
Abstract
Drug-induced hepatotoxicity constitutes a major reason for non-approval and post-marketing withdrawal of pharmaceuticals. In many cases, preclinical models lack predictive capacity for hepatic damage in humans. A vital concern is the integration of immune system effects in preclinical safety assessment. The immune-related Adverse Outcome Pathway (irAOP) approach, which is applied within the Immune Safety Avatar (imSAVAR) consortium, presents a novel method to understand and predict immune-mediated adverse events elicited by pharmaceuticals and thus targets this issue. It aims to dissect the molecular mechanisms involved and identify key players in drug-induced side effects. As irAOPs are still in their infancy, there is a need for a model irAOP to validate the suitability of this tool. For this purpose, we developed a hepatotoxicity-based model irAOP for recombinant human IL-2 (aldesleukin). Besides producing durable therapeutic responses against renal cell carcinoma and metastatic melanoma, the boosted immune activation upon IL-2 treatment elicits liver damage. The availability of extensive data regarding IL-2 allows both the generation of a comprehensive putative irAOP and to validate the predictability of the irAOP with clinical data. Moreover, IL-2, as one of the first cancer immunotherapeutics on the market, is a blueprint for various biological and novel treatment regimens that are under investigation today. This review provides a guideline for further irAOP-directed research in immune-mediated hepatotoxicity.
Drug-induced liver injury
Drug-induced liver injury (DILI) is the most important cause for liver failure. It is an adverse reaction to a variety of drugs and other agents such as herbal products and dietary supplements (Hoofnagle and Björnsson Citation2019). The most common drugs leading to DILI include antimicrobials such as amoxicillin-clavulanate, central nervous system drugs such as valproate, cardiovascular drugs such as statins, and immunomodulatory agents such as tumor necrosis factor (TNF) inhibitors (Francis and Navarro Citation2022).
Diagnosis of DILI is difficult due to the facets of this condition and relies mostly on the exclusion of other entities. Commonly, it is associated with an increase in liver enzymes in the blood (Hoofnagle and Björnsson Citation2019). Other clinical symptoms may include weakness, poor appetite, jaundice, icteric sclera, dark urine, nausea, abdominal discomfort, fever, itchy skin, and ascites (Lu et al. Citation2016). It is crucial to detect DILI early and to halt drug exposure in order to avoid progression toward chronic disease or acute liver failure (David and Hamilton Citation2010).
Various studies report the epidemiology of DILI. Variations in epidemiological findings are evident, influenced by factors such as study design and the geographical location in which the research was undertaken. Altogether, the prevalence of DILI is rare in the general population, but it increases in hospitalized patients. The epidemiology of DILI has been thoroughly examined in previous literature (Li et al. Citation2022). In the general population of Western countries, incidences for DILI range from 2.3 cases per 100,000 persons/year in Sweden (retrospective cohort study (Valle et al. Citation2006)) or 2.7 per 100,000 persons/year in the United States (prospective cohort study (Vega et al. Citation2017)), to 19.1 per 100,000 persons/year in Iceland (prospective cohort study (Björnsson et al. Citation2013)). In Eastern countries, China estimates a rate of DILI cases in the general population of 23.8 per 100,000 persons/year (retrospective cohort study (Shen et al. Citation2019)) and in South Korea, the incidence of DILI patients that require hospitalization was estimated to be 12 per 100,000 persons/year (prospective case-series study (Suk et al. Citation2012)). There are various confounding factors, which complicate direct comparisons of epidemiological data (Li et al. Citation2022).
DILI can be classified into different patterns of injury, mostly hepatocellular, cholestatic, and mixed hepatocellular-cholestatic. These patterns refer to histologic features of injury with different location and degree of inflammation, necrosis, and apoptosis (Kleiner Citation2014). Mechanisms of DILI come in various shapes: direct (intrinsic), idiosyncratic, and indirect. The direct form of DILI is associated with the intrinsic toxicity of drugs, which causes liver injury by directly damaging liver cells or interfering with biliary excretion (Lu et al. Citation2016). It presents the most frequent DILI subtype, is dose-dependent, and predictable in animals as it is induced by compounds with immanent hepatotoxic effects, such acetaminophen (Gerussi et al. Citation2021). In contrast, idiosyncratic DILI is caused by the action of the drug and not by the toxic properties of the drug itself. Possible mechanisms for idiosyncratic DILI involve the host immune response and genetic and metabolic factors of the patients, for instance defects in detoxification pathways. It is often accompanied by allergic and autoimmune reactions. Therefore, idiosyncratic DILI is less common than direct DILI, unpredictable, and neither dose-dependent nor detectable in animal models (Fontana Citation2014; Hoofnagle and Björnsson Citation2019). Indirect DILI presents a recently emerging type of drug-induced hepatotoxicity. It is caused by the action of the active substance and both, induction of liver injury and aggravation of already existing conditions, are mechanisms of indirect DILI (Hoofnagle and Björnsson Citation2019). Immune-mediated liver injury is a subclass of indirect DILI which is primarily induced by immunomodulatory agents, TNF antagonists, and checkpoint inhibitors (Hoofnagle and Björnsson Citation2019). Although the mechanisms remain to be elucidated, blocking the immune checkpoints in a tolerogenic organ such as the liver presumably leads to immune-mediated adverse events. Important mechanisms discussed are the decrease of self-tolerance and activation of cytotoxic T-lymphocytes (Shojaie et al. Citation2021).
High dose recombinant human interleukin = (IL)-2, also known as the pharmaceutical aldesleukin, is applied to promote the immune response in the context of renal cell cancer and malignant melanoma (Boyman et al. Citation2006). It was observed to induce mild-to-moderate alterations in liver enzymes in at least half of the patients. In certain patients, elevations exceeding 5-=10-times the upper limit of normal was measured, resulting in the need for early cessation or postponement of further therapy (Bethesda Citation2018). We hypothesize that the liver damage observed in aldesleukin treated patients might be classified as indirect, immune-mediated liver DILI. For a better understanding of the liver immune system, the hepatic immune landscape will be described next.
DILI is a major risk in drug development and the main cause for withdrawal of drugs from the market (Meunier and Larrey Citation2019). Due to the small number of cases, it is difficult to rule DILI out with certainty before a new drug is approved for the market and widely available (Babai et al. Citation2021).
The hepatic immune landscape
The liver is constantly exposed to intestine-derived bacterial products and food-derived antigens. Under homeostatic conditions, a complex hepatic immune landscape with different cell types contributes to hepatic tolerance and prevents aberrant immune activation. Central immune cells in the healthy liver are myeloid macrophages, dendritic cells and myeloid-derived suppressor cells as well as lymphoid natural killer (NK), NKT, and T-cells (Robinson et al. Citation2016). Tissue-resident macrophages, also known as Kupffer cells (KC) represent the largest myeloid population in the human body and are implicated in both liver tolerance induction and in the pathogenesis of liver diseases. Other cell types which modulate the hepatic immune response include liver sinusoidal endothelial cells (LSEC) (Crispe Citation2003; Tsuchida and Friedman Citation2017; Kolios et al. Citation2006). The various roles of the LSEC layer in antigen presentation and tolerance induction and its morphology allowing transmigration of cells from sinusoids to the liver parenchyma, have been extensively reviewed (Guidotti and Iannacone Citation2013; DeLeve and Maretti-Mira Citation2017; Kubes and Jenne Citation2018; Wilkinson et al. Citation2020). Prominent non-parenchymal cell types are hepatic stellate cells (HSC). Activated HSC are major drivers of fibrosis during liver injury, but may also counteract CD8+ T-cell activation (Schildberg et al. Citation2011; Tsuchida and Friedman Citation2017).
Immune cell subsets differ both within the liver and in relation to those in the peripheral blood. In the healthy human liver, a reversed CD4+/CD8+ T cell ratio compared to the peripheral blood can be found (in peripheral blood 2:1, in liver 1:3.5), while the NK cell fraction is significantly enhanced (12.8% in the peripheral blood, in the liver 30.6%). CD3+ lymphocytes are predominantly located close to the portal tracts, others being dispersed throughout the parenchyma (Norris et al. Citation1998; Tu et al. Citation2007). In homeostasis, interaction of lymphocytes with liver-resident cells confers tolerance (see above), as regulatory T (Treg)-cells are expanded and autoreactive CD8+ T-cells are inactivated or deleted. Under inflammatory conditions, both T-cell subtypes can be activated and contribute to both, immunity to fight infections and liver damage. There is little interaction between tissue resident NK cells and the liver non-parenchymal cells in homeostasis, but during chronic hepatic inflammation, resident NK cells release immunomodulatory cytokines, thus becoming liver-protective (Heymann and Tacke Citation2016).
The intrahepatic lymphocyte compartment is generally in a more activated state than the peripheral blood. For instance, intrahepatic lymphocytes show increased expression of activation markers such as the immune checkpoint CD27, the co-stimulatory molecule CD28, and the early activation antigen CD69. Further, intrahepatic CD4+ and CD8+ cells show elevated CD95 (Fas receptor) expression (Tu et al. Citation2007). The chemokine receptor CXCR6 is expressed on activated intrahepatic lymphocytes and its ligand, CXCL16, is constitutively expressed on LSECs (Heesch et al. Citation2014). In vitro IL-12/IL-18 stimulated CXCR6+ liver resident NK and T cells show enhanced expression of IFNγ and LAMP-1 (CD107a), a marker for cytotoxic T- and NK cell degranulation, compared to their CXCR6- counterparts (Zhao, et al. Citation2020a). Reflecting a potential role of CXCR6+ liver resident NK and T-cells in Hepatitis B virus (HBV) associated liver disease, these cell types occur with increased frequency in HBV-infected livers. Moreover, single cell RNA sequencing identified distinct liver resident CD14+ monocyte, CD16+ monocyte and macrophage populations. These tissue-resident cell types may aid the maintenance of immune tolerance as gene set enrichment analysis showed downregulation of pro-inflammatory responses, the complement pathway, as well as IL-2-JAK-STAT5, IL-6-JAK-STAT3 and p53 signaling pathways (Zhao et al. Citation2020a). Intrahepatic lymphocytes play an important role in various liver diseases (Wang and Zhang Citation2019), as discussed below. In summary, the hepatic immune cell landscape differs at least partially from the peripheral blood immune cell compartment, especially in T- and NK cell composition, activation status, and surface marker expression.
IL-2 as a cancer immunotherapeutic
IL-2 is a human cytokine produced by macrophages and lymphocytes that induces the proliferation and maturation of T-cells. A recombinant form of IL-2 (aldesleukin) has been shown to be active against renal cell cancer and malignant melanoma (Bethesda Citation2018). The high dose IL-2 regimen for the treatment of cancer is usually 600,000 IU/kg, administered intravenously every 8 h for up to 14 doses over 5 d (Fisher et al. Citation1989; Atkins et al. Citation1999). Upon completion of infusion, approximately 30% of injected aldesleukin is detectable in the plasma. Common side effects include fatigue, fever, chills, nausea, diarrhea, and capillary leak syndrome leading to edema, hypotension, renal insufficiency, and pulmonary edema. Less common side effects include shock, anaphylaxis, severe infections, autoimmune conditions, and neurologic complications including somnolence, stupor, and coma (Bethesda Citation2018). In clinical trials it was also found to increase liver enzymes in serum and treatment might lead to cholestasis in certain patients (Bethesda Citation2018). Most of the adverse effects are reversible and manageable today (Atkins et al. Citation1999; Dutcher et al. Citation2014).
IL-2-mediated hepatotoxicity
Various clinical studies report adverse effects of high dose IL-2 on the liver during treatment of renal cell carcinoma or malignant melanoma. For renal cell carcinoma patients, hepatic dysfunctions have been reported in 11% of patients treated with high dose aldesleukin. These effects were not dose-limiting but reversible upon completion of the treatment (Fyfe et al. Citation1995). Grade 3 and 4 liver toxicities were similarity observed in a clinical study comparing high dose IL-2 treatment with IL-2/IFNγ combination treatment in patients suffering from metastatic renal cell carcinoma (McDermott et al. Citation2005). IL-2 therapy is linked to impaired liver function, manifesting in jaundice, right upper quadrant tenderness, hepatomegaly, nausea, anorexia, and pruritus (Fisher et al. Citation1989; Dutcher et al. Citation2001). The most common hepatic function abnormalities comprise hyperbilirubinemia with bilirubin levels rising to 10 mg/dl, and low albumin with levels dropping to 2 g/dl. Five- to ten-fold increases in hepatic enzyme levels have been reported and aldesleukin therapy is discontinued in some centers if such elevations are observed (Dutcher et al. Citation2014). Hepatobiliary scans of patients with different tumor entities treated with high dose IL-2 showed delayed liver clearance as determined by biliary excretion, quantitative analyses also pointing toward delayed uptake. Interestingly, these effects were not restricted to patients with a specific bilirubin level threshold. Ultrasound detected an increased distance of hepatobiliary and vascular structures from the parenchyma, suggesting hepatic edema or infiltrative processes (Fisher et al. Citation1989). For the management of high dose IL-2 related toxicities, therapy can be interrupted when bilirubin or hepatic transaminases are significantly augmented, albumin is dramatically decreased, and prothrombin time is prolonged. Upon cessation of high-dose IL-2 therapy, liver damage reverses as liver function parameters normally return to baseline within 5 to 6 days (Schwartz et al. Citation2002). In clinical trials, low-dose IL-2 treatment was well tolerated (Mahmoudpour et al. Citation2019) and no adverse effects on liver function were indicated (Todd et al. Citation2016).
The initially proposed mechanism for IL-2-mediated hepatotoxicity was activation of tissue-resident macrophages, which produce TNFα and trigger activation of the local endothelium leading to recruitment of peripheral immune cells that mediate liver damage (Nakagawa et al. Citation1996). This hypothesis has been extended to include various other IL-2 receptor (IL-2R) expressing cell types, as well as novel mechanisms induced by IL-2, as described below. In a literature-based approach, we identified key players and biorelevant molecules for IL-2-mediated hepatotoxicity. For this, we employed the Adverse Outcome Pathway (AOP) concept.
Immune-related Adverse Outcome Pathways
The AOP concept provides a framework for the collection, evaluation and integration of available data to understand the adverse events induced by specific mechanisms of action (Ankley et al. Citation2010). Extended to immunological processes, the immune-related AOP (irAOP) concept presents an overall AOP to describe immune-mediated adverse effects (Villeneuve et al. Citation2018). AOPs are anchored between two Key Events (KE), which are the Molecular Initiating Event (MIE) and the Adverse Outcome (AO). These are connected by a stringent order of KE depicting unique and measurable states of a system, linked to each other by KE relationships (KER). Starting with the MIE at the molecular level, the connecting KE ascend through cellular to organ levels, with the AO becoming apparent in vivo on the organ or organism level (Knapen et al. Citation2015). This approach enables, e.g., the identification of key molecules in biologically-relevant immune processes in the diagnosis or prognosis of hepatotoxicity and aid development of specific in vitro tests (Vinken Citation2020). Moreover, AOP can identify knowledge gaps to be filled with experimental evidence, rendering an AOP a living document (OECD Series on Adverse Outcome Pathways Citation2018). In a recent article, Ball et al. (Citation2021) illustrate how AOP, knowledge drawn from assays, and structure activity relationship predictions can be combined to select predictive assays for compound toxicities and to link assay outcomes to AO (Ball et al. Citation2021).
In the past, the AOP concept was mainly used for answering toxicological questions of chemicals. For some time now, however, the AOP concept has been adapted to biomedical research questions and shown to have great value for biological research and drug discovery (Bal-Price and Meek Citation2017). Ultimately, the concept can help to understand the pathomechanisms of human diseases. This information can subsequently be used to develop novel and safer drugs. In this review, we aim to provide a prototype for further evaluation of immune-mediated liver damage evoked by novel IL-2-based therapies or of biological investigative approaches with a similar mode of action. For the generation of such a potential irAOP, published AOP may serve as template. There are already several AOP leading to liver injury published in the open-source AOP Wiki, and some of them account for immunotoxicity mechanisms. These include, for example, AOPs describing chemically induced immune-mediated hepatitis (AOP 362), immune-mediated liver fibrosis (AOP 38), AOP for liver cholestasis (e.g. AOP 27), inflammation (e.g. AOP 144), and cancer (e.g. AOP 220) (Gijbels and Vinken Citation2017).
In this review, we generate a putative irAOP for IL-2 induced DILI based on relevant literature data, to provide an overview of possible mechanisms, key players, and knowledge gaps in IL-2-mediated hepatotoxicity.
Overview of the model irAOP for IL-2-mediated hepatotoxicity
Based on literature review, we developed an irAOP framework for IL-2-mediated hepatotoxicity. For this, we collected both direct evidence for IL-2-mediated hepatotoxicity and for general mechanisms for immune-mediated injury.
High dose aldesleukin treatment supplies the circulation with a surplus of IL-2. Therefore, aldesleukin is not entirely consumed by Treg cells, but presumably also bound by cells with a lower affinity toward IL-2 such as cytotoxic T- and NK cells, that are subsequently activated, which is the principle of IL-2 therapy (Abbas et al. Citation2018). Aldesleukin is also distributed to the liver (Proleukin® Product Monograph Citation2018), where it may act on local IL-2R expressing cells. We hypothesize that this binding of IL-2 to liver resident cells depicts the starting point (MIE) of the model irAOP. We suggest that due to the availability of high IL-2 concentrations, the tolerogenic environment of the liver is overcome and cytotoxic cells are activated to create a pro-inflammatory environment (KE1) which leads subsequently to recruitment/infiltration of (activated) immune cells to the liver (KE2). Further, we theorize that the exacerbated inflammation then leads to damage of the liver (KE3), and finally to immune-mediated hepatitis with impaired liver function (AO, ). Within our irAOP model, we identified biorelevant molecules and mechanisms, which are listed in . More, several knowledge gaps were identified, which are highlighted in the appropriate section and summarized in with a selection of models that could be used to fill the gaps and render the picture of the model irAOP more complete.
Figure 1. Illustrated potential immune-related Adverse Outcome Pathway for IL-2-mediated hepatotoxicity. Binding of IL-2 to IL-2R expressing cells initiates the AOP (MIE). IL-2 binding induces signaling and proliferation (a.), receptor upregulation (b.), cytokine expression (c.) or interaction with other immune cells (d.; KE1). Consequently, activated immune cells are recruited to the liver sinusoids and adhere to LSECs and/or transmigrate into the perisinusoidal space (KE2). Liver damage occurs by direct or indirect cytotoxic effects or interaction of recruited immune cells with hepatocytes or by impairing the blood flow in the sinusoid lumen (KE3). Finally, impaired liver function or killing of hepatocytes triggers the AO hepatotoxicity (not depicted). Black arrows mark interactions, dashed lines indicate possible interaction points which lack of literature evidence.
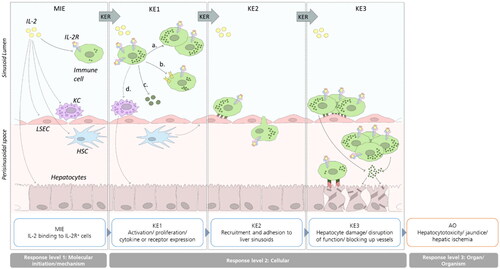
Table 1. Biorelevant molecules and mechanisms related to IL-2 according to the irAOP.
Table 2. Knowledge gaps identified from the irAOP and suggested experimental approaches to fill the gaps.
Human genetic polymorphisms and murine knock out experiments provide valuable information about the impact of IL-2 on immune-mediated disease and can aid to identify key players and mechanisms in IL-2-mediated hepatotoxicity. In a patient with IL-2Rα deficiency due to a frameshift mutation, lymphocytic infiltrates in various tissues, inter alia in the liver, were observed and the authors suspect this autoimmunity arises from the lack of self-tolerance (Roifman Citation2000). Treg cell targeted IL-2Rα conditional knockout mice lack their regulatory function and thus, autoimmune features are observed, such as lymphocytic organ infiltration and liver inflammation (Toomer et al. Citation2019). Clear evidence for the importance of IL-2Rα on immune-mediated hepatotoxicity may be the occurrence of primary biliary cirrhosis, an autoimmune liver disease in which the host immune system attacks biliary cells, in a 5-year-old with IL-2Rα deficiency. Liver biopsy assessment showed lymphocyte infiltrates in the liver and no signs of infection. The authors speculate the disease to be promoted by autoreactive T-cells via two mechanisms: the up-regulation of the anti-apoptotic protein B-cell lymphoma 2 (bcl-2) during T-cell development and the lack of Treg cells which normally suppress autoreactive T cells (Aoki et al. Citation2006). Studies with IL-2Rα deficient mice were applied to gain a closer insight into primary biliary cirrhosis. The livers of the animals showed CD4+ T- and predominantly CD8+ T-cell infiltrates, while NK and NKT cells were decreased compared to control animals. Regarding cytokines, pro-inflammatory TH1 cytokines (TNFα, IL-2, IL-12p40, IFNγ) and IL-6 were increased in the serum (Wakabayashi et al. Citation2006). Regarding the IL-2Rβ chain, an IL-2RB missense mutation (L77P) hinders the egress of IL-2Rβ from the endoplasmic reticulum and consequently, humans with this mutation show autoimmune disease also affecting the liver. Patients with this mutation lack IL-2Rβ surface expression on T-cells while they retain moderate IL-2Rβ expression on NK cells. The CD56bright NK cell compartment is enhanced and acts cytotoxic upon IL-2 stimulation. As patients lack high-affinity IL-2R expressing T-cells (Treg cells), this loss of tolerance with concomitant NK cell activation may lead to the observed dysregulations of the immune system (Zhang et al. Citation2019). This renders NK cells and especially CD56bright NK cells an interesting cell type to address in the context of aldesleukin-mediated hepatotoxicity. Deficiency in IL-2Rγ leads to X-linked severe combined immunodeficiency and about 200 different mutations have been characterized so far, leading to different severities of the disease (Lim et al. Citation2019; Belaid et al. Citation2021).
These publications highlight the importance of a functional Treg cell compartment to counteract self-targeted immune cells. Simultaneously, a concomitant downmodulation of regulatory function (immune tolerance) with consistent or even promoted effector cell (T-cell and/or NK cell) function might favor immune-mediated diseases such as immune-mediated hepatotoxicity. This is well exemplified by depletion or functional inhibition of Treg cells in mice with a human immune system: Low dose IL-2 treatment of these animals caused weight loss and pulmonary edema, which are toxicities seen in high-dose IL-2-treated mice with functional Treg cells (Li et al. Citation2017).
IL-2 binding (MIE)
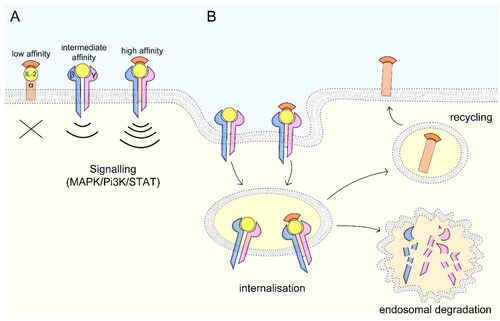
IL-2 binds to the IL-2R. Other binding modalities are unknown. The IL-2R is generally expressed on various cell types such as lymphocytes, neutrophils, and monocytes (David et al. Citation1998; Malek Citation2008; Boyman and Sprent Citation2012) (). The IL-2R constitutes one to three subunits that are differentially expressed and exhibit different IL-2 binding affinities (Stauber et al. Citation2006). The low-affinity receptor consists of IL-2Rα (CD25; Kd ∼10−8 M) only and does not generate a signal due to lack of intracellular signaling domains. Both membrane-bound and soluble IL-2Rα (sCD25) can act as decoy receptors. The intermediate-affinity receptor is composed of IL-2Rβ (CD122) and -γ (CD132, Kd ∼10−9 M) and the high-affinity receptor of IL-2Rα, -β, and -γ (Kd ∼10−11 M). Both intermediate- and high-affinity IL-2Rs have intracellular signaling domains, therefore enabling signaling capacity (). IL-2 binding to the intermediate-affinity receptor induces IL-2Rα production and subsequent formation of the high-affinity receptor (Baume et al. Citation1992; Grant et al. Citation1992; Spolski et al. Citation2018). Upon binding, the IL-2-IL-2R complex is internalized and the IL-2Rβ and -γ subunits are degraded in endosomes while the -α subunit is recycled to the cell surface (Hémar et al. Citation1995) ().
Figure 2. IL-2 receptor signaling and mechanisms of internalization. (A) The low-affinity receptor consists of IL-2Rα, the intermediate-affinity receptor consists of IL-2Rβ and -γ and the high-affinity receptor of all three subunits. While the low-affinity receptor lacking intracellular signaling domains can act as decoy receptor, the intermediate- and high-affinity receptors signal via MAPK, Pi3K and STAT pathways. (B) Upon IL-2 binding, the ligand-receptor complex is internalized and the IL-2Rβ and –γ subunits are targeted for endosomal degradation, while the –α subunit is recycled to the cell surface. Figure adapted after Spolski et al. (Citation2018).
As described, important immune cells in the healthy liver are myeloid macrophages, lymphoid NK, NKT, and T-cells (Robinson et al. Citation2016) and for neutrophils, a role in cancer immunotherapy is attributed (Siwicki et al. Citation2021). Therefore, we give an overview of these cell types in the following.
Murine KCs express all three IL-2R subunits on a protein level and respond to IL-2 treatment in vitro (Simone et al. Citation2021). In human, neutrophils, monocytes, and differentiated macrophages express the intermediate-affinity IL-2R, while low-affinity IL-2Rα is absent (Scheibenbogen et al. Citation1992; Djeu et al. Citation1993; Girard et al. Citation1995; Abdel-Salam and Ebaid Citation2014). Notably, IFNγ is a more potent inducer of the IL-2Rγ chain than IL-2 in human monocytes (Bosco et al. Citation1994b).
Peripheral blood-derived human NK cells express the intermediate-affinity IL-2R, but only a small fraction expresses the α-chain. Expression of the high-affinity receptor occurs mostly in CD56bright NK cells (Baume et al. Citation1992; David et al. Citation1998; Rudnicka et al. Citation2015) (). Upon lipopolysaccharide (LPS) activation, IL-2Rα is upregulated on CD56+ NK cells (Rudnicka et al. Citation2015).
NKT cells express both NK cell markers and T-cell receptor-αβ chains representing a bridge between innate and adaptive immunity. During liver inflammation, Type I and Type II NKT cells confer protective and damaging roles, respectively (Bandyopadhyay et al. Citation2016). Human peripheral blood NKT cells express surface IL-2Rβ more on the mature CD4- than on the CD4+ subpopulation (Baev et al. Citation2004). In contrast, on in vitro expanded human peripheral blood-derived NKT cells, IL-2Rα expression was observed in a greater fraction of CD4+ than of CD4-CD8- double negative NKT cells. Analogous to CD4+CD25+ Treg cells, a regulatory function for CD4+ NKT cells is suggested, as they express FoxP3 and inhibit antigen-specific CD8+ T-cells (Osada et al. Citation2005). Surface expression of IL-2Rα in peripheral blood NKT cells was confirmed, but NKT cell subtypes were not assessed (López et al. Citation2018). On resting murine NKT cells, all three subunits of the IL-2R were detectable (Jukes et al. Citation2012) (). These findings indicate that, as IL-2Rα is expressed at least on several NKT cell subtypes, this cell type might present a primary target of IL-2 and be implicated in the fine-tuning of IL-2 response by immune-activating and -inhibiting actions.
In human peripheral blood-derived T-cells, the intermediate-affinity IL-2R is more distinctly expressed on resting CD8+ than on CD4+ T-cells (David et al. Citation1998). The level of IL-2Rα expression influences the differentiation of CD8+ T-cells. While IL-2Rαlow CD8+ T-cells differentiate to memory-phenotype cells, IL-2Rαhigh cells give rise to effector T-cells (Spolski et al. Citation2018). Further, murine memory-phenotype CD8+ T-cells require IL-2 signal for re-activation (Blachère et al. Citation2006). Taken together, IL-2R expression plays a critical role in CD8+ T-cell function, development, and reactivation and due to differing expression of the IL-2R subunits, potentially diverging responsiveness for aldesleukin treatment exist.
Treg cells control T-cell-mediated immune responses and have important functions in preventing autoimmunity. They act by expressing anti-inflammatory cytokines such as IL-10 and IL-35, via cytolysis and by modulating dendritic cell responses (Vignali et al. Citation2008). Treg cells are characterized as high-affinity IL-2R expressing cells (Malek Citation2008). Treg cells expressing the trimeric IL-2R have the capability to consume local IL-2, thereby exerting a suppressive function termed metabolic disruption which involves the deprivation of the surrounding effector cells of IL-2 (Vignali et al. Citation2008). In contrast, in vitro, sCD25 containing supernatant does not interfere with the proliferation of CD4+CD25- T-cells or deplete IL-2. The presence of Treg cells may therefore be necessary to mediate effector T-cell suppression (Pedersen and Lauritsen Citation2009). Such a synergistic effect between Treg cells and sCD25, especially at supraphysiologic concentrations of this molecule, is supported in patients with hepatocellular carcinoma (Cabrera et al. Citation2010a). In these patients, mean serum levels of sCD25 are increased 5-7-fold compared to healthy controls and high sCD25 levels are associated with low CD4+ T-cell reactivity as shown in ex vivo assays. These findings support a possible role for this soluble receptor in immune-inhibition (Cabrera et al. Citation2010b). In patients with chronic liver disease, sCD25 concentrations positively correlate with pro-inflammatory cytokine/chemokine expression as well as disease progression, thereby qualifying as a potential marker for immune cell activation in this condition (Seidler et al. Citation2012). Clearly, it would be helpful to understand the role of sCD25 in IL-2-induced hepatotoxicity, which seems not yet to have been addressed ().
In-depth knowledge about the IL-2 receptor expression in the human liver cell subtypes would be crucial to understand which cells possibly respond directly to high dose aldesleukin treatment. Transcriptomic analysis of human liver specimens showed all three subunits of IL-2R to be expressed (Fagerberg et al. Citation2013). However, no cell type specific values were indicated (). In murine KC, all three IL-2R subunits could be detected in flow cytometry analyses (Simone et al. Citation2021).
Activation of cells by IL-2 (KE1) and release of pro-inflammatory cytokines
Activation of Kupffer cells
In many cases, data about the IL-2 response of hepatic immune cells are missing. The liver contains a high density of myeloid cell types in homeostasis. Among them, KC as liver resident macrophages constitute the largest population of hepatic immune cells. High concentrations of IL-2 prime and trigger KCs to suppress protein synthesis (Curran et al. Citation1988). Murine KC can be activated by IL-2 to cross-present hepatic antigens to T-cells, thus promoting an immune response (Simone et al. Citation2021).
Activation of monocytes and neutrophils
Human monocytes are activated by IL-2, which leads to enhanced cytotoxicity (Espinoza-Delgado et al. Citation1990) as well as cytokine, growth factor, and growth factor receptor expression as previously reviewed (Espinoza‐Delgado et al. Citation1995). Furthermore, IL-2 induces upregulation of IL-2Rβ and -γ, but not of -α mRNA in these cells (Espinoza‐Delgado et al. Citation1992; Bosco et al. Citation1994a), indicating the promotion of intermediate-affinity IL-2R expression. Protein tyrosine kinase activation plays a central role in IL-2 signaling, mediating effector and secretory functions (Bosco et al. Citation2000). Human peripheral blood monocytes express the pro-inflammatory cytokines IL-1β, −6, −8, and TNFα after IL-2 stimulation (Bosco et al. Citation2000). In human neutrophils, IL-2 stimulation induces TNFα mRNA (Wei et al. Citation1993).
Activation of NK cells
In primary human NK cells, IL-2 signaling is well documented. Upon IL-2 binding, high-affinity IL-2R expression and STAT1, −3, −4, and −5 phosphorylation are induced (Wang et al. Citation1999). Generally, IL-2 stimulation confers protection from apoptosis mediated by upregulation of the anti-apoptotic protein bcl-2 (Armant et al. Citation1995). IL-2 leads to an increased secretory and cytotoxic response in human NK cells by directly acting on intracellular signaling (Bonnema et al. Citation1994) which manifests in IFN-γ and TNF-α production, while IL-1β protein is not upregulated (Young and Ortaldo Citation1987; Numerof et al. Citation1990). In human NK cells, IL-2 favors proliferation of the immature CD56bright compared to the cytotoxic CD56dim population (Baume et al. Citation1992). IL-2 up-regulates the expression of the activating receptor killer cell immunoglobulin-like receptor 2DL4 (KIR2DL4) on CD56high cells (Kikuchi-Maki et al. Citation2003). These findings suggest a high responsiveness of this NK cell sub-population toward IL-2. This may have relevance for hepatic toxicities after IL-2 treatment as in humans, immature NK cells constitute nearly 50% of intrahepatic NK cells (Hudspeth et al. Citation2016). Moreover, the presence of certain NK cell subpopulations might be predictive for liver injury: Comparison of NK cell subpopulation occurrence from healthy versus diseased liver and found a positive correlation between integrin α-1/IL-2Rα expressing (CD49a+CD25+) NK cells and alanine aminotransferase levels, indicating liver damage. In vitro, the CD49a+CD25+ NK cell population proliferates in response to low dose IL-2, indicating a lower activation threshold than in peripheral blood-derived NK cells (Martrus et al. Citation2017). Elucidation of IL-2 signaling threshold in hepatic NK cells would provide more information on the main IL-2 responders in the liver ().
Activation of T lymphocytes
IL-2 signaling in T-cells has been extensively reviewed (Benczik and Gaffen Citation2004; Beyer et al. Citation2011; Liao et al. Citation2011a, Citation2011b; Ross and Cantrell Citation2018). Rollings et al. (Citation2018) compared the proteome of murine effector CD8+ T-cells maintained in IL-2 with samples deprived of IL-2. The authors state that an IL-2 dependent increase in effector molecules goes hand in hand with an upregulation of proteins that control these molecules, thereby regulating T-cell activity. Based on proteomic data gained on cytotoxic lymphocytes, IL-2 mediates cell cycle progression and proliferation. IL-2 sustains IL-2Rα expression in T-cell receptor activated effector CD8+ T-cells and IL-2 deprivation leads to downregulation of expression of all three IL-2R subunits. The authors highlight the fine-tuning of IL-2 responses and the critical role of IL-2 signaling not only for the maintenance of T-cell responses in an activated state but also in mediating responsiveness of quiescent T-cells and cell cycle progression (Rollings et al. Citation2018). With regard to T-cell activation, IL-2 reduces the T-cell receptor signaling threshold in CD8+ T- but not in CD4+ T-cells. The decreased T-cell receptor threshold in CD8+ T-cells may be enabled by increased expression of the cell cycle regulator c-Myc, possibly transforming sub-optimal T-cell receptor stimulation to an immune response to antigen (Au-Yeung et al. Citation2017). This increased response of CD8+ T-cells toward IL-2 may be crucial for the induction of cytotoxicity in the liver by this cell type.
Secondary antigen encounter of CD8+ T-cells primed in the absence of CD4+ TH cells leads to CD8+ T-cell death. Addition of IL-2 rescues CD8+ T-cells by modulating the apoptosis relevant genes bcl-2, Flip, Trail, and DR-5, thereby downregulating the sensitivity to DR-5-mediated death. Moreover, IL-2 restores effector functions and enables secondary expansion. Therefore, IL-2 enables full effector function of CD8+ T-cells in the absence of CD4+ help (Wolkers et al. Citation2011). In a murine model in which T-cell priming against HBV is restricted to the liver, primed CD8+ T-cells led to dysfunctional cells without cytotoxic potential. In vivo administration of an IL-2/IL-2-antibody complex rescued the defective CD8+ T-cells by restoring gene expression levels, measured in wild type control mice. This gene regulation was only observed for antigen specific cells. In human CD8+ T-cells isolated from patients with chronic HBV infection, T-cells from immune tolerant patients but not T-cells from immune active patients, showed restored cytotoxic functions when expanded after IL-2 incubation, suggesting a similar mechanism to that in the mouse (Bénéchet et al. Citation2019). Thus, in the setting of HBV infection, exogenous IL-2 can shift the T-cell response from immune tolerant to immune active. The question remains whether such a mechanism might be implicated in IL-2-mediated hepatotoxicity in cancer patients in whom immune tolerance also arises (). Overall, the literature points toward a pro-immunogenic role of IL-2 by increasing not only proliferation, but also providing anti-apoptotic cues to helpless T-cells and up-regulating cytotoxic potential by decreasing T-cell receptor stimulation threshold and shifting the immune status from tolerant to active.
The balance of TH cell subsets (TH1 versus TH2) is attributed a role in autoimmune hepatitis (Behfarjam et al. Citation2017). IL-2 has a multitude of roles in regulating differentiation of TH1, TH2, and TH17 cells. IL-2 induces IL-12Rβ2, enhances responsiveness to IL-12, thereby promoting TH1 cell differentiation, but down-regulates IL-6Rα and gp130, resulting in decreased IL-6 responsiveness and inhibition of TH17 cells in primary murine cells. IL-2 regulates TH2 cell differentiation in a STAT5-dependent manner, acting early at the Il4ra locus to induce IL-4Rα expression (Liao et al. Citation2008; Citation2011b). In primary murine CD4+ T-cells, IL-2 helps maintain transcriptional flexibility of gene expression in T-follicular helper cells (Oestreich et al. Citation2012). Therefore, it would be interesting to assess the role of exogenous IL-2 in TH cell response or in polarization of local cell populations ().
IL-2 is central to Treg cell homeostasis by maintaining lineage-specific FoxP3 expression (Zorn et al. Citation2006). IL-2R signals via STAT5 in Treg cells. In vitro analysis of Treg cell function with regard to suppression of effector T-cell proliferation reveals that low dose IL-2 enhances while high dose IL-2 abrogates suppressive capacity of Treg cells, suggesting a concentration window for optimal Treg cell stimulation (Moon et al. Citation2015). In a murine model of biliary fibrosis, depletion of intrahepatic Treg cells increased intrahepatic CD8+ T- cells and aggravated fibrosis, and administration of low dose IL-2/anti-IL-2-antibody complexes led to Treg cell expansion and suppressed proliferation of CD8+ T-cells, ameliorating hepatobiliary injury (Taylor et al. Citation2018). Whether there is an IL-2 concentration limit in the liver above which tolerance induction is overcome, remains unclear ().
In NKT cells isolated from PBMC, IL-2 signals via STAT3, −4, −5, and −6 as well as p38 mitogen-activated protein kinase (MAPK) pathway. IL-2 induces NKT proliferation and expression of IFNγ and IL-4, as well as granulocyte-macrophage colony-stimulating factor (GM-CSF) and perforin (Hou et al. Citation2003; Bessoles et al. Citation2008). In NKT cells isolated from murine spleen and liver, anergic NKT cells downregulate IL-2Rα expression. Exogenous IL-2 rescues proliferative capacity and reconstitutes expression of IL-4 but not IFNγ (Iyoda et al. Citation2010). This indicates that exogenous IL-2 is implicated in the fine-tuning of NKT cell response, which might be a critical point for this irAOP.
Hepatic cells. Binding of IL-2 to IL-2R+ cells induces IL-2R signaling (KE1). Such IL-2 binding potentially occurs directly in the liver as well, as indicated by rapid uptake of radiolabelled IL-2 in rats (Proleukin® Product Monograph Citation2018). Although no specific literature on the effect of IL-2 on LSEC could be identified, information on other endothelial cell types is available providing potential hints for the situation in the liver. Assessment of human and bovine endothelial cell layers indicates enhanced permeability upon IL-2 incubation and up-regulation of IL-2Rα expression (Downie et al. Citation1992). The human brain microvascular endothelial cell line hCMEC/D3 expresses IL-2Rβ and -γ subunits (mRNA and protein) and -α is inducible upon IL-2 stimulation, leading to expression of pro-inflammatory IL-6 and Monocyte Chemoattractant Protein-1 (MCP-1) (Wylezinski and Hawiger Citation2016). As LSECs are specialized endothelial cells with distinct functions and properties, similar experiments to those on hCMEC/D3 would be crucial for understanding IL-2 effects in the liver ().
Inflammatory processes can suppress cytochrome P450 (CYP) enzymes, thereby interfering with drug metabolism and can depict an additional readout for liver damage (de Jong et al. Citation2020). In freshly isolated rat hepatocytes, IL-2 stimulation downregulates CYP2C11 and CYP3A2 mRNA (Tinel et al. Citation1995). Non-parenchymal cell fractions such as KC were not addressed in these studies. However, KC are presumably implicated in IL-2-mediated CYP downregulation, as shown in primary human hepatocyte/KC co-cultures (Sunman et al. Citation2004). Curran et al. (Citation1988) showed that treatment with IL-2 enhances KC response to LPS but is also sufficient to activate KCs at doses > 1 x 104 U/mL (Curran et al. Citation1988). In high dose IL-2 treated mice, increased activity of phagocytes was measured after 2 h, and these cells were identified to be KC (Nakagawa et al. Citation1996).
Lipid-lipid interactions laterally organize plasma membranes of cells into microdomains called lipid rafts which can influence membrane-bound receptor signaling by retaining or freeing them from lipid raft domains (reviewed in Varshney et al. (Citation2016) and Levental et al. (Citation2020)). In the murine T cell line CTLL-2, IL-2Rα locates to lipid rafts while IL-2Rβ and –γ are enriched in the soluble phase. The trimeric IL-2R locates to the soluble phase, indicating that IL-2R signaling occurs in the soluble membrane regions (Marmor and Julius Citation2001). In contrast, in PHA-activated primary human T-cells, the IL-2Rβ predominantly locates to lipid rafts while the –α and –γ subunit are enriched in the plasma membrane. Upon IL-2 stimulation, the IL-2Rβ translocates to the plasma membrane fraction, indicating that the formation of the trimeric receptor is induced (Goebel et al. Citation2002). These findings indicate that lipid rafts play a role in IL-2R signaling and, potentially, patient response to therapeutic IL-2. Additionally, the differing results obtained by Marmor and Julius (Citation2001) and Goebel et al. (Citation2002) emphasize that the correct cell source should be chosen to establish novel in vitro models translatable to the human setting.
In summary, there are rich data for IL-2-mediated immune cell activation in the periphery and there is information about KC response to IL-2. For a better understanding of the processes in the liver, addressing hepatic immune cells specifically would be crucial (). We presume that immune cells recruited from the periphery might play a role in IL-2-mediated hepatotoxicity. Therefore, in the next section mechanisms for the recruitment of peripheral immune cells to the liver are addressed.
Recruitment of immune cells to the liver (KE2)
We speculate that IL-2 promotes the infiltration of leukocytes into the liver by locally upregulating chemokines and adhesion molecules (). In mice, IL-2 treatment induced significant leukocyte infiltration (Lentsch et al. Citation1997).
In humans, IL-2 signaling and adhesion processes are presumably closely connected. In the human T-lymphoma cell line HuT-102-B2 that constitutively expresses the high-affinity IL-2R, ICAM-1 and IL-2R are situated in close proximity (Burton et al. Citation1990) and in super antigen activated CD4+ T cells, ICAM-1 co-stimulation induces IL-2 expression (Labuda et al. Citation1998).
The liver presents unique mechanisms of leukocyte adhesion because selectin-dependent rolling does not precede adhesion in the sinusoids but plays a role in post-sinusoidal venules. In contrast, concomitant P-selectin and ICAM-1 deficiency in mice abolishes lymphocyte adhesion to the liver sinusoids. This not only highlights the differences from other endothelial cell types, but also argues against the hypothesis that trapping of lymphocytes in the sinusoids solely follows sheer physical trapping (Wong et al. Citation1997).
Immune cell recruitment to the liver has been addressed in various murine settings. Murine in vivo studies showed that under non-inflamed conditions primarily NK, NKT, and memory T-cells migrate to the liver, mirroring populations identified as intrahepatic lymphocytes under homeostatic conditions (Klugewitz et al. Citation2004). Upon intraperitoneal IL-2 injection in mice, leukocytes, neutrophils, and platelet adhesion occur to liver venules besides leucocytosis and increased neutrophil counts in the circulation. Moreover, in IL-2 treated mice, VCAM-1 and ICAM-1 were upregulated and their binding partners, LFA-1 and VLA-4, on lymphocytes increased (Anderson et al. Citation1996; Nakagawa et al. Citation1996; Lentsch et al. Citation1997). In vivo immunotoxic effects of high dose IL-2 were evaluated histologically in humanized mice and a massive leukocyte infiltration in the liver was found, as well as systemic immune cell activation (Li et al. Citation2017; Yong et al. Citation2020). CD4+ and CD8+ T-cells might also be important in IL-2-mediated toxicities, as depletion of these cell types increases survival (Li et al. Citation2017). Translation of these effects to humans and dissection of the implicated immune cell types would enhance understanding of the hepatotoxic mechanisms of IL-2 ().
In liver sinusoids of mice treated with IL-2, adherent lymphocytes, platelets, and neutrophils can be found, which presumably leads to impaired blood flow in the liver (Nakagawa et al. Citation1996). Generally, the infiltration of neutrophils is associated with liver inflammation (Heymann and Tacke Citation2016). In the literature, L-selectin and CD11b (integrin α-M)/CD18 (integrin β-2) expression on neutrophils are attributed to an important role in the adhesion and migration of the cells across endothelial cell layers. Human neutrophils upregulate CD11b and CD18 expression upon incubation with IL-2 and adhere to human umbilical vein endothelial cells (HUVECs) (Li et al. Citation1996; Abdel-Salam and Ebaid Citation2014). Interestingly, neutrophils isolated from patients after the first and second course of IL-2 therapy showed normal random migration but impaired chemotaxis toward the chemotactic peptide FMLP or zymosan-activated serum. One week after completion of the IL-2 treatment, values reverted to normal. Superoxide production, phagocytosis, bactericidal activity and granule secretion were not influenced (Klempner et al. Citation1990). These results point toward a potential role of neutrophils in vascular injury (including liver), supporting the findings in the murine setting, but there is a knowledge gap because experiments with LSEC are missing ().
In IL-2-activated human PBMC, the NK cell fraction adheres to endothelial cells. This effect is directly attributed to IL-2, as blocking of the pro-inflammatory secondary cytokines TNFα, IL-1α or -β by antisera does not inhibit this adhesion (Aronson et al. Citation1988). Analysis of adhesion molecule expression on IL-2 treated human NK cells indicated up-regulation of CD11a, CD2 (LFA-3R), CD44, CD54 (ICAM-1), and CD58 (LFA-3) (Mäenpää et al. Citation1993) with phosphorylation and thus activation of CD18 signaling (Umehara et al. Citation1993). IL-2 activated primary human NK cells also exhibit chemotaxis to MCP-1 and, to a less extent, MCP-2 and MCP-3 (Allavena et al. Citation1994). This finding is underpinned by increased expression of the MCP-1 receptor CCR2 on IL-2 stimulated NK cells (Polentarutti et al. Citation1997).
In primary human memory-phenotype CD45RO+ lymphocytes, IL-2 induces expression of the chemokine receptors MCP-1 and RANTES and increases chemotactic responses toward MCP-1, −2, −3, MIP-1α, -β, and RANTES (Loetscher et al. Citation1996).
Tlymphocytes. While trapping of activated CD8+ T-cells in the hepatic sinusoids in the context of inflammation is widely documented, there is little known about recruitment of IL-2 activated CD8+ T-cells to the liver (). Murine models indicate that in the context of inflammation, activated CD8+ T-cells are preferentially trapped in the liver and undergo consequent apoptosis, thus clearing them from any immune responses (Mehal et al. Citation1999, Citation2001). In an adoptive transfer model using transgenic T-cell receptor (TCR) OT1 cells, bearing an ovalbumin (OVA) peptide specific TCR restricted to MHC I, ICAM-1/LFA-1, and VCAM-1/VLA-4 interactions enable CD8+ T-cell trapping in the liver. In contrast to ICAM-1-mediated trapping of CD8+ T-cells, in which antigen presentation by hepatocytes or LSEC is central, VCAM-1-induced trapping occurs independently of antigen presentation. The authors hypothesize that antigen recognition might enhance affinity of T-cell LFA-1 for ICAM-1 via T-cell receptor signaling. They suggest a model for trapping activated T-cells in the liver initiated by passive interactions of VCAM-1/VLA-4 or VAP-1/VAP-1R, following increased adhesion mediated by antigen stabilized ICAM-1/LFA-1 interaction (John and Crispe Citation2004). An additional mechanism for CD8+ T-cell arrest in the liver sinusoids is provided by non-antigen specific binding to adherent platelets which provide a docking site for T-cells (Guidotti et al. Citation2015). This might involve a link to IL-2, as increased platelet adhesion to liver sinusoids was measured in IL-2 treated mice (Nakagawa et al. Citation1996).
In human Treg cells, IL-2 promotes tissue homing by increasing α4- and β7-integrin expression (Hsu et al. Citation2018). Assessment of surface marker expression on liver-infiltrating T-cells in inflamed human livers showed that nearly all CCR10+CD4+ T-cells expressed α4β1- integrin and subpopulations expressed α4β7- integrin (Eksteen et al. Citation2006). A further role for α4- integrin in liver inflammation involving TH cells has been shown in murine Concanavalin A liver inflammation. TH1 cells adhere to post sinusoidal venules via α4 integrin while TH2 cells rely on VAP-1 (Bonder et al. Citation2005). In vitro polarized and in vivo injected murine TH1 and, to a less extent, TH2 cells adhere to the liver sinusoids in large fractions and locate preferentially to the periportal field (Klugewitz et al. Citation2002).
Little is known regarding IL-2-induced recruitment of NKT cells to the human liver (). In this process, LFA-1 expression on non-lymphoid cells plays an essential role, as suggested by LFA-1-deficient mice (Emoto et al. Citation1999). Excluding ICAM-1 as an interaction partner of LFA-1 on NKT cells, the authors did not provide evidence for alternative LFA-1 ligands. CD1d-reactive NKT cells patrolling the liver sinusoids also express CXCR6, which binds to CXCL16 expressed on LSEC. CXCR6-deficient mice exhibit reduced NKT numbers in the liver sinusoids and diminished severity of Concanavalin A-mediated hepatitis (Geissmann et al. Citation2005). IL-2 was shown to up-regulate CXCR6 surface expression on CD3+ PBMCs (Perna et al. Citation2009).
Adhesion to LSEC. As described above, on lymphocytes, the adhesion molecules LFA-1, VLA-4 and on neutrophils, CD11b and CD18 are up-regulated upon IL-2 stimulation (Anderson et al. Citation1996; Abdel-Salam and Ebaid Citation2014). Consequently, it is essential to address the expression of important immune cell binding partners in the liver, specifically in the LSEC layer. To our knowledge, no information exists on direct effects of IL-2 on LSEC adhesion marker expression. However, LSEC adhesion markers have been described. Human LSEC express VAP-1, while VAP-1 is not present on parenchymal or bile duct cells (Salmi et al. Citation1993; Lalor et al. Citation2007). Additionally, upon activation of LSEC with pro-inflammatory TNFα, VCAM-1 and E-selectin are induced, and ICAM-1 is increased. Under static conditions, CD11a, CD18, and VAP-1 mediate adhesion of peripheral lymphocytes, while VCAM-1, ICAM-1, and E-selectin are implicated to a less extent. Upon activation of LSEC with TNFα, additionally ICAM-1 and VCAM-1 confer adhesion. Under physiological flow conditions, few lymphocytes adhere to sinusoidal cells. Pro-inflammatory activation of LSEC leads to increased lymphocyte adhesion with shear-stress dependent increase in adhesion. This adhesion process is only marginally mediated by rolling, rather than immediate arrest, which is most likely conferred by VAP-1, with its monoamine oxidase activity (Lalor et al. Citation2002). Upon binding of the VAP-1 enzymatic substrate benzylamine, the NF-κB pathway is induced and CXCL8, E-selectin, and ICAM-1 are up-regulated (Lalor et al. Citation2007).
In the context of IL-2, lymphocyte migration across the LSEC layer has not been addressed to our knowledge (). Patten et al. (Citation2016) have provided a comprehensive assessment of lymphocyte migration through the primary human endothelial cell layer in vitro. They demonstrated that intracellular crawling represents an important mechanism during transendothelial migration of lymphocytes across the LSEC layer, migration of lymphocytes being facilitated by formation of intracellular vesicles. Antibody blocking experiments showed that ICAM-1 and stabilin-1, as well as PD-L1 but not CXCR3, have important roles in intracellular migration of lymphocytes. Regarding junctional molecules, LSEC express VE-cadherin, JAM-A, and ZO-1, while occludin, claudin-1, and E-cadherin are undetectable. Combination treatment with IFNγ and TNFα leads to a reduction in junctional molecule levels as seen by increased permeability upon concurrent IFNγ and TNFα stimulation. Between healthy and chronic-disease liver tissue, junctional molecule expression is comparable but differs strongly from HUVEC. Expression analyses confirm the differences between LSEC and HUVEC at both baseline and upon IFNγ and TNF-α treatment (Patten et al. Citation2016).
Taken together, these data provide a rich source of information on lymphocyte recruitment and adhesion to, as well as migration through the liver sinusoids that differs from mechanisms observed in HUVEC. Further research is needed to assess the roles of the respective molecules secondarily induced upon IL-2 stimulation ().
Immune-mediated hepatocyte damage (KE3)
IL-2 induced damage of hepatocytes constitutes the final key event of the irAOP that precedes the adverse outcome hepatotoxicity, which has already been described.
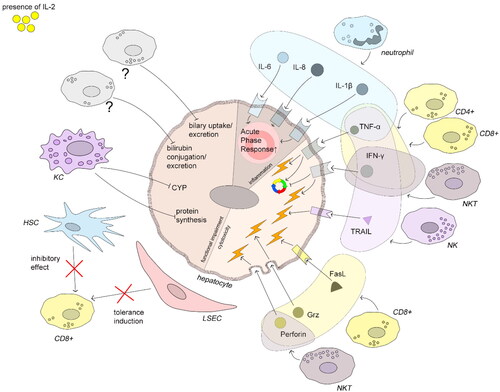
Inflammatory cytokines TNFα is a pleiotropic cytokine implicated in liver homeostasis and contributes to both hepatocyte injury and proliferation. In rats, high amounts of TNFα, produced during LPS-induced liver injury, act pro-apoptotically, and low concentrations released during acetaminophen-induced liver injury promote cell survival. The authors identified the Hippo/Yap pathway as a central regulator of these pleiotropic effects (Zhao et al. Citation2020b). Moreover, in mice challenged with different pro-inflammatory stimuli, both the presence and the cell type producing the TNFα determine its pathophysiological effects. Thus, TNFα produced by macrophages and neutrophils, but not from T- and B-cells, was detrimental in septic shock (Grivennikov et al. Citation2005). IL-6 also has pleiotropic roles in the liver, promoting liver regeneration but also inflammation and cancer (Schmidt-Arras and Rose-John Citation2016). IL-8 is a pro-inflammatory cytokine and chemoattractant and is up-regulated systemically and locally in the livers of patients with chronic liver diseases (Zimmermann et al. Citation2011). In human hepatocytes, both IL-6 and IL-8 induce the expression of acute phase proteins and show additive effects, while IL-1β and TNFα lead to a weaker response (Castell et al. Citation1989; Wigmore et al. Citation1997). IL-1 type cytokines rather modulate than induce an acute phase response (Bode et al. Citation2012). Moreover, it was proposed that IL-1β plays a role in liver damage activating local leukocytes and recruiting further immune cells (Barbier et al. Citation2019) (). IFNγ induces cell cycle arrest and p53-independent apoptosis of primary murine hepatocytes (Kano et al. Citation1997).
Figure 3. Immune-mediated hepatocyte damage induced by IL-2. All depicted mechanisms follow IL-2 stimulation. Neutrophils produce the cytokines IL-1β, -6, and -8 which increase acute phase protein production. TNFα can act pro-apoptotically but also promotes liver regeneration under certain conditions (see text). TNFα is produced by neutrophils, CD4+ T-, CD8+ T-, NKT, and NK cells. IFNγ inhibits cell cycle progression and induces apoptosis and is produced by CD4+ T-, CD8+ T-, and NK cells. NK cells induce hepatocyte apoptosis by TRAIL-TRAIL-R interaction. CD8+ T-cells induce hepatocyte damage by FasL-Fas interaction and by production of perforin and granzymes. Exogenous IL-2 can both overcome the tolerance induction and impair the inhibitory effect of HSCs. KCs downmodulate protein synthesis and CYP activity. Impaired bilirubin and biliary acid metabolism are an AO of IL-2 therapy in humans, but the key players are not evident from literature. The figure compiles human, murine, and rat data.
PBMCs. Early studies by Mier et al. (Citation1987) on incubation of Hep3B cells with supernatants of IL-2 stimulated PBMCs showed an IL-2 concentration-dependent increase in acute phase protein synthesis, only with unfractionated PBMC and not when incubating Hep3B cells with IL-2 alone. IL-1β was identified as the mediator of an indirect effect of IL-2 on acute phase protein production in liver cells, while roles of IL-1α and TNFα could be excluded by incubation with antisera (Mier et al. Citation1987). Contrasting to the results obtained by Mier et al. (Citation1987), Wigmore et al. (Citation2002) did not find significant influences of IL-2 incubation on IL-6 and TNFα increase in PBMC supernatants and no effect of these supernatants on hepatic acute phase protein synthesis (Wigmore et al. Citation2002). Despite comparable IL-2 concentrations and incubation times, three apparent experimental differences may explain the different outcomes. Firstly, Mier et al. (Citation1987) incubated PBMC at a 2-fold higher density (2 x 106 cells/mL compared to 1 x 106 cells.ml). Culturing PBMC at high density has been shown to enhance T- cell responses, for instance toward the CD28 superagonist TGN1412 (Römer et al. Citation2011). This might also apply to IL-2 stimulated cells. Secondly, Mier et al. (Citation1987) incubated the Hep3B cell line with 1:3 diluted PBMC supernatant, while Wigmore et al. (Citation2002) diluted supernatant 1:10. Lastly, the use of cell lines by Mier et al. (Citation1987) versus primary human hepatocytes by Wigmore et al. (Citation2002) could be a factor. Comparison of these data shows that there is a need to identify the correct culture and stimulation conditions for modeling immune-mediated hepatotoxicity. Additionally, the impact of immune cell subtypes on the observed effects remains elusive ().
Tlymphocytes. In both primary human CD4+ and CD8+ T-cells, IL-2 induces IFNγ expression in a dose-dependent manner (Kasahara et al. Citation1983). Activated CD8+ T-cells express the cytotoxic surface molecule FasL and binding FasL on activated T-cells induces apoptosis on Fas expressing hepatocytes. For this antigen-independent hepatocyte killing mechanism, the term collateral damage was proposed (Ando et al. Citation1997; Crispe et al. Citation2000; Mehal et al. Citation2001). In murine naïve CD8+ T-cells, there is a dose-dependent increase in perforin and granzyme A, B, and C mRNA with IL-2 stimulation, accompanied by increased cytolytic activity (). The up-regulation of mRNA expression is independent of the anti-apoptotic and proliferative effects of IL-2 (Janas et al. Citation2005) ().
Also, in antigen-specific responses, Fas/FasL-mediated apoptosis by infiltrating CD8+ T-cells plays an important role in hepatotoxicity. In OT-1 mice, Fas-expressing mice showed hepatotoxicity upon OVA peptide activation, while in Fas-deficient OT-1lpr mice, lymphocyte infiltration was comparable but hepatocyte damage was nearly undiscernible. Isolated liver CD8+ T-cells are cytotoxic against the hepatoma cell line Hepa1.6 and express high levels of TNFα, perforin, and FasL, highlighting the enhanced cytotoxic activity of liver infiltrating immune cells (Kennedy et al. Citation2001). The Fas/FasL system supposedly plays a major role in inflammatory conditions such as sepsis. In cecal ligation and puncture induced sepsis in mice, inhibition of Fas/FasL signaling by a Fas fusion protein restored hepatic blood flow, lessened liver damage, and decreased IL-10 production (Chung et al. Citation2001). These data emphasize the role of CD4+ T-cell-derived TNF-α and IFN-γ, and CD8+ T-cell FasL-mediated apoptosis in IL-2-induced hepatic injury.
NKT cells contribute to liver injury by influencing the local immune milieu through IL-4, −5, and −13 release, directly acting on hepatocytes via IFNγ and TNFα production, as well as by killing hepatocytes through cell-cell contact dependent mechanisms (Gao et al. Citation2009). FasL/Fas interaction plays an important role in NKT cell-mediated hepatocyte killing, as shown in murine Concanavalin A-induced liver injury (Takeda et al. Citation2000). Notably, NKT cells can be non-specifically activated by DMSO to express granzyme B and IFNγ and locate to the liver (Masson et al. Citation2008). These findings point toward a contribution of antigen-independently activated NKT cells to liver damage, comparable to the collateral damage hypothesis mentioned earlier which remains to be tested for IL-2 ().
Kupffer cells. KC themselves influence hepatocyte functions, as high IL-2 concentrations added to primary rat KC suppress protein synthesis in rat hepatocytes in co-culture (Curran et al. Citation1988). In primary human hepatocyte cultures, there is a concentration-dependent decrease in CYP activity in hepatocyte/KC co-cultures stimulated with IL-2, without direct effects of IL-2 (Kashuba et al. Citation2000; Sunman et al. Citation2004). Therefore, it would be favorable to assess, whether CYP activity could be an additional marker for IL-2-mediated hepatic injury (). In hepatocyte/KC co-cultures, IL-2 only produces negligible effects on pro-inflammatory cytokines (Nguyen et al., Citation2015). Furthermore, data from in vitro co-culture experiments of primary murine KC and lymphocytes under Concanavalin A stimulation suggest that these cell types act synergistically and enhance cytokine expression compared to the isolated cell types (Gantner et al. Citation1996), but it is not clear how these findings translate to IL-2 (). These results point toward a complex interaction of pro-inflammatory responses in the liver resident immune cell populations. It is probable that lessons can be learned from interaction of macrophages with lymphocytes since KCs are specialized macrophages and antigen-presenting cells. Therefore, the KC activation state would likely affect the outcome of any IL-2 exposure.
NK cells. IL-2 induces lytic granule convergence in human NK cells - an early step in NK cell cytotoxicity - in a Src kinase dependent manner (James et al. Citation2013) (). TNF-Related Apoptosis-Inducing Ligand (TRAIL) binds to the death receptor DR5 which induces apoptosis (Yuan et al. Citation2018). Liver NK cells, but not peripheral blood NK cells, express low TRAIL, which can be induced with IL-2. Concomitantly, IL-2 stimulated liver NK cells exert cytotoxicity against the HepG2 cell line. Stimulation with IL-2 leads to maintenance of the Killer-cell inhibitory receptors CD158a, -b, and the C-type lectin receptor CD94 in liver and peripheral blood NK cells. Well-differentiated hepatocellular carcinoma cells weakly express the death inducing receptors TRAIL-R1/DR4 and -R2/DR5 as well as the non-signaling –R3/DcR1 and –R4/DcR2, while normal hepatocytes lack these receptors. At the same time, endothelial cells express these receptors more abundantly. Poorly differentiated hepatocellular carcinoma and HepG2 cells express the TRAIL-R1/–DR4 and TRAIL-R2/–DR5 but lack expression of the non-signaling death receptors (Ishiyama et al. Citation2006). As described above, CD56bright NK cells are a major NK cell sub-population in the human liver (Hudspeth et al. Citation2016). In chronic HBV-related liver cirrhosis, TRAIL+CD56bright NK cells are associated with the degree of fibrosis and CXCR3-related chemokine levels (Jiang et al. Citation2021) highlighting the hepatotoxic relevance of this cell type (). These findings indicate that IL-2 may activate liver NK cells to promote hepatic inflammation and kill hepatocytes, mainly via TRAIL.
LSEC exert antigen-presenting cell functions and despite being highly capable of taking up antigens, they are alone poor stimulators of an immune response (Katz et al. Citation2004). LSEC rather promote liver tolerance by PD-L1 interaction with PD-1 on T-cells. This tolerance can be overcome by anti-CD28 antibody addition (Diehl et al. Citation2008). In primary murine OT-1 restricted T-cells, exogenous IL-2 can overcome the tolerance induction by LSECs during first contact with naïve CD8+ T-cells and during T-cell priming, even in the absence of co-stimulation by CD80/CD86, resulting in CD8+ T-cell effector function, such as IFNγ production () (Schurich et al. Citation2010). Whether IL-2 can break the LSEC tolerance in humans remains unclear (). Furthermore, with regard to CD4+ T-cells, murine LSEC induce differentiation of naïve CD4+ T-cells to TH0 cells but not to TH1 cells, thereby contributing to liver immune tolerance. This effect is not dominant as it can be overcome by other antigen-presenting cells, for instance in the case of non-specific lymphocyte recruitment to the liver (Knolle et al. Citation1999). In a murine antigen-specific TH1 cell-inducible liver injury model, the late effector phase of acute liver injury by TH1 cells is mediated by TNFα and IFNγ. Fas and perforin were not found to play a role in this hepatitis model (Ohta et al. Citation2000). Translation of these effects to IL-2 would indicate whether TH1 cell-mediated liver injury also plays a role in this irAOP ().
HSC down-modulate CD8+ T-cell activation by trans-interaction (i.e., as third-party modulators) of ICAM-1 on HSC with its ligand LFA-1 on CD8+ T-cells. Knockout experiments in mice indicate that ICAM-1 is up-regulated on HSC and mediates down-regulation of IL-2Rα expression and thus, the IL-2 response. Exogenous IL-2 antagonizes this inhibitory effect of HSC in a dose-dependent manner (Schildberg et al. Citation2011) (). These results indicate that IL-2 can contribute to breaking the tolerogenic potential of the liver, but further research remains to provide evidence in the human setting ().
In conclusion, there are a variety of possible mechanisms for IL-2-induced immune-mediated hepatic damage (). Research is needed to characterize specifically the effect of IL-2 in these processes, which would aid to validate or reject the hypotheses made in this putative irAOP and to sharpen the picture of the irAOP. Regarding the mechanisms retrieved from the literature, IL-2-mediated hepatotoxicity can be assigned to indirect DILI. Possibly, the elucidation of mechanisms in IL-2-induced liver damage can be extrapolated to checkpoint-inhibitor-mediated indirect DILI and vice-versa.
Identification of knowledge gaps and biorelevant molecules with the irAOP approach
The development of a hypothetical irAOP for IL-2-mediated hepatotoxicity unveiled several knowledge gaps of central importance. Addressing these gaps is essential, as it would not only enhance our comprehension of the underlying mechanisms but is also pivotal for validating this potential irAOP. In , the identified gaps and potential methods that could be applied to fill the gaps, are summarized. Primarily, there is a lack of data that can only be generated with experiments on human hepatocytes and/or liver resident cells (e.g., LSEC, KC), which is however difficult to achieve due to the low availability of these primary human cell types. Central experiments to the validation of the irAOP would be the elucidation of IL-2R expression on non-parenchymal cells in the liver to identify cells capable of responding directly to IL-2 signaling. Secondly, the literature does not provide sufficient information on IL-2-mediated recruitment and migration of immune cells to liver sinusoids and into the perisinusoidal space. To achieve this, use of human LSEC and IL-2-activated immune cells, especially in flow-based settings, could provide valuable information on the situation in the liver. For this, more sophisticated experimental approaches recapitulating the physiology of the liver would be desirable, such as organ-on-a-chip models with perfused immune cells. Setups are already available that model liver sinusoids (Du et al. Citation2017) or that have been used for determination of liver dysfunction upon treatment with a sepsis-related protein (Blaurock-Möller et al. Citation2019). These models highlight the capacity of liver microphysiological systems to model liver toxicities. However, such organ-on-a-chip approaches would need adaptation to fill the knowledge gaps by selecting adequate readouts provided by the irAOP () and by application of primary human material - ideally, the use of induced pluripotent stem cells would allow fully autologous microphysiological systems (Ramme et al. Citation2019). Regarding the latter KE of the irAOP, it would be helpful to compare healthy human liver samples to resections from IL-2 treated patients who have clear hepatotoxicity. High-definition spatial transcriptomic profiling combines single cell RNA sequencing with positional information in tissues in situ (Vickovic et al. Citation2019). This method could provide valuable information, not only on the transcriptional landscape, but could also aid identification of the location of immune cell sub-populations in the liver. Overall, it seems that irAOPs are a suitable tool to identify knowledge gaps and biorelevant molecules and to link AO to both their MIE and the associated KE for IL-2-mediated hepatotoxicity (, ).
Besides IL-2-specific mechanisms, the hypothetical irAOP presented here also highlights common hepatotoxicity mechanisms (e.g., acute phase protein production, hepatocyte damage by cytotoxic molecules, impaired bilirubin or bile acid metabolism). Hence, models suggested here can potentially be used as blueprints for the evaluation of novel IL-2 drug candidates or IL-2 combination therapies with similar AOs. This irAOP concept may also be applicable for other biologicals, low molecular-weight therapeutics, or RNA-based drugs, if these molecules share common KEs with those described in this putative irAOP. The next step would be to translate this irAOP to other drugs known to cause immune-mediated hepatotoxicity, such as TNFα inhibitors (French et al. Citation2016) or checkpoint inhibitors (Zen and Yeh Citation2018) and to evaluate to what extent transferability is possible. The overall aim would be to generate a compound-agnostic irAOP for immune-mediated DILI.
Conclusion
IL-2 is a well-characterized molecule with known mechanisms of action. Nevertheless, many missing links hinder the extensive understanding of IL-2-mediated hepatotoxicity. The irAOP concept described here helps to identify these knowledge gaps and provides starting points for further research. The relevance of such attempts becomes clear as currently, modifications of IL-2 and/or IL-2 combination therapies are under investigation to increase efficacy and reduce toxicity of conventional IL-2 therapy (Overwijk et al. Citation2021). To augment the chances of success for such therapies, understanding and correct modeling of IL-2-mediated hepatic toxicities is crucial. Therefore, the inclusion of relevant immune cell fractions and exploitation of key event-defining molecules as readouts would provide meaningful and targeted in vitro hepatotoxicity models which could facilitate reduced use of in vivo preclinical models.
| Abbreviations | ||
| AO | = | Adverse Outcome |
| Bcl-2 | = | B-cell lymphoma 2 |
| CYP | = | Cytochrome P450 |
| DILI | = | Drug-induced liver injury |
| Eomes | = | Eomesodermin |
| FPKM | = | Fragments Per Kilobase Million |
| GM-CSF | = | Granulocyte-macrophage colony-stimulating factor |
| HBV | = | Hepatitis B virus |
| HSC | = | Hepatic Stellate Cell |
| HUVEC | = | Human Umbilical Vein Endothelial Cell |
| IL-2 | = | Interleukin-2 |
| IL-2R | = | IL-2 receptor |
| imSAVAR | = | Immune Safety Avatar |
| irAOP | = | Immune-related Adverse Outcome Pathway |
| KE | = | Key Event |
| KIR2DL4 | = | Killer cell immunoglobulin-like receptor 2DL4 |
| KC | = | Kupffer Cell |
| LPS | = | Lipopolysaccharide |
| LSEC | = | Liver Sinusoidal Endothelial Cell |
| MAPK | = | Mitogen-Activated Protein Kinase |
| MCP-1 | = | Monocyte Chemoattractant Protein-1 |
| MIE | = | Molecular Initiating Event |
| NKG2D | = | Natural Killer Group 2 Member D |
| NKT | = | Natural Killer T Cell |
| OVA | = | Ovalbumin |
| PBMC | = | Peripheral blood mononuclear cell |
| sCD25 | = | soluble IL-2Rα |
| TCR | = | T cell receptor |
| Th | = | T helper |
| TNF-α | = | Tumor necrosis factor alpha |
| TRAIL | = | TNF-Related Apoptosis-Inducing Ligand |
| Treg | = | CD4+ regulatory T |
Acknowledgements
The JU receives support from the European Union′s Horizon 2020 research and innovation programme and EFPIA and JDRF INTERNATIONAL. The project was supported by the Landesoffensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz (LOEWE) Centre Novel Drug Targets against Poverty-Related and Neglected Tropical Infectious Diseases (DRUID), the LOEWE Centre Translational Biodiversity Genomics (TBG) and the Fraunhofer Cluster of Excellence Immune-mediated diseases (CIMD).
Disclosure statement
MJP is an employee of EpiEndo Pharmaceuticals ehf.
Correction Statement
This article was originally published with errors, which have now been corrected in the online version. Please see Correction (http://dx.doi.org/10.1080/1547691X.2024.2342736)
Additional information
Funding
References
- Abbas AK, Trotta E, R Simeonov D, Marson A, Bluestone JA. 2018. Revisiting IL-2: biology and therapeutic prospects. Sci Immunol. 3(25):eaat1482. doi:10.1126/sciimmunol.aat1482.
- Abdel-Salam BK, Ebaid H. 2014. Expression of CD11b and CD18 on polymorphonuclear neutrophils stimulated with interleukin-2. Cent Eur J Immunol. 39(2):209–215. doi:10.5114/ceji.2014.43725.
- Allavena P, Bianchi G, Zhou D, van Damme J, Jílek P, Sozzani S, Mantovani A. 1994. Induction of natural killer cell migration by monocyte chemotactic protein − 1, −2 and −3. Eur J Immunol. 24(12):3233–3236. doi:10.1002/eji.1830241249.
- Anderson JA, Lentsch AB, Hadjiminas DJ, Miller FN, Martin AW, Nakagawa K, Edwards MJ. 1996. The role of cytokines, adhesion molecules, and chemokines in interleukin-2-induced lymphocytic infiltration in C57BL/6 mice. J Clin Invest. 97(8):1952–1959. doi:10.1172/JCI118627.
- Ando K, Hiroishi K, Kaneko T, Moriyama T, Muto Y, Kayagaki N, Yagita H, Okumura K, Imawari M. 1997. Perforin, Fas/Fas ligand, and TNF-alpha pathways as specific and bystander killing mechanisms of hepatitis C virus-specific human CTL. J Immunol. 158(11):5283–5291.
- Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, Johnson RD, Mount DR, Nichols JW, Russom CL, Schmieder PK, et al. 2010. Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem. 29(3):730–741. doi:10.1002/etc.34.
- Aoki CA, Roifman CM, Lian Z-X, Bowlus CL, Norman GL, Shoenfeld Y, Mackay IR, Gershwin ME. 2006. IL-2 receptor alpha deficiency and features of primary biliary cirrhosis. J Autoimmun. 27(1):50–53. doi:10.1016/j.jaut.2006.04.005.
- Armant M, Delespesse G, Sarfati M. 1995. IL-2 and IL-7 but not IL-12 protect natural killer cells from death by apoptosis and up-regulate bcl-2 expression. Immunology. 85(2):331–337.
- Aronson FR, Libby P, Brandon EP, Janicka MW, Mier JW. 1988. IL-2 rapidly induces natural killer cell adhesion to human endothelial cells. A potential mechanism for endothelial injury. J Immunol. 141(1):158–163. doi:10.4049/jimmunol.141.1.158.
- Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, et al. 1999. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 17(7):2105–2116. doi:10.1200/JCO.1999.17.7.2105.
- Au-Yeung B, Smith GA, Mueller JL, Heyn CS, Jaszczak RG, Weiss A, Zikherman J. 2017. IL-2 modulates the TCR signaling threshold for CD8 but not CD4 T cell proliferation on a single cell level. J Immunol. 198(6):2445–2456. doi:10.4049/jimmunol.1601453.
- Babai S, Auclert L, Le-Louët H. 2021. Safety data and withdrawal of hepatotoxic drugs. Therapie. 76(6):715–723. doi:10.1016/j.therap.2018.02.004.
- Baev DV, Peng X-H, Song L, Barnhart JR, Crooks GM, Weinberg KI, Metelitsa LS. 2004. Distinct homeostatic requirements of CD4+ and CD4- subsets of Valpha24-invariant natural killer T cells in humans. Blood. 104(13):4150–4156. doi:10.1182/blood-2004-04-1629.
- Ball T, Barber CG, Cayley A, Chilton ML, Foster R, Fowkes A, Heghes C, Hill E, Hill N, Kane S, et al. 2021. Beyond adverse outcome pathways: making toxicity predictions from event networks, SAR models, data and knowledge. Toxicol Res. 10(1):102–122. doi:10.1093/toxres/tfaa099.
- Bal-Price A, Meek MEB. 2017. Adverse outcome pathways: application to enhance mechanistic understanding of neurotoxicity. Pharmacol Ther. 179:84–95. doi:10.1016/j.pharmthera.2017.05.006.
- Bandyopadhyay K, Marrero I, Kumar V. 2016. NKT cell subsets as key participants in liver physiology and pathology. Cell Mol Immunol. 13(3):337–346. doi:10.1038/cmi.2015.115.
- Barbier L, Ferhat M, Salamé E, Robin A, Herbelin A, Gombert J-M, Silvain C, Barbarin A. 2019. Interleukin-1 family cytokines: keystones in liver inflammatory diseases. Front Immunol. 10:2014. doi:10.3389/fimmu.2019.02014.
- Baume DM, Robertson MJ, Levine H, Manley TJ, Schow PW, Ritz J. 1992. Differential responses to interleukin 2 define functionally distinct subsets of human natural killer cells. Eur J Immunol. 22(1):1–6. doi:10.1002/eji.1830220102.
- Behfarjam F, Sanati MH, Nasseri Moghaddam S, Ataei M, Nikfam S, Jadali Z. 2017. Role of Th1/Th2 cells and related cytokines in autoimmune hepatitis. Turk J Gastroenterol. 28(2):110–114. doi:10.5152/tjg.2017.17501.
- Belaid B, Lamara Mahammed L, Mohand Oussaid A, Migaud M, Khadri Y, Casanova JL, Puel A, Ben Halla N, Djidjik R. 2021. Case report: interleukin-2 receptor common gamma chain defect presented as a hyper-IgE syndrome. Front Immunol. 12:696350. doi:10.3389/fimmu.2021.696350.
- Benczik M, Gaffen SL. 2004. The interleukin (IL)-2 family cytokines: survival and proliferation signaling pathways in T lymphocytes. Immunol Invest. 33(2):109–142. doi:10.1081/imm-120030732.
- Bénéchet AP, Simone G d, Di Lucia P, Cilenti F, Barbiera G, Le Bert N, Fumagalli V, Lusito E, Moalli F, Bianchessi V, et al. 2019. Dynamics and genomic landscape of CD8+ T cells undergoing hepatic priming. Nature. 574(7777):200–205. doi:10.1038/s41586-019-1620-6.
- Bessoles S, Fouret F, Dudal S, Besra GS, Sanchez F, Lafont V. 2008. IL‐2 triggers specific signaling pathways in human NKT cells leading to the production of pro‐ and anti‐inflammatory cytokines. J Leukoc Biol. 84(1):224–233. doi:10.1189/jlb.1007669.
- Bethesda. 2018. Aldesleukin: liverTox: clinical and research information on drug-induced liver injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases.
- Beyer T, Busse M, Hristov K, Gurbiel S, Smida M, Haus U-U, Ballerstein K, Pfeuffer F, Weismantel R, Schraven B, et al. 2011. Integrating signals from the T-cell receptor and the interleukin-2 receptor. PLoS Comput Biol. 7(8):e1002121. doi:10.1371/journal.pcbi.1002121.
- Björnsson ES, Bergmann OM, Björnsson HK, Kvaran RB, Olafsson S. 2013. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 144(7):1419–1425.e3. doi:10.1053/j.gastro.2013.02.006.
- Blachère NE, Morris HK, Braun D, Saklani H, Di Santo JP, Darnell RB, Albert ML. 2006. IL-2 is required for the activation of memory CD8+ T cells via antigen cross-presentation. J Immunol. 176(12):7288–7300. doi:10.4049/jimmunol.176.12.7288.
- Blaurock-Möller N, Gröger M, Siwczak F, Dinger J, Schmerler D, Mosig AS, Kiehntopf M. 2019. CAAP48, a new sepsis biomarker, induces hepatic dysfunction in an in vitro liver-on-chip model. Front Immunol. 10:273. doi:10.3389/fimmu.2019.00273.
- Bode JG, Albrecht U, Häussinger D, Heinrich PC, Schaper F. 2012. Hepatic acute phase proteins–regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur J Cell Biol. 91(6-7):496–505. doi:10.1016/j.ejcb.2011.09.008.
- Bonder CS, Norman MU, Swain MG, Zbytnuik LD, Yamanouchi J, Santamaria P, Ajuebor M, Salmi M, Jalkanen S, Kubes P. 2005. Rules of recruitment for Th1 and Th2 lymphocytes in inflamed liver: a role for alpha-4 integrin and vascular adhesion protein-1. Immunity. 23(2):153–163. doi:10.1016/j.immuni.2005.06.007.
- Bonnema JD, Rivlin KA, Ting AT, Schoon RA, Abraham RT, Leibson PJ. 1994. Cytokine-enhanced NK cell-mediated cytotoxicity. Positive modulatory effects of IL-2 and IL-12 on stimulus-dependent granule exocytosis. J Immunol. 152(5):2098–2104.
- Bosco MC, Curiel RE, Zea AH, Malabarba MG, Ortaldo JR, Espinoza-Delgado I. 2000. IL-2 signaling in human monocytes involves the phosphorylation and activation of p59hck. J Immunol. 164(9):4575–4585. doi:10.4049/jimmunol.164.9.4575.
- Bosco MC, Espinoza-Delgado I, Schwabe M, Gusella GL, Longo DL, Sugamura K, Varesio L. 1994a. Regulation by interleukin-2 (IL-2) and interferon gamma of IL-2 receptor gamma chain gene expression in human monocytes. Blood. 83(10):2995–3002. doi:10.1182/blood.V83.10.2995.2995.
- Bosco MC, Espinoza-Delgado I, Schwabe M, Russell SM, Leonard WJ, Longo DL, Varesio L. 1994b. The gamma subunit of the interleukin-2 receptor is expressed in human monocytes and modulated by interleukin-2, interferon gamma, and transforming growth factor beta 1. Blood. 83(12):3462–3467. doi:10.1182/blood.V83.12.3462.bloodjournal83123462.
- Boyman O, Sprent J. 2012. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 12(3):180–190. doi:10.1038/nri3156.
- Boyman O, Surh CD, Sprent J. 2006. Potential use of IL-2/anti-IL-2 antibody immune complexes for the treatment of cancer and autoimmune disease. Expert Opin Biol Ther. 6(12):1323–1331. doi:10.1517/14712598.6.12.1323.
- Burton J, Goldman CK, Rao P, Moos M, Waldmann TA. 1990. Association of intercellular adhesion molecule 1 with the multichain high-affinity interleukin 2 receptor. Proc Natl Acad Sci U S A. 87(18):7329–7333. doi:10.1073/pnas.87.18.7329.
- Cabrera R, Ararat M, Cao M, Xu Y, Wasserfall C, Atkinson MA, Liu C, Nelson DR. 2010a. Hepatocellular carcinoma immunopathogenesis: clinical evidence for global T cell defects and an immunomodulatory role for soluble CD25 (sCD25). Dig Dis Sci. 55(2):484–495. doi:10.1007/s10620-009-0955-5.
- Cabrera R, Ararat M, Eksioglu EA, Cao M, Xu Y, Wasserfall C, Atkinson MA, Liu C, Nelson DR. 2010b. Influence of serum and soluble CD25 (sCD25) on regulatory and effector T-cell function in hepatocellular carcinoma. Scand J Immunol. 72(4):293–301. doi:10.1111/j.1365-3083.2010.02427.x.
- Castell JV, Gómez-Lechón MJ, David M, Andus T, Geiger T, Trullenque R, Fabra R, Heinrich PC. 1989. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett. 242(2):237–239. doi:10.1016/0014-5793(89)80476-4.
- Chung CS, Yang S, Song GY, Lomas J, Wang P, Simms HH, Chaudry IH, Ayala A. 2001. Inhibition of Fas signaling prevents hepatic injury and improves organ blood flow during sepsis. Surgery. 130(2):339–345. doi:10.1067/msy.2001.116540.
- Crispe IN, Dao T, Klugewitz K, Mehal WZ, Metz DP. 2000. The liver as a site of T-cell apoptosis: graveyard, or killing field? Immunol Rev. 174(1):47–62. doi:10.1034/j.1600-0528.2002.017412.x.
- Crispe IN. 2003. Hepatic T cells and liver tolerance. Nat Rev Immunol. 3(1):51–62. doi:10.1038/nri981.
- Curran RD, Billiar TR, West MA, Bentz BG, Simmons RL. 1988. Effect of interleukin 2 on Kupffer cell activation. Interleukin 2 primes and activates Kupffer cells to suppress hepatocyte protein synthesis in vitro. Arch Surg. 123(11):1373–1378. doi:10.1001/archsurg.1988.01400350087013.
- David D, Bani L, Moreau J-L, Demaison C, Sun K, Salvucci O, Nakarai T, Montalembert M d, Chouaı¨b S, Joussemet M, et al. 1998. Further analysis of interleukin-2 receptor subunit expression on the different human peripheral blood mononuclear cell subsets. Blood. 91(1):165–172. doi:10.1182/blood.V91.1.165.165_165_172.
- David S, Hamilton JP. 2010. Drug-induced liver injury. US Gastroenterol Hepatol Rev. 6:73–80.
- de Jong LM, Jiskoot W, Swen JJ, Manson ML. 2020. Distinct effects of inflammation on cytochrome P450 regulation and drug metabolism: lessons from experimental models and a potential role for pharmacogenetics. Genes. 11(12):1509. doi:10.3390/genes11121509.
- DeLeve LD, Maretti-Mira AC. 2017. Liver sinusoidal endothelial cell: an update. Semin Liver Dis. 37(4):377–387. doi:10.1055/s-0037-1617455.
- Diehl L, Schurich A, Grochtmann R, Hegenbarth S, Chen L, Knolle PA. 2008. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology. 47(1):296–305. doi:10.1002/hep.21965.
- Djeu JY, Liu JH, Wei S, Rui H, Pearson CA, Leonard WJ, Blanchard DK. 1993. Function associated with IL-2 receptor-beta on human neutrophils. Mechanism of activation of antifungal activity against Candida albicans by IL-2. J Immunol. 150(3):960–970. doi:10.4049/jimmunol.150.3.960.
- Downie GH, Ryan US, Hayes BA, Friedman M. 1992. Interleukin-2 directly increases albumin permeability of bovine and human vascular endothelium in vitro. Am J Respir Cell Mol Biol. 7(1):58–65. doi:10.1165/ajrcmb/7.1.58.
- D'Souza WN, Lefrançois L. 2003. IL-2 is not required for the initiation of CD8 T cell cycling but sustains expansion. J Immunol. 171(11):5727–5735. doi:10.4049/jimmunol.171.11.5727.
- D'Souza WN, Schluns KS, Masopust D, Lefrançois L. 2002. Essential role for IL-2 in the Regulation of antiviral extralymphoid CD8 T cell responses. J Immunol. 168(11):5566–5572. doi:10.4049/jimmunol.168.11.5566.
- Du Y, Li N, Yang H, Luo C, Gong Y, Tong C, Gao Y, Lü S, Long M. 2017. Mimicking liver sinusoidal structures and functions using a 3D-configured microfluidic chip. Lab Chip. 17(5):782–794. doi:10.1039/c6lc01374k.
- Dutcher J, Atkins MB, Margolin K, Weiss G, Clark J, Sosman J, Logan T, Aronson F, Mier J. 2001. Kidney cancer: the cytokine working group experience (1986–2001). Med Oncol. 18(3):209–219. doi:10.1385/MO:18:3:209.
- Dutcher JP, Schwartzentruber DJ, Kaufman HL, Agarwala SS, Tarhini AA, Lowder JN, Atkins MB. 2014. High dose interleukin-2 (Aldesleukin) - expert consensus on best management practices-2014. J Immunother Cancer. 2(1):26. doi:10.1186/s40425-014-0026-0.
- Eksteen B, Miles A, Curbishley SM, Tselepis C, Grant AJ, Walker LSK, Adams DH. 2006. Epithelial inflammation is associated with CCL28 production and the recruitment of regulatory T cells expressing CCR10. J Immunol. 177(1):593–603. doi:10.4049/jimmunol.177.1.593.
- Emoto M, Mittrücker HW, Schmits R, Mak TW, Kaufmann SH. 1999. Critical role of leukocyte function-associated antigen-1 in liver accumulation of CD4 + NKT cells. J Immunol. 162(9):5094–5098.
- Espinoza‐Delgado I, Bosco MC, Musso T, Gusella GL, Longo DL, Varesio L. 1995. Interleukin‐2 and human monocyte activation. J Leukoc Biol. 57(1):13–19. doi:10.1002/jlb.57.1.13.
- Espinoza‐Delgado I, Longo D, Gusella G, Varesio L. 1992. Regulation of IL-2 receptor subunit genes in human monocytes. Differential effects of IL-2 and IFN-gamma. J Immunol. 149(9):2961–2968.
- Espinoza-Delgado I, Longo DL, Gusella GL, Varesio L. 1990. IL-2 enhances c-fms expression in human monocytes. J Immunol. 145(4):1137–1143.
- Esser MT, Dinglasan RD, Krishnamurthy B, Gullo CA, Graham MB, Braciale VL. 1997. IL-2 induces Fas ligand/Fas (CD95L/CD95) cytotoxicity in CD8+ and CD4+ T lymphocyte clones. J Immunol. 158(12):5612–5618.
- Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, et al. 2013. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics*. Mol Cell Proteomics. 13(2):397–406. doi:10.1074/mcp.M113.035600.
- Fisher B, Keenan AM, Garra BS, Steinberg SM, White DE, DiBisceglie AM, Hoofnagle JH, Yolles P, Rosenberg SA, Lotze MT. 1989. Interleukin-2 induces profound reversible cholestasis: a detailed analysis in treated cancer patients. J Clin Oncol. 7(12):1852–1862. doi:10.1200/JCO.1989.7.12.1852.
- Fontana RJ. 2014. Pathogenesis of idiosyncratic drug-induced liver injury and clinical perspectives. Gastroenterology. 146(4):914–928. doi:10.1053/j.gastro.2013.12.032.
- Francis P, Navarro VJ. 2022. Drug induced hepatotoxicity. St. Petersburg (FL): StatPearls Publishing.
- French JB, Bonacini M, Ghabril M, Foureau D, Bonkovsky HL. 2016. Hepatotoxicity associated with the use of anti-TNF-α agents. Drug Saf. 39(3):199–208. doi:10.1007/s40264-015-0366-9.
- Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, Louie AC. 1995. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. 13(3):688–696. doi:10.1200/JCO.1995.13.3.688.
- Gantner F, Leist M, Küsters S, Vogt K, Volk HD, Tiegs G. 1996. T cell stimulus-induced crosstalk between lymphocytes and liver macrophages results in augmented cytokine release. Exp Cell Res. 229(1):137–146. doi:10.1006/excr.1996.0351.
- Gao B, Radaeva S, Park O. 2009. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 86(3):513–528. doi:10.1189/jlb.0309135.
- Geissmann F, Cameron TO, Sidobre S, Manlongat N, Kronenberg M, Briskin MJ, Dustin ML, Littman DR. 2005. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 3(4):e113. doi:10.1371/journal.pbio.0030113.
- Gerussi A, Natalini A, Antonangeli F, Mancuso C, Agostinetto E, Barisani D, Di Rosa F, Andrade R, Invernizzi P. 2021. Immune-mediated drug-induced liver injury: immunogenetics and experimental models. Int J Mol Sci. 22(9):4557. doi:10.3390/ijms22094557.
- Gijbels E, Vinken M. 2017. An update on adverse outcome pathways leading to liver injury. Appl in Vitro Toxicol. 3(4):283–285. doi:10.1089/aivt.2017.0027.
- Girard D, Gosselin J, Heitz D, Paquin R, Beaulieu AD. 1995. Effects of interleukin-2 on gene expression in human neutrophils. Blood. 86(3):1170–1176.
- Goebel J, Forrest K, Morford L, Roszman TL. 2002. Differential localization of IL-2- and -15 receptor chains in membrane rafts of human T cells. Journal of Leukocyte Biology. 72(1):199–206. doi:10.1189/jlb.72.1.199.
- Grant AJ, Roessler E, Ju G, Tsudo M, Sugamura K, Waldmann TA. 1992. The interleukin 2 receptor (IL-2R): the IL-2R alpha subunit alters the function of the IL-2R beta subunit to enhance IL-2 binding and signaling by mechanisms that do not require binding of IL-2 to IL-2R alpha subunit. Proc Natl Acad Sci U S A. 89(6):2165–2169. doi:10.1073/pnas.89.6.2165.
- Grivennikov SI, Tumanov AV, Liepinsh DJ, Kruglov AA, Marakusha BI, Shakhov AN, Murakami T, Drutskaya LN, Förster I, Clausen BE, et al. 2005. Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: protective and deleterious effects. Immunity. 22(1):93–104. doi:10.1016/j.immuni.2004.11.016.
- Guidotti LG, Iannacone M. 2013. Effector CD8 T cell trafficking within the liver. Mol Immunol. 55(1):94–99. doi:10.1016/j.molimm.2012.10.032.
- Guidotti LG, Inverso D, Sironi L, Di Lucia P, Fioravanti J, Ganzer L, Fiocchi A, Vacca M, Aiolfi R, Sammicheli S, et al. 2015. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell. 161(3):486–500. doi:10.1016/j.cell.2015.03.005.
- Heesch K, Raczkowski F, Schumacher V, Hünemörder S, Panzer U, Mittrücker H-W. 2014. The function of the chemokine receptor CXCR6 in the T cell response of mice against Listeria monocytogenes. PLoS One. 9(5):e97701. doi:10.1371/journal.pone.0097701.
- Hémar A, Subtil A, Lieb M, Morelon E, Hellio R, Dautry-Varsat A. 1995. Endocytosis of interleukin 2 receptors in human T lymphocytes: distinct intracellular localization and fate of the receptor alpha, beta, and gamma chains. J Cell Biol. 129(1):55–64. doi:10.1083/jcb.129.1.55.
- Heymann F, Tacke F. 2016. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 13(2):88–110. doi:10.1038/nrgastro.2015.200.
- Hoofnagle JH, Björnsson ES. 2019. Drug-induced liver injury - types and phenotypes. N Engl J Med. 381(3):264–273. doi:10.1056/NEJMra1816149.
- Hou R, Goloubeva O, Neuberg DS, Strominger JL, Wilson SB. 2003. Interleukin-12 and interleukin-2-induced invariant natural killer T-cell cytokine secretion and perforin expression independent of T-cell receptor activation. Immunology. 110(1):30–37. doi:10.1046/j.1365-2567.2003.01701.x.
- Hsu PS, Lai CL, Hu M, Santner-Nanan B, Dahlstrom JE, Lee CH, Ajmal A, Bullman A, Arbuckle S, Al Saedi A, et al. 2018. IL-2 enhances gut homing potential of human naive regulatory T cells early in life. J Immunol. 200(12):3970–3980. doi:10.4049/jimmunol.1701533.
- Hudspeth K, Donadon M, Cimino M, Pontarini E, Tentorio P, Preti M, Hong M, Bertoletti A, Bicciato S, Invernizzi P, et al. 2016. Human liver-resident CD56(bright)/CD16(neg) NK cells are retained within hepatic sinusoids via the engagement of CCR5 and CXCR6 pathways. J Autoimmun. 66:40–50. doi:10.1016/j.jaut.2015.08.011.
- Ishiyama K, Ohdan H, Ohira M, Mitsuta H, Arihiro K, Asahara T. 2006. Difference in cytotoxicity against hepatocellular carcinoma between liver and periphery natural killer cells in humans. Hepatology. 43(2):362–372. doi:10.1002/hep.21035.
- Iyoda T, Ushida M, Kimura Y, Minamino K, Hayuka A, Yokohata S, Ehara H, Inaba K. 2010. Invariant NKT cell anergy is induced by a strong TCR-mediated signal plus co-stimulation. Int Immunol. 22(11):905–913. doi:10.1093/intimm/dxq444.
- James AM, Hsu H-T, Dongre P, Uzel G, Mace EM, Banerjee PP, Orange JS. 2013. Rapid activation receptor- or IL-2-induced lytic granule convergence in human natural killer cells requires Src, but not downstream signaling. Blood. 121(14):2627–2637. doi:10.1182/blood-2012-06-437012.
- Janas ML, Groves P, Kienzle N, Kelso A. 2005. IL-2 regulates perforin and granzyme gene expression in CD8+ T cells independently of its effects on survival and proliferation. J Immunol. 175(12):8003–8010. doi:10.4049/jimmunol.175.12.8003.
- Jiang Y, Qin S, Wei X, Liu X, Guan J, Zhu H, Chang G, Chen Y, Lu H, Qian J, et al. 2021. Highly activated TRAIL + CD56bright NK cells are associated with the liver damage in HBV-LC patients. Immunol Lett. 232:9–19. doi:10.1016/j.imlet.2020.12.008.
- John B, Crispe IN. 2004. Passive and active mechanisms trap activated CD8+ T cells in the liver. J Immunol. 172(9):5222–5229. doi:10.4049/jimmunol.172.9.5222.
- Jukes J-P, Wood KJ, Jones ND. 2012. Bystander activation of iNKT cells occurs during conventional T-cell alloresponses. Am J Transplant. 12(3):590–599. doi:10.1111/j.1600-6143.2011.03847.x.
- Kano A, Watanabe Y, Takeda N, Aizawa S, Akaike T. 1997. Analysis of IFN-gamma-induced cell cycle arrest and cell death in hepatocytes. J Biochem. 121(4):677–683. doi:10.1093/oxfordjournals.jbchem.a021639.
- Kasahara T, Hooks JJ, Dougherty SF, Oppenheim JJ. 1983. Interleukin 2-mediated immune interferon (IFN-gamma) production by human T cells and T cell subsets. J Immunol. 130(4):1784–1789. doi:10.4049/jimmunol.130.4.1784.
- Kashuba ADM, Hawke RL, Tonkin J, LeCluyse EL. 2000. Direct exposure to interleukins (IL)-2, -10 and -12 does not significantly suppress CYP3A activity in human hepatocytes. American Society for Clinical Pharmacology and Therapeutics Annual Meeting, Los Angeles, CA; 67:100.
- Katz SC, Pillarisetty VG, Bleier JI, Shah AB, DeMatteo RP. 2004. Liver sinusoidal endothelial cells are insufficient to activate T cells. J Immunol. 173(1):230–235. doi:10.4049/jimmunol.173.1.230.
- Kennedy NJ, Russell JQ, Michail N, Budd RC. 2001. Liver damage by infiltrating CD8+ T cells is Fas dependent. J Immunol. 167(11):6654–6662. doi:10.4049/jimmunol.167.11.6654.
- Kikuchi-Maki A, Yusa S, Catina TL, Campbell KS. 2003. KIR2DL4 is an IL-2-regulated NK cell receptor that exhibits limited expression in humans but triggers strong IFN-gamma production. J Immunol. 171(7):3415–3425. doi:10.4049/jimmunol.171.7.3415.
- Kleiner DE. 2014. Liver histology in the diagnosis and prognosis of drug-induced liver injury. Clin Liver Dis. 4(1):12–16. doi:10.1002/cld.371.
- Klempner MS, Noring R, Mier JW, Atkins MB. 1990. An acquired chemotactic defect in neutrophils from patients receiving interleukin-2 immunotherapy. N Engl J Med. 322(14):959–965. doi: 10.1056/NEJM199004053221404.
- Klugewitz K, Blumenthal-Barby F, Eulenburg K, Emoto M, Hamann A. 2004. The spectrum of lymphoid subsets preferentially recruited into the liver reflects that of resident populations. Immunol Lett. 93(2-3):159–162. doi:10.1016/j.imlet.2004.03.007.
- Klugewitz K, Topp SA, Dahmen U, Kaiser T, Sommer S, Kury E, Hamann A. 2002. Differentiation-dependent and subset-specific recruitment of T-helper cells into murine liver. Hepatology. 35(3):568–578. doi:10.1053/jhep.2002.31310.
- Knapen D, Vergauwen L, Villeneuve DL, Ankley GT. 2015. The potential of AOP networks for reproductive and developmental toxicity assay development. Reprod Toxicol. 56:52–55. doi:10.1016/j.reprotox.2015.04.003.
- Knolle PA, Schmitt E, Jin S, Germann T, Duchmann R, Hegenbarth S, Gerken G, Lohse A. 1999. Induction of cytokine production in naive CD4+ T cells by antigen-presenting murine liver sinusoidal endothelial cells but failure to induce differentiation toward Th1 cells. Gastroenterology. 116(6):1428–1440. doi:10.1016/S0016-5085(99)70508-1.
- Kogure T, Fujinaga H, Niizawa A, Hai LX, Shimada Y, Ochiai H, Terasawa K. 1999. Killer-cell inhibitory receptors, CD158a/b, are upregulated by interleukin-2, but not interferon-gamma or interleukin-4. Mediators Inflamm. 8(6):313–318. doi:10.1080/09629359990324.
- Kolios G, Valatas V, Kouroumalis E. 2006. Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol. 12(46):7413–7420. doi:10.3748/wjg.v12.i46.7413.
- Konjević G, Mirjačić Martinović K, Vuletić A, Radenković S. 2010. Novel aspects of in vitro IL-2 or IFN-α enhanced NK cytotoxicity of healthy individuals based on NKG2D and CD161 NK cell receptor induction. Biomed Pharmacother. 64(10):663–671. doi:10.1016/j.biopha.2010.06.013.
- Kubes P, Jenne C. 2018. Immune responses in the liver. Annu Rev Immunol. 36(1):247–277. doi:10.1146/annurev-immunol-051116-052415.
- Labuda T, Wendt J, Hedlund G, Dohlsten M. 1998. ICAM-1 costimulation induces IL-2 but inhibits IL-10 production in superantigen-activated human CD4+ T cells. Immunology. 94(4):496–502. doi:10.1046/j.1365-2567.1998.00540.x.
- Lalor PF, Edwards S, McNab G, Salmi M, Jalkanen S, Adams DH. 2002. Vascular adhesion protein-1 mediates adhesion and transmigration of lymphocytes on human hepatic endothelial cells. J Immunol. 169(2):983–992. doi:10.4049/jimmunol.169.2.983.
- Lalor PF, Sun PJ, Weston CJ, Martin‐Santos A, Wakelam MJO, Adams DH. 2007. Activation of vascular adhesion protein‐1 on liver endothelium results in an NF‐κB–dependent increase in lymphocyte adhesion. Hepatology. 45(2):465–474. doi:10.1002/hep.21497.
- Lentsch AB, Miller FN, Edwards MJ. 1997. Interleukin-2-induced hepatic injury involves temporal patterns of cell adhesion in the microcirculation. Am J Physiol. 272(4 Pt 1):G727–31. doi:10.1152/ajpgi.1997.272.4.G727.
- Levental I, Levental KR, Heberle FA. 2020. Lipid rafts: controversies resolved, mysteries remain. Trends Cell Biol. 30(5):341–353. doi:10.1016/j.tcb.2020.01.009.
- Li J, Gyorffy S, Lee S, Kwok CS. 1996. Effect of recombinant human interleukin 2 on neutrophil adherence to endothelial cells in vitro. Inflammation. 20(4):361–372. doi:10.1007/BF01486739.
- Li X, Tang J, Mao Y. 2022. Incidence and risk factors of drug-induced liver injury. Liver Int. 42(9):1999–2014. doi:10.1111/liv.15262.
- Li Y, Strick-Marchand H, Lim AI, Ren J, Masse-Ranson G, Dan L, Jouvion G, Rogge L, Lucas S, Bin L, Di Santo JP. Regulatory T cells control toxicity in a humanized model of IL-2 therapy. Nat Commun. 2017;8(1):1762. doi:10.1038/s41467-017-01570-9.
- Liao W, Lin J-X, Leonard WJ. 2011a. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol. 23(5):598–604. doi:10.1016/j.coi.2011.08.003.
- Liao W, Lin J-X, Wang L, Li P, Leonard WJ. 2011b. Cytokine receptor modulation by interleukin-2 broadly regulates T helper cell lineage differentiation. Nat Immunol. 12(6):551–559. doi:10.1038/ni.2030.
- Liao W, Schones DE, Oh J, Cui Y, Cui K, Tae-Young R, Zhao K, Leonard WJ. 2008. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of IL-4 receptor α chain expression. Nat Immunol. 9(11):1288–1296. doi:10.1038/ni.1656.
- Lim CK, Abolhassani H, Appelberg SK, Sundin M, Hammarström L. 2019. IL2RG hypomorphic mutation: identification of a novel pathogenic mutation in exon 8 and a review of the literature. Allergy Asthma Clin Immunol. 15(1):2. doi:10.1186/s13223-018-0317-y.
- Loetscher P, Seitz M, Baggiolini M, Moser B. 1996. Interleukin-2 regulates CC chemokine receptor expression and chemotactic responsiveness in T lymphocytes. J Exp Med. 184(2):569–577. doi:10.1084/jem.184.2.569.
- López S, García-Serrano S, Gutierrez-Repiso C, Rodríguez-Pacheco F, Ho-Plagaro A, Santiago-Fernandez C, Alba G, Cejudo-Guillen M, Rodríguez-Cañete A, Valdes S, et al. 2018. Tissue-Specific Phenotype and Activation of iNKT Cells in Morbidly Obese Subjects: interaction with Adipocytes and Effect of Bariatric Surgery. Obes Surg. 28(9):2774–2782. doi:10.1007/s11695-018-3215-y.
- Lu R-J, Zhang Y, Tang F-L, Zheng Z-W, Fan Z-D, Zhu S-M, Qian X-F, Liu N-N. 2016. Clinical characteristics of drug-induced liver injury and related risk factors. Exp Ther Med. 12(4):2606–2616. doi:10.3892/etm.2016.3627.
- Mäenpää A, Jääskeläinen J, Carpén O, Patarroyo M, Timonen T. 1993. Expression of integrins and other adhesion molecules on NK cells; impact of IL-2 on short- and long-term cultures. Int J Cancer. 53(5):850–855. doi:10.1002/ijc.2910530524.
- Mahmoudpour SH, Jankowski M, Valerio L, Becker C, Espinola-Klein C, Konstantinides S, Quitzau K, Barco S. 2019. Safety of low-dose subcutaneous recombinant interleukin-2: systematic review and meta-analysis of randomized controlled trials. Sci Rep. 9(1):7145. doi:10.1038/s41598-019-43530-x.
- Malek TR. 2008. The biology of interleukin-2. Annu Rev Immunol. 26(1):453–479. doi:10.1146/annurev.immunol.26.021607.090357.
- Marmor MD, Julius M. 2001. Role for lipid rafts in regulating interleukin-2 receptor signaling. Blood. 98(5):1489–1497. doi:10.1182/blood.V98.5.1489.
- Martrus G, Kautz T, Lunemann S, Richert L, Glau L, Salzberger W, Goebels H, Langeneckert A, Hess L, Poch T, et al. 2017. Proliferative capacity exhibited by human liver-resident CD49a + CD25+ NK cells. PLoS One. 12(8):e0182532. doi:10.1371/journal.pone.0182532.
- Masson MJ, Carpenter LD, Graf ML, Pohl LR. 2008. Pathogenic role of natural killer T and natural killer cells in acetaminophen-induced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology. 48(3):889–897. doi:10.1002/hep.22400.
- McDermott DF, Regan MM, Clark JI, Flaherty LE, Weiss GR, Logan TF, Kirkwood JM, Gordon MS, Sosman JA, Ernstoff MS, et al. 2005. Randomized phase III trial of high-dose interleukin-2 versus subcutaneous interleukin-2 and interferon in patients with metastatic renal cell carcinoma. J Clin Oncol. 23(1):133–141. doi:10.1200/JCO.2005.03.206.
- Mehal WZ, Azzaroli F, Crispe IN. 2001. Antigen presentation by liver cells controls intrahepatic T cell trapping, whereas bone marrow-derived cells preferentially promote intrahepatic T cell apoptosis. J Immunol. 167(2):667–673. doi:10.4049/jimmunol.167.2.667.
- Mehal WZ, Juedes AE, Crispe IN. 1999. Selective retention of activated CD8+ T cells by the normal liver. J Immunol. 163(6):3202–3210. doi:10.4049/jimmunol.163.6.3202.
- Meunier L, Larrey D. 2019. Drug-induced liver injury: biomarkers, requirements, candidates, and validation. Front Pharmacol. 10:1482. doi:10.3389/fphar.2019.01482.
- Mier JW, Dinarello CA, Atkins MB, Punsal PI, Perlmutter DH. 1987. Regulation of hepatic acute phase protein synthesis by products of interleukin 2 (IL 2)-stimulated human peripheral blood mononuclear cells. J Immunol. 139(4):1268–1272.
- Moon B-I, Kim TH, Seoh J-Y. 2015. Functional modulation of regulatory T cells by IL-2. PLoS One. 10(11):e0141864. doi:10.1371/journal.pone.0141864.
- Nakagawa K, Miller FN, Sims DE, Lentsch AB, Miyazaki M, Edwards MJ. 1996. Mechanisms of interleukin-2-induced hepatic toxicity. Cancer Res. 56(3):507–510.
- Nguyen TV, Ukairo O, Khetani SR, McVay M, Kanchagar C, Seghezzi W, Ayanoglu G, Irrechukwu O, Evers R. 2015. Establishment of a hepatocyte-Kupffer cell coculture model for assessment of proinflammatory cytokine effects on metabolizing enzymes and drug transporters. Drug Metab Dispos. 43(5):774–785. doi:10.1124/dmd.114.061317.
- Norris S, Collins C, Doherty DG, Smith F, McEntee G, Traynor O, Nolan N, Hegarty J, O'Farrelly C. 1998. Resident human hepatitis lymphocytes are phenotypically different from circulating lymphocytes. J Hepatol. 28(1):84–90. doi:10.1016/S0168-8278(98)80206-7.
- Numerof RP, Kotik AN, Dinarello CA, Mier JW. 1990. Pro-interleukin-1β production by a subpopulation of human T cells, but not NK cells, in response to interleukin-2. Cell Immunol. 130(1):118–128. doi:10.1016/0008-8749(90)90166-O.
- OECD Series on Adverse Outcome Pathways. 2018. doi:10.1787/5jlv1m9d1g32-en.
- Oestreich KJ, Mohn SE, Weinmann AS. 2012. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat Immunol. 13(4):405–411. doi:10.1038/ni.2242.
- Ohta A, Sekimoto M, Sato M, Koda T, Nishimura S, Iwakura Y, Sekikawa K, Nishimura T. 2000. Indispensable role for TNF-alpha and IFN-gamma at the effector phase of liver injury mediated by Th1 cells specific to hepatitis B virus surface antigen. J Immunol. 165(2):956–961. doi:10.4049/jimmunol.165.2.956.
- Osada T, Morse MA, Lyerly HK, Clay TM. 2005. Ex vivo expanded human CD4+ regulatory NKT cells suppress expansion of tumor antigen-specific CTLs. Int Immunol. 17(9):1143–1155. doi:10.1093/intimm/dxh292.
- Overwijk WW, Tagliaferri MA, Zalevsky J. 2021. Engineering IL-2 to give new life to T cell immunotherapy. Annu Rev Med. 72(1):281–311. doi:10.1146/annurev-med-073118-011031.
- Patten DA, Wilson GK, Bailey D, Shaw RK, Jalkanen S, Salmi M, Rot A, Weston CJ, Adams DH, Shetty S. 2016. Human liver sinusoidal endothelial cells promote intracellular crawling of lymphocytes during recruitment: a new step in migration. Hepatology. 65(1):294–309. doi:10.1002/hep.28879.
- Pedersen AE, Lauritsen JP. 2009. CD25 shedding by human natural occurring CD4 + CD25+ regulatory T cells does not inhibit the action of IL-2. Scand J Immunol. 70(1):40–43. doi:10.1111/j.1365-3083.2009.02268.x.
- Perna A, Yang X, Li L. 2009. Effects of IL-2 and IL-15 on CXCR6 expression and cytokine production by human lymphocytes. J Immunol. 182(1_Supplement):98.26. doi:10.4049/jimmunol.182.Supp.98.26.
- Polentarutti N, Allavena P, Bianchi G, Giardina G, Basile A, Sozzani S, Mantovani A, Introna M. 1997. IL-2-regulated expression of the monocyte chemotactic protein-1 receptor (CCR2) in human NK cells: characterization of a predominant 3.4-kilobase transcript containing CCR2B and CCR2A sequences. J Immunol. 158(6):2689–2694.
- Proleukin® Product Monograph. 2018. Product monograph: proleukin (Aldesleukin). Submission Control No: 217665.
- Ramme AP, Koenig L, Hasenberg T, Schwenk C, Magauer C, Faust D, Lorenz AK, Krebs A-C, Drewell C, Schirrmann K, et al. 2019. Autologous induced pluripotent stem cell-derived four-organ-chip. Future Sci OA. 5(8):FSO413. doi:10.2144/fsoa-2019-0065.
- Robinson MW, Harmon C, O'Farrelly C. 2016. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. 13(3):267–276. doi:10.1038/cmi.2016.3.
- Roifman CM. 2000. Human IL-2 receptor alpha chain deficiency. Pediatr Res. 48(1):6–11. doi:10.1203/00006450-200007000-00004.
- Rollings CM, Sinclair LV, Brady HJM, Cantrell DA, Ross SH. 2018. Interleukin-2 shapes the cytotoxic T cell proteome and immune environment-sensing programs. Sci Signal. 11(526):eaap8112. doi:10.1126/scisignal.aap8112.
- Römer PS, Berr S, Avota E, Na S-Y, Battaglia M, Berge I t, Einsele H, Hünig T. 2011. Preculture of PBMCs at high cell density increases sensitivity of T-cell responses, revealing cytokine release by CD28 superagonist TGN1412. Blood. 118(26):6772–6782. doi:10.1182/blood-2010-12-319780.
- Ross SH, Cantrell DA. 2018. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu Rev Immunol. 36(1):411–433. doi:10.1146/annurev-immunol-042617-053352.
- Rudnicka K, Matusiak A, Chmiela M. 2015. CD25 (IL-2R) expression correlates with the target cell induced cytotoxic activity and cytokine secretion in human natural killer cells. Acta Biochim Pol. 62(4):885–894. doi:10.18388/abp.2015_1152.
- Salmi M, Kalimo K, Jalkanen S. 1993. Induction and function of vascular adhesion protein-1 at sites of inflammation. J Exp Med. 178(6):2255–2260. doi:10.1084/jem.178.6.2255.
- Scheibenbogen C, Keilholz U, Richter M, Andreesen R, Hunstein W. 1992. The interleukin-2 receptor in human monocytes and macrophages: regulation of expression and release of the α and β chains (p55 and p75). Res Immunol. 143(1):33–37. doi:10.1016/0923-2494(92)80077-X.
- Schildberg FA, Wojtalla a, Siegmund SV, Endl E, Diehl L, Abdullah Z, Kurts C, Knolle PA. 2011. Murine hepatic stellate cells veto CD8 T cell activation by a CD54‐dependent mechanism. Hepatology. 54(1):262–272. doi:10.1002/hep.24352.
- Schmidt-Arras D, Rose-John S. 2016. IL-6 pathway in the liver: from physiopathology to therapy. J Hepatol. 64(6):1403–1415. doi:10.1016/j.jhep.2016.02.004.
- Schurich A, Berg M, Stabenow D, Böttcher J, Kern M, Schild H-J, Kurts C, Schuette V, Burgdorf S, Diehl L, et al. 2010. Dynamic regulation of CD8 T cell tolerance induction by liver sinusoidal endothelial cells. J Immunol. 184(8):4107–4114. doi:10.4049/jimmunol.0902580.
- Schwartz RN, Stover L, Dutcher JP. 2002. Managing toxicities of high-dose interleukin-2. Oncology. 11(Suppl 13):11–20.
- Seidler S, Zimmermann HW, Weiskirchen R, Trautwein C, Tacke F. 2012. Elevated circulating soluble interleukin-2 receptor in patients with chronic liver diseases is associated with non-classical monocytes. BMC Gastroenterol. 12(1):38. doi:10.1186/1471-230X-12-38.
- Setoguchi R, Hori S, Takahashi T, Sakaguchi S. 2005. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 201(5):723–735. doi:10.1084/jem.20041982.
- Sharma R, Das A. 2018. IL-2 mediates NK cell proliferation but not hyperactivity. Immunol Res. 66(1):151–157. doi:10.1007/s12026-017-8982-3.
- Shen T, Liu Y, Shang J, Xie Q, Li J, Yan M, Xu J, Niu J, Liu J, Watkins PB, et al. 2019. Incidence and etiology of drug-induced liver injury in Mainland China. Gastroenterology. 156(8):2230–2241.e11. doi:10.1053/j.gastro.2019.02.002.
- Shojaie L, Ali M, Iorga A, Dara L. 2021. Mechanisms of immune checkpoint inhibitor-mediated liver injury. Acta Pharm Sin B. 11(12):3727–3739. doi:10.1016/j.apsb.2021.10.003.
- Simone G d, Andreata F, Bleriot C, Fumagalli V, Laura C, Garcia-Manteiga JM, Di Lucia P, Gilotto S, Ficht X, Ponti FFd, et al. 2021. Identification of a Kupffer cell subset capable of reverting the T cell dysfunction induced by hepatocellular priming. Immunity. 54(9):2089–2100.e8. doi:10.1016/j.immuni.2021.05.005.
- Siwicki M, Gort-Freitas NA, Messemaker M, Bill R, Gungabeesoon J, Engblom C, Zilionis R, Garris C, Gerhard GM, Kohl A, et al. 2021. Resident Kupffer cells and neutrophils drive liver toxicity in cancer immunotherapy. Sci Immunol. 6(61):eabi7083. doi:10.1126/sciimmunol.abi7083.
- Spolski R, Li P, Leonard WJ. 2018. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. 18(10):648–659. doi:10.1038/s41577-018-0046-y.
- Stauber DJ, Debler EW, Horton PA, Smith KA, Wilson IA. 2006. Crystal structure of the IL-2 signaling complex: paradigm for a heterotrimeric cytokine receptor. Proc Natl Acad Sci U S A. 103(8):2788–2793. doi:10.1073/pnas.0511161103.
- Suk KT, Kim DJ, Kim CH, Park SH, Yoon JH, Kim YS, Baik GH, Kim JB, Kweon YO, Kim BI, et al. 2012. A prospective nationwide study of drug-induced liver injury in Korea. Am J Gastroenterol. 107(9):1380–1387. doi:10.1038/ajg.2012.138.
- Sunman JA, Hawke RL, LeCluyse EL, Kashuba ADM. 2004. Kupffer cell-mediated IL-2 suppression of CYP3A activity in human hepatocytes. Drug Metab Dispos. 32(3):359–363. doi:10.1124/dmd.32.3.359.
- Takeda K, Hayakawa Y, van Kaer L, Matsuda H, Yagita H, Okumura K. 2000. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci U S A. 97(10):5498–5503. doi:10.1073/pnas.040566697.
- Taylor AE, Carey AN, Kudira R, Lages CS, Shi T, Lam S, Karns R, Simmons J, Shanmukhappa K, Almanan M, et al. 2018. Interleukin 2 promotes hepatic regulatory T cell responses and protects from biliary fibrosis in murine sclerosing cholangitis. Hepatology. 68(5):1905–1921. doi:10.1002/hep.30061.
- Tinel M, Robin M-A, Doostzadeh J, Maratrat M, Ballet F, Fardel N, Kahwaji JE, Beaune P, Daujat M, Labbe G, et al. 1995. The interleukin-2 receptor down-regulates the expression of cytochrome P450 in cultured rat hepatocytes. Gastroenterology. 109(5):1589–1599. doi:10.1016/0016-5085(95)90648-7.
- Todd JA, Evangelou M, Cutler AJ, Pekalski ML, Walker NM, Stevens HE, Porter L, Smyth DJ, Rainbow DB, Ferreira RC, et al. 2016. Regulatory T cell responses in participants with type 1 diabetes after a single dose of interleukin-2: a non-randomised, open label, adaptive dose-finding trial. PLoS Med. 13(10):e1002139. doi:10.1371/journal.pmed.1002139.
- Toomer KH, Lui JB, Altman NH, Ban Y, Chen X, Malek TR. 2019. Essential and non-overlapping IL-2Rα-dependent processes for thymic development and peripheral homeostasis of regulatory T cells. Nat Commun. 10(1):1037. doi:10.1038/s41467-019-08960-1.
- Tsuchida T, Friedman SL. 2017. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 14(7):397–411. doi:10.1038/nrgastro.2017.38.
- Tu Z, Bozorgzadeh A, Crispe IN, Orloff MS. 2007. The activation state of human intrahepatic lymphocytes. Clin Exp Immunol. 149(1):186–193. doi:10.1111/j.1365-2249.2007.03415.x.
- Umehara H, Takashima A, Minami Y, Bloom ET. 1993. Signal transduction via phosphorylated adhesion molecule, LFA-1 beta (CD18), is increased by culture of natural killer cells with IL-2 in the generation of lymphokine-activated killer cells. Int Immunol. 5(1):19–27. doi:10.1093/intimm/5.1.19.
- Valle MBd, Av Klinteberg V, Alem N, Olsson R, Björnsson E. 2006. Drug-induced liver injury in a Swedish University hospital out-patient hepatology clinic. Aliment Pharmacol Ther. 24(8):1187–1195. doi:10.1111/j.1365-2036.2006.03117.x.
- Varshney P, Yadav V, Saini N. 2016. Lipid rafts in immune signalling: current progress and future perspective. Immunology. 149(1):13–24. doi:10.1111/imm.12617.
- Vega M, Verma M, Beswick D, Bey S, Hossack J, Merriman N, Shah A, Navarro V. 2017. The incidence of drug- and herbal and dietary supplement-induced liver injury: preliminary findings from gastroenterologist-based surveillance in the population of the state of Delaware. Drug Saf. 40(9):783–787. doi:10.1007/s40264-017-0547-9.
- Vickovic S, Eraslan G, Salmén F, Klughammer J, Stenbeck L, Schapiro D, Äijö T, Bonneau R, Bergenstråhle L, Navarro JF, et al. 2019. High-definition spatial transcriptomics for in situ tissue profiling. Nat Methods. 16(10):987–990. doi:10.1038/s41592-019-0548-y.
- Vignali DAA, Collison LW, Workman CJ. 2008. How regulatory T cells work. Nat Rev Immunol. 8(7):523–532. doi:10.1038/nri2343.
- Villeneuve DL, Landesmann B, Allavena P, Ashley N, Bal-Price A, Corsini E, Halappanavar S, Hussell T, Laskin D, Lawrence T, et al. 2018. Representing the process of inflammation as key events in adverse outcome pathways. Toxicol Sci. 163(2):346–352. doi:10.1093/toxsci/kfy047.
- Vinken M. 2020. Liver adverse outcome pathways: what’s in for the hepatologist? J Gastrointestin Liver Dis. 29(4):659–664. doi:10.15403/jgld-2809.
- Wakabayashi K, Lian Z-X, Moritoki Y, Lan RY, Tsuneyama K, Chuang Y-H, Yang G-X, Ridgway W, Ueno Y, Ansari AA, et al. 2006. IL-2 receptor alpha(-/-) mice and the development of primary biliary cirrhosis. Hepatology. 44(5):1240–1249. doi:10.1002/hep.21385.
- Wang KS, Ritz J, Frank DA. 1999. IL-2 induces STAT4 activation in primary NK Cells and NK Cell lines, but not in T Cells. The Journal of Immunology. 162(1):299–304. doi:10.4049/jimmunol.162.1.299.
- Wang Y, Zhang C. 2019. The roles of liver-resident lymphocytes in liver diseases. Front Immunol. 10:1582. doi:10.3389/fimmu.2019.01582.
- Wei S, Blanchard DK, Liu JH, Leonard WJ, Djeu JY. 1993. Activation of tumor necrosis factor-alpha production from human neutrophils by IL-2 via IL-2-R beta. J Immunol. 150(5):1979–1987. doi:10.4049/jimmunol.150.5.1979.
- Wigmore SJ, Fearon KC, Maingay JP, Lai PB, Ross JA. 1997. Interleukin-8 can mediate acute-phase protein production by isolated human hepatocytes. Am J Physiol. 273(4):E720–6. doi:10.1152/ajpendo.1997.273.4.E720.
- Wigmore SJ, Fearon KCH, Maingay JP, Garden OJ, Ross JA. 2002. Effect of interleukin-2 on peripheral blood mononuclear cell cytokine production and the hepatic acute phase protein response. Clin Immunol. 104(2):174–182. doi:10.1006/clim.2002.5244.
- Wilkinson AL, Qurashi M, Shetty S. 2020. The role of sinusoidal endothelial cells in the axis of inflammation and cancer within the liver. Front Physiol. 11:990. doi:10.3389/fphys.2020.00990.
- Wolkers MC, Bensinger SJ, Green DR, Schoenberger SP, Janssen EM. 2011. Interleukin-2 rescues helpless effector CD8+ T cells by diminishing the susceptibility to TRAIL mediated death. Immunol Lett. 139(1–2):25–32. doi:10.1016/j.imlet.2011.04.011.
- Wong J, Johnston B, Lee SS, Bullard DC, Smith CW, Beaudet AL, Kubes P. 1997. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J Clin Invest. 99(11):2782–2790. doi:10.1172/JCI119468.
- Wylezinski LS, Hawiger J. 2016. Interleukin 2 activates brain microvascular endothelial cells resulting in destabilization of adherens junctions*. J Biol Chem. 291(44):22913–22923. doi:10.1074/jbc.M116.729038.
- Yong KSM, Her Z, Tan SY, Tan WWS, Liu M, Lai F, Heng SM, Fan Y, Chang KTE, Wang C-I, et al. 2020. Humanized mouse as a tool to predict immunotoxicity of human biologics. Front Immunol. 11:553362. doi:10.3389/fimmu.2020.553362.
- Young HA, Ortaldo JR. 1987. One-signal requirement for interferon-gamma production by human large granular lymphocytes. J Immunol. 139(3):724–727. doi:10.4049/jimmunol.139.3.724.
- Yuan X, Gajan A, Chu Q, Xiong H, Wu K, Wu GS. 2018. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev. 37(4):733–748. doi:10.1007/s10555-018-9728-y.
- Zen Y, Yeh MM. 2018. Hepatotoxicity of immune checkpoint inhibitors: a histology study of seven cases in comparison with autoimmune hepatitis and idiosyncratic drug-induced liver injury. Mod Pathol. 31(6):965–973. doi:10.1038/s41379-018-0013-y.
- Zhang X, Sun S, Hwang I, Tough DF, Sprent J. 1998. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 8(5):591–599. doi:10.1016/S1074-7613(00)80564-6.
- Zhang Z, Gothe F, Pennamen P, James JR, McDonald D, Mata CP, Modis Y, Alazami AM, Acres M, Haller W, et al. 2019. Human interleukin-2 receptor β mutations associated with defects in immunity and peripheral tolerance. J Exp Med. 216(6):1311–1327. doi:10.1084/jem.20182304.
- Zhao J, Zhang S, Liu Y, He X, Qu M, Xu G, Wang H, Huang M, Pan J, Liu Z, et al. 2020a. Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Discov. 6(1):22. doi:10.1038/s41421-020-0157-z.
- Zhao S, Jiang J, Jing Y, Liu W, Yang X, Hou X, Gao L, Wei L. 2020b. The concentration of tumor necrosis factor-α determines its protective or damaging effect on liver injury by regulating Yap activity. Cell Death Dis. 11(1):70. doi:10.1038/s41419-020-2264-z.
- Zhou W, Zhang F, Aune TM. 2003. Either IL-2 or IL-12 is sufficient to direct Th1 differentiation by nonobese diabetic T cells. J Immunol. 170(2):735–740. doi:10.4049/jimmunol.170.2.735.
- Zimmermann HW, Seidler S, Gassler N, Nattermann J, Luedde T, Trautwein C, Tacke F. 2011. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS One. 6(6):e21381. doi:10.1371/journal.pone.0021381.
- Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, et al. 2006. IL-2 regulates FOXP3 expression in human CD4+ CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 108(5):1571–1579. doi:10.1182/blood-2006-02-004747.