson L, Temoche-Diaz MM, et al. Low-bias ncRNA libraries using ordered two-template relay: serial template jumping by a modified retroelement reverse transcriptase. Proc Natl Acad Sci, USA. 2021;118(42):118. doi: 10.1073/pnas.2107900118
- Itokawa K. Illumina TruSeq Library quantification with qPCR probe method. https://protocols.io/view/illumina-truseq-library-quantification-with-qpcr-p-bnpamdie:protocols.io. 2020.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12. doi: 10.14806/ej.17.1.200
- Smith T, Heger A, Sudbery I. UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 2017;27(3):491–499. doi: 10.1101/gr.209601.116
- Holmes AD, Howard JM, Chan PP, et al. tRNA Analysis of eXpression (tRAX): a tool for integrating analysis of tRnas, tRNA-derived small RNAs, and tRNA modifications. bioRxiv. 2022;1–55. doi: 10.1101/2022.07.02.498565
- Cappannini A, Ray A, Purta E, et al. MODOMICS: a database of RNA modifications and related information. 2023 update. Nucleic Acids Res. 2024;52(D1):D239–D44. doi: 10.1093/nar/gkad1083
- Brachmann CB, Davies A, Cost GJ, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14(2):115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115:AID-YEA204>3.0.CO;2-2
- Hughes TR, Marton MJ, Jones AR, et al. Functional discovery via a compendium of expression profiles. Cell. 2000;102(1):109–126. doi: 10.1016/S0092-8674(00)00015-5
- Berg MD, Genereaux J, Zhu Y, et al. Acceptor stem differences contribute to species-specific use of yeast and human tRNA(Ser). Genes (Basel). 2018;9(12):612. doi: 10.3390/genes9120612
- Sprouffske K, Wagner A. Growthcurver: an R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinf. 2016;17(1):172. doi: 10.1186/s12859-016-1016-7
- Hoffman KS, Duennwald ML, Karagiannis J, et al. Saccharomyces cerevisiae Tti2 regulates PIKK proteins and stress response. G3 (Bethesda). 2016;6(6):1649–1659. doi: 10.1534/g3.116.029520
- Berg MD, Hoffman KS, Genereaux J, et al. Evolving mistranslating tRNAs through a phenotypically ambivalent intermediate in Saccharomyces cerevisiae. Genetics. 2017;206(4):1865–1879. doi: 10.1534/genetics.117.203232
- Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308
- Turowski TW, Tollervey D. Transcription by RNA polymerase III: insights into mechanism and regulation. Biochem Soc Trans. 2016;44(5):1367–1375. doi: 10.1042/BST20160062
- Roura Frigole H, Camacho N, Castellvi Coma M, et al. tRNA deamination by ADAT requires substrate-specific recognition mechanisms and can be inhibited by tRfs. RNA. 2019;25(5):607–619. doi: 10.1261/rna.068189.118
- Söll D, Jones DS, Ohtsuka E, et al. Specificity of sRNA for recognition of codons as studied by the ribosomal binding technique. J Mol Biol. 1966;19(2):556–573. doi: 10.1016/S0022-2836(66)80023-2
- Torres AG, Pineyro D, Rodriguez-Escriba M, et al. Inosine modifications in human tRNAs are incorporated at the precursor tRNA level. Nucleic Acids Res. 2015;43(10):5145–5157. doi: 10.1093/nar/gkv277
- Clark WC, Evans ME, Dominissini D, et al. tRNA base methylation identification and quantification via high-throughput sequencing. RNA. 2016;22(11):1771–1784. doi: 10.1261/rna.056531.116
- Harada F, Matsubara M, Kato N. Stable tRNA precursors in HeLa cells. Nucleic Acids Res. 1984;12(24):9263–9269. doi: 10.1093/nar/12.24.9263
- Geslain R, Cubells L, Bori-Sanz T, et al. Chimeric tRNAs as tools to induce proteome damage and identify components of stress responses. Nucleic Acids Res. 2010;38(5):e30. doi: 10.1093/nar/gkp1083
- Shcherbakov D, Teo Y, Boukari H, et al. Ribosomal mistranslation leads to silencing of the unfolded protein response and increased mitochondrial biogenesis. Commun Biol. 2019;2(1):381. doi: 10.1038/s42003-019-0626-9
- Cowan JL, Morley SJ. The proteasome inhibitor, MG132, promotes the reprogramming of translation in C2C12 myoblasts and facilitates the association of hsp25 with the eIF4F complex. Eur J Biochem. 2004;271(17):3596–3611. doi: 10.1111/j.0014-2956.2004.04306.x
- Cozma E, Rao M, Dusick M, et al. Anticodon sequence determines the impact of mistranslating tRNAAla variants. RNA Biol. 2023;20(1):791–804. doi: 10.1080/15476286.2023.2257471
- Kim D, Johnson J. Construction, expression, and function of a new yeast amber suppressor, tRNATrpA. J Biol Chem. 1988;263(15):7316–7321. doi: 10.1016/S0021-9258(18)68644-2
- Hancock SM, Uprety R, Deiters A, et al. Expanding the genetic code of yeast for incorporation of diverse unnatural amino acids via a pyrrolysyl-tRNA synthetase/tRNA pair. J Am Chem Soc. 2010;132(42):14819–14824. doi: 10.1021/ja104609m
- Blanchet S, Cornu D, Argentini M, et al. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic Acids Res. 2014;42(15):10061–10072. doi: 10.1093/nar/gku663
- Dewe JM, Whipple JM, Chernyakov I, et al. The yeast rapid tRNA decay pathway competes with elongation factor 1A for substrate tRNAs and acts on tRNAs lacking one or more of several modifications. RNA. 2012;18(10):1886–1896. doi: 10.1261/rna.033654.112
- Whipple JM, Lane EA, Chernyakov I, et al. The yeast rapid tRNA decay pathway primarily monitors the structural integrity of the acceptor and T-stems of mature tRNA. Genes Dev. 2011;25(11):1173–1184. doi: 10.1101/gad.2050711
- Haig D, Hurst LD. A quantitative measure of error minimization in the genetic code. J Mol Evol. 1991;33(5):412–417. doi: 10.1007/BF02103132
- Caldararo F, Di Giulio M. The genetic code is very close to a global optimum in a model of its origin taking into account both the partition energy of amino acids and their biosynthetic relationships. Biosystems. 2022;214:104613. doi: 10.1016/j.biosystems.2022.104613
- Haumont E, Fournier M, de Henau S, et al. Enzymatic conversion of adenosine to inosine in the wobble position of yeast tRNAAsp: the dependence on the anticodon sequence. Nucleic Acids Res. 1984;12(6):2705–2715. doi: 10.1093/nar/12.6.2705
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci, USA. 1992;89(22):10915–10919. doi: 10.1073/pnas.89.22.10915
- Vetsigian K, Woese C, Goldenfeld N. Collective evolution and the genetic code. Proc Natl Acad Sci, USA. 2006;103(28):10696–10701. doi: 10.1073/pnas.0603780103
- Woese CR, Dugre DH, Dugre SA, et al. On the fundamental nature and evolution of the genetic code. Cold Spring Harb Symp Quant Biol. 1966;31:723–736. doi: 10.1101/SQB.1966.031.01.093
- Then A, Macha K, Ibrahim B, et al. A novel method for achieving an optimal classification of the proteinogenic amino acids. Sci Rep. 2020;10(1):15321. doi: 10.1038/s41598-020-72174-5
- Cooley RB, Arp DJ, Karplus PA. Evolutionary origin of a secondary structure: π-Helices as Cryptic but widespread insertional variations of α-Helices that enhance protein functionality. J Mol Biol. 2010;404(2):232–246. doi: 10.1016/j.jmb.2010.09.034
- Fodje MN, Al-Karadaghi S. Occurrence, conformational features and amino acid propensities for the π-helix. Protein Engineering, Design And Selection. 2002;15(5):353–358. doi: 10.1093/protein/15.5.353
- Hopper AK, Nostramo RT. tRNA processing and subcellular trafficking proteins multitask in pathways for other RNAs. Front Genet. 2019;10:96. doi: 10.3389/fgene.2019.00096
- Mohler K, Mann R, Bullwinkle TJ, et al. Editing of misaminoacylated tRNA controls the sensitivity of amino acid stress responses in Saccharomyces cerevisiae. Nucleic Acids Res. 2017;45(7):3985–3996. doi: 10.1093/nar/gkx077
- Paredes JA, Carreto L, Simoes J, et al. Low level genome mistranslations deregulate the transcriptome and translatome and generate proteotoxic stress in yeast. BMC Biol. 2012;10(1):55. doi: 10.1186/1741-7007-10-55
- Ling J, Reynolds N, Ibba M. Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol. 2009;63(1):61–78. doi: 10.1146/annurev.micro.091208.073210
- Aerni HR, Shifman MA, Rogulina S, et al. Revealing the amino acid composition of proteins within an expanded genetic code. Nucleic Acids Res. 2015;43(2):e8. doi: 10.1093/nar/gku1087
- Dana A, Tuller T. The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res. 2014;42(14):9171–9181. doi: 10.1093/nar/gku646
- Hirsh D. Tryptophan transfer RNA as the UGA suppressor. J Mol Biol. 1971;58(2):439–458. doi: 10.1016/0022-2836(71)90362-7
- Schmeing TM, Voorhees RM, Kelley AC, et al. How mutations in tRNA distant from the anticodon affect the fidelity of decoding. Nat Struct Mol Biol. 2011;18(4):432–436. doi: 10.1038/nsmb.2003
- Tittle JM, Schwark DG, Biddle W, et al. Impact of queuosine modification of endogenous E. coli tRNAs on sense codon reassignment. Front Mol Biosci. 2022;9:938114. doi: 10.3389/fmolb.2022.938114
- Guo LT, Wang YS, Nakamura A, et al. Polyspecific pyrrolysyl-tRNA synthetases from directed evolution. Proc Natl Acad Sci, USA. 2014;111(47):16724–16729. doi: 10.1073/pnas.1419737111
- LaRiviere FJ, Wolfson AD, Uhlenbeck OC. Uniform binding of aminoacyl-tRNAs to elongation factor tu by thermodynamic compensation. Science. 2001;294(5540):165–168. doi: 10.1126/science.1064242
- Guo Q, Gong Q, Tong KL, et al. Recognition by tryptophanyl-tRNA synthetases of discriminator base on tRNATrp from three biological domains. J Biol Chem. 2002;277(16):14343–14349. doi: 10.1074/jbc.M111745200
- Xu F, Chen X, Xin L, et al. Species-specific differences in the operational RNA code for aminoacylation of tRNA(Trp). Nucleic Acids Res. 2001;29(20):4125–4133. doi: 10.1093/nar/29.20.4125
- Asahara H, Himeno H, Tamura K, et al. Recognition nucleotides of Escherichia coli tRnaleu and its elements facilitating discrimination from tRnaser and tRNATyr. J Mol Biol. 1993;231(2):219–229. doi: 10.1006/jmbi.1993.1277
- Giegé R, Eriani G. Transfer RNA Recognition and Aminoacylation by Synthetases. London, England: Encyclopedia of Life Sciences: Macmillan Publishers Ltd, Nature Publishing Group; 2021.
- Johnson DB, Xu J, Shen Z, et al. RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat Chem Biol. 2011;7(11):779–786. doi: 10.1038/nchembio.657
- O’Donoghue P, Ling J, Wang YS, et al. Upgrading protein synthesis for synthetic biology. Nat Chem Biol. 2013;9(10):594–598. doi: 10.1038/nchembio.1339
- Peeler JC, Falco JA, Kelemen RE, et al. Generation of recombinant mammalian selenoproteins through genetic code expansion with photocaged selenocysteine. ACS Chem Biol. 2020;15(6):1535–1540. doi: 10.1021/acschembio.0c00147
- Zheng Y, Mukherjee R, Chin MA, et al. Expanding the scope of single- and double-noncanonical amino acid mutagenesis in mammalian cells using orthogonal polyspecific leucyl-tRNA synthetases. Biochemistry. 2018;57(4):441–445. doi: 10.1021/acs.biochem.7b00952
- Brocker MJ, Ho JM, Church GM, et al. Recoding the genetic code with selenocysteine. Angew Chem Int Ed Engl. 2014;53(1):319–323. doi: 10.1002/anie.201308584
- Biddle W, Schwark DG, Schmitt MA, et al. Directed evolution pipeline for the improvement of orthogonal translation machinery for genetic code expansion at sense codons. Front Chem. 2022;10:815788. doi: 10.3389/fchem.2022.815788
- Zuko A, Mallik M, Thompson R, et al. tRNA overexpression rescues peripheral neuropathy caused by mutations in tRNA synthetase. Science. 2021;373(6559):1161–1166. doi: 10.1126/science.abb3356
- Dolgin E. tRNA therapeutics burst onto startup scene. Nat Biotechnol. 2022;40(3):283–286. doi: 10.1038/s41587-022-01252-y
- Hou Y, Zhang W, McGilvray PT, et al. Engineered mischarged transfer RNAs for correcting pathogenic missense mutations. Mol Ther. 2023;32(2):352–371. doi: 10.1016/j.ymthe.2023.12.014
- Mort M, Ivanov D, Cooper DN, et al. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29(8):1037–1047. doi: 10.1002/humu.20763
- Lee HL, Dougherty JP. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol Ther. 2012;136(2):227–266. doi: 10.1016/j.pharmthera.2012.07.007
- Morkous SS. Treatment with ataluren for Duchenne Muscular Dystrophy. Pediatr Neurol Briefs. 2020;34:12. doi: 10.15844/pedneurbriefs-34-12
- Lueck JD, Yoon JS, Perales-Puchalt A, et al. Engineered transfer RNAs for suppression of premature termination codons. Nat Commun. 2019;10(1):822. doi: 10.1038/s41467-019-08329-4
- Roy B, Friesen WJ, Tomizawa Y, et al. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc Natl Acad Sci, USA. 2016;113(44):12508–12513. doi: 10.1073/pnas.1605336113
- Martins-Dias P, Romao L. Nonsense suppression therapies in human genetic diseases. Cell Mol Life Sci. 2021;78(10):4677–4701. doi: 10.1007/s00018-021-03809-7
- Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6(7):1044–1055. doi: 10.1017/S1355838200000716
- Heinemann IU, Rovner AJ, Aerni HR, et al. Enhanced phosphoserine insertion during Escherichia coli protein synthesis via partial UAG codon reassignment and release factor 1 deletion. FEBS Lett. 2012;586(20):3716–3722. doi: 10.1016/j.febslet.2012.08.031
- Wang J, Zhang Y, Mendonca CA, et al. AAV-delivered suppressor tRNA overcomes a nonsense mutation in mice. Nature. 2022;604(7905):343–348. doi: 10.1038/s41586-022-04533-3
- Bily TMI, Heinemann IU, O’Donoghue P. Missense suppressor tRNA therapeutics correct disease-causing alleles by misreading the genetic code. Mol Ther. 2024;32(2):273–274. doi: 10.1016/j.ymthe.2024.01.001
- Isaacson JR, Berg MD, Charles B, et al. A novel mistranslating tRNA model in drosophila melanogaster has diverse, sexually dimorphic effects. G3 (Bethesda). 2022;12(5):12. doi: 10.1093/g3journal/jkac035
- Lant JT, Berg MD, Sze DHW, et al. Visualizing tRNA-dependent mistranslation in human cells. RNA Biol. 2018;15(4–5):567–575. doi: 10.1080/15476286.2017.1379645
- Grosjean H, Westhof E. An integrated, structure- and energy-based view of the genetic code. Nucleic Acids Res. 2016;44(17):8020–8040. doi: 10.1093/nar/gkw608