
ABSTRACT
The endoplasmic reticulum (ER) serves as a hub for various cellular processes, and maintaining ER homeostasis is essential for cell function. Reticulophagy is a selective process that removes impaired ER subdomains through autophagy-mediatedlysosomal degradation. While the involvement of ubiquitination in autophagy regulation is well-established, its role in reticulophagy remains unclear. In this study, we screened deubiquitinating enzymes (DUBs) involved in reticulophagy and identified USP20 (ubiquitin specific peptidase 20) as a key regulator of reticulophagy under starvation conditions. USP20 specifically cleaves K48- and K63-linked ubiquitin chains on the reticulophagy receptor RETREG1/FAM134B (reticulophagy regulator 1), thereby stabilizing the substrate and promoting reticulophagy. Remarkably, despite lacking a transmembrane domain, USP20 is recruited to the ER through its interaction with VAPs (VAMP associated proteins). VAPs facilitate the recruitment of early autophagy proteins, including WIPI2 (WD repeat domain, phosphoinositide interacting 2), to specific ER subdomains, where USP20 and RETREG1 are enriched. The recruitment of WIPI2 and other proteins in this process plays a crucial role in facilitating RETREG1-mediated reticulophagy in response to nutrient deprivation. These findings highlight the critical role of USP20 in maintaining ER homeostasis by deubiquitinating and stabilizing RETREG1 at distinct ER subdomains, where USP20 further recruits VAPs and promotes efficient reticulophagy.
Abbreviations: ACTB actin beta; ADRB2 adrenoceptor beta 2; AMFR/gp78 autocrine motility factor receptor; ATG autophagy related; ATL3 atlastin GTPase 3; BafA1 bafilomycin A1; BECN1 beclin 1; CALCOCO1 calcium binding and coiled-coil domain 1; CCPG1 cell cycle progression 1; DAPI 4’,6-diamidino-2-phenylindole; DTT dithiothreitol; DUB deubiquitinating enzyme; EBSS Earle’s Balanced Salt Solution; FFAT two phenylalanines (FF) in an acidic tract; GABARAP GABA type A receptor-associated protein; GFP green fluorescent protein; HMGCR 3-hydroxy-3-methylglutaryl-CoA reductase; IL1B interleukin 1 beta; LIR LC3-interacting region; MAP1LC3/LC3 microtubule associated protein 1 light chain 3; PIK3C3/Vps34 phosphatidylinositol 3-kinase catalytic subunit type 3; RB1CC1/FIP200 RB1 inducible coiled-coil 1; RETREG1/FAM134B reticulophagy regulator 1; RFP red fluorescent protein; RHD reticulon homology domain; RIPK1 receptor interacting serine/threonine kinase 1; RTN3L reticulon 3 long isoform; SEC61B SEC61 translocon subunit beta; SEC62 SEC62 homolog, preprotein translocation factor; SIM super-resolution structured illumination microscopy; SNAI2 snail family transcriptional repressor 2; SQSTM1/p62 sequestosome 1; STING1/MITA stimulator of interferon response cGAMP interactor 1; STX17 syntaxin 17; TEX264 testis expressed 264, ER-phagy receptor; TNF tumor necrosis factor; UB ubiquitin; ULK1 unc-51 like autophagy activating kinase 1; USP20 ubiquitin specific peptidase 20; USP33 ubiquitin specific peptidase 33; VAMP8 vesicle associated membrane protein 8; VAPs VAMP associated proteins; VMP1 vacuole membrane protein 1; WIPI2 WD repeat domain, phosphoinositide interacting 2; ZFYVE1/DFCP1 zinc finger FYVE-type containing 1.
Introduction
Macroautophagy/autophagy is a conservative eukaryotic cellular process that delivers protein aggregates, damaged organelles and invaded pathogens to lysosome for degradation [Citation1–3]. It is essential for survival, differentiation, development and homeostasis, defects of which are linked to various diseases such as neurodegenerative diseases, inflammatory disorders and cancers [Citation4,Citation5]. Autophagy is initiated by the formation of a specific double-membrane organelle called the autophagosome, which engulfs cargo to lysosome for destruction. It is a multistep process involving several key ATG proteins and signaling complexes. Among them, the ULK1 (unc-51 like autophagy activating kinase 1)/ULK2 complex, composed of ULK1/ULK2 kinase, ATG13 (autophagy related 13), RB1CC1/FIP200, and ATG101 (autophagy related 101), is essential for autophagy initiation [Citation6–8].
The endoplasmic reticulum (ER) is a membrane-bound organelle comprising the nuclear envelope and peripheral sheets and tubules. The integrity of ER is essential for protein and lipid synthesis, protein quality control, calcium homeostasis and inter-organelle communication [Citation9,Citation10]. In multicellular organisms, the ER serves as a platform where autophagy-related proteins are recruited for autophagosome formation [Citation11]. The ER-resident tail-anchored VAPs (VAMP associated proteins), VAPA and VAPB, use their MSP domain to interact with the FFAT (two phenylalanines [FF] in an acidic tract) motif present in both RB1CC1 and ULK1. These interactions are crucial for recruiting ULK1 and stabilizing the ULK1 complex at sites where autophagosomes form on the ER [Citation12]. VAPs also interact with WIPI2 (WD repeat domain, phosphoinositide interacting 2) to tether the ER-phagophore for phagophore expansion [Citation12]. The ER is not only a key site for the initiation of autophagy but also a substrate of autophagy [Citation11,Citation13]. To maintain ER homeostasis, certain ER fractions can be selectively degraded through a pathway known as reticulophagy/ER-phagy [Citation13–15]. Mutations in genes encoding reticulophagy components have been associated with specific neurodegenerative diseases [Citation16,Citation17].
Macro-reticulophagy relies on the activation of the reticulophagy receptors, which bridge damaged ER fractions and core autophagy components. Multiple reticulophagy receptors, including RETREG1/FAM134B (reticulophagy regulator 1), RTN3L (reticulon 3 long isoform), ATL3 (atlastin GTPase 3), SEC62 (SEC62 homolog, preprotein translocation factor), CCPG1 (cell cycle progression 1), TEX264 (testis expressed 264, ER-phagy receptor), and CALCOCO1 (calcium binding and coiled-coil domain 1), have been reported in mammals [Citation18–25]. These receptors play distinct roles in recognizing and targeting specific ER subdomains for degradation. Most reticulophagy receptors contain transmembrane domains that locate them on the ER, while only CALCOCO1 is located in the cytosol but can interact with ER-resident proteins, VAPs, to mediate ER fraction degradation [Citation25]. These receptors contain at least one MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) (or GABARAP [GABA type A receptor-associated protein])-interacting region (LIR or GIR) that can bind to Atg8 family proteins. RETREG1 is the first identified receptor responsible for the degradation of sheet ER. It contains reticulon homology domains (RHDs) that comprise two short hairpin-like transmembrane domains, enabling the generation of membrane curvature [Citation18,Citation26]. It has a LIR motif in the C terminus to interact with LC3. Recent research has revealed that RETREG1 undergoes oligomerization prior to binding with LC3 [Citation27]. Additionally, the oligomerization process of RETREG1 is facilitated by phosphorylation and acetylation of RHDs [Citation28]. However, detail molecular mechanisms governing reticulophagy under stress remain insufficiently understood.
Ubiquitination is a critical post-translational modification involved in multiple cellular pathways [Citation29,Citation30]. The ubiquitination process, relying on ATP, involves a cascade of enzymatic steps, starting with activation of ubiquitin by an E1 enzyme, transfer to an E2 enzyme, and finally, facilitation of attachment to the target protein by an E3 enzyme [Citation29]. Different types of polyubiquitin chains mediate distinct signaling pathways, ultimately determining the fate of the substrate protein [Citation29,Citation30]. Conversely, deubiquitinating enzymes (DUBs) catalyze the removal of ubiquitin from target proteins, allowing for precise temporal and spatial control of protein ubiquitination [Citation31]. E3 ubiquitin ligases and DUBs work in concert to regulate the ubiquitin chain assembly on specific substrates, thereby influencing various biological processes in organisms. Recent studies have shown their close association with autophagy, influencing the regulation of various autophagy-related proteins and impacting the autophagy flux [Citation32–40]. However, whether ubiquitination regulates reticulophagy has not been cleared elucidated.
In this study, we screened a DUB library and identified USP20 as a key factor regulating reticulophagy under starvation conditions. USP20 specifically cleaves K48- and K63-linked ubiquitin chain on the reticulophagy receptor RETREG1. The deubiquitination of RETREG1 results in protein stabilization, which subsequently drives reticulophagy to resolve ER stress. Despite lacking a transmembrane domain, USP20 is consistently recruited to the ER through its interaction with ER-resident VAPs. The interaction of USP20 with VAPs facilitates VAPs localization to specific ER subdomains under conditions of nutrient deprivation, where USP20 forms puncta with the reticulophagy receptor RETREG1. Concurrently, VAPs recruit autophagy initiation proteins, including WIPI2, to these subdomains, further enhancing reticulophagy. These findings highlight the crucial role of USP20 in promoting reticulophagy by deubiquitinating RETREG1 at specific ER subdomains. Additionally, USP20 facilitates the assembly of autophagy initiation proteins, thus significantly contributing to the maintenance of ER homeostasis.
Results
USP20 promotes autophagy at multiple steps
To investigate the role of ubiquitin-dependent regulation in autophagy, we conducted an unbiased screen by individually overexpressing each of the DUBs (with 32 DUBs well-expressed) in cells and assessing the changes of LC3B. We were particularly interested in USP20 among the candidate DUBs, as its overexpression led to an increased ratio of LC3B-II to LC3B-I compared to the control under starvation conditions using EBSS medium (Figure S1A-Gi). The RFP-GFP-LC3B serves as an autophagic indicator, enabling the characterization of different stages of the autophagy pathway. It allows the visualization of autophagosomes (RFP+ GFP+, appearing as yellow puncta) and autolysosomes (RFP+ GFP−, appearing as red puncta) due to the unique property of GFP being quenched under acidic conditions [Citation41] (). Compared to the control cells, knockdown of USP20 resulted in a reduction in autophagy flux, as evidenced by an increase in the number of autophagosomes (yellow puncta) and a decrease in the number of autolysosomes (red puncta) when cultured in EBSS (, quantification in 1C-E). Consistently, under starvation conditions, the size of autophagosomes (yellow puncta) was increased in USP20 knockdown cells compared to the control (). Furthermore, when cells with USP20 knockdown were cultured in EBSS supplemented with the lysosomal inhibitor bafilomycin A1 (BafA1), a mild but not significant decrease in the number of autophagosomes and an increase in their size were observed, compared to the control (, quantification in 1C and F). These findings strongly indicate that knockdown of USP20 inhibits autophagy flux. Furthermore, the CRISPR-Cas9-mediated knockout of USP20 resulted in a significant reduction in the number of LC3B puncta and the level of LC3B-II compared to the control, both under basal and starvation conditions (). These results suggest that USP20 has the potential to influence both autophagy flux and autophagy initiation.
Figure 1. Depletion of USP20 impairs the autophagy pathway. (A) The construction of a dual fluorescent RFP-GFP-LC3B enables the characterization of different stages of the autophagy pathway. Autophagosomes are represented in yellow (RFP+ GFP+), while autolysosomes appear in red (RFP+ GFP−) due to the quenching of GFP under acidic conditions in the lysosome. (B) HeLa cells stably expressing RFP-GFP-LC3B were transfected with either scramble shRNA or USP20 shRNA. After 72 h of transfection, cells were treated with EBSS or EBSS+BafA1 (100 nM) for 4 h. Scale bar: 10 μm. (C,D) The number of autophagosomes (GFP+ RFP+, yellow puncta) (C) or autolysosome (RFP+ GFP−, red puncta) (D) per cell from (B) was quantified. (E) the percentage of autophagosomes (yellow) and autolysosomes (red) in a cell under starvation conditions using EBSS medium from (B) was determined. (F) the size of autophagosomes (GFP+ RFP+, yellow puncta) in (B) was quantified. (G) USP20 knockout inhibits autophagy. U-2 OS wild-type and USP20 knockout cells were subjected to DMEM or EBSS treatment for 4 h prior to fixation. Subsequently, cells were immunostained with LC3B (red), and the nuclei were labeled with DAPI (blue). Scale bar: 10 μm. (H) The number of LC3B puncta per cell from (G) was quantified. (I) Immunoblotting was conducted to assess the knockdown efficiency of USP20 and to analyze the ratio of LC3B-II to ACTB under normal and starvation conditions. ACTB was used as a loading control. Statistical analyses were performed on data from three independent experiments, with counts of more than 100 cells. Error bars represent SEM. The significance levels are indicated as n.s., not significant, **p < 0.01, and ***p < 0.001 (one-way ANOVA with Tukey’s test).
To investigate the specific stage of the autophagy process in which USP20 is involved, we analyzed multiple complexes that have functional roles in autophagy regulation. These complexes include the ULK1 complex and the class III PtdIns3K complex involved in autophagy initiation, the LC3 lipidation-related enzymes playing roles in autophagosome formation, specific ER-localized proteins that have been shown to facilitate autophagy initiation, and the SNARE complex mediating the fusion of autophagosome with lysosome. By employing immunoprecipitation to identify USP20 interacting proteins, we discovered that USP20 interacts with various proteins involved in different stages of the autophagy process. It interacts with ATG101 in the ULK1 complex, ATG14 (autophagy related 14) and PIK3C3/Vps34 (phosphatidylinositol 3-kinase catalytic subunit type 3) in the class III PtdIns3K complex, VAPA, VAPB, and VMP1 (vacuole membrane protein 1) on the ER membrane responsible for autophagosome membrane initiation, ATG4B (autophagy related 4B cysteine peptidase) in the enzyme cascade for LC3 lipidation, STX17 (syntaxin 17) and VAMP8 (vesicle associated membrane protein 8) involved in autophagosome and lysosome fusion, as well as the well-known autophagy receptor SQSTM1/p62 (sequestosome 1) (Figure S2A-I). However, under our experimental conditions, we did not detect its interaction with ULK1, BECN1 (beclin 1), WIPI2, or ZFYVE1/DFCP1 (zinc finger FYVE-type containing 1) (Figure S2I). These findings suggest that USP20 may have an extensive role in the regulation of the autophagy pathway.
USP20 participates in the reticulophagy process
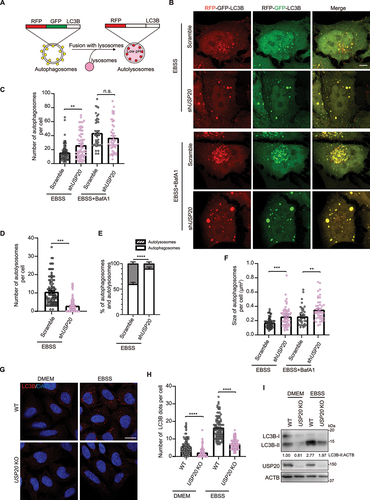
Consistent with previous reports [Citation42], our fluorescence imaging results confirm the colocalization of FLAG-USP20 with the ER marker RFP-SEC61B (SEC61 translocon subunit beta), despite the absence of a transmembrane domain (). This localization pattern suggests a potential association of USP20 with ER-related functions or interactions. Based on this, we further investigated whether USP20 plays a role in regulating reticulophagy, a process involved in the degradation of ER components. To assess the role of USP20 in regulating reticulophagy, we first employed a GFP-SEC61B cleavage strategy. Our results demonstrated a significant decrease in the cleaved GFP band in cells with USP20 knockdown, indicating the inhibition of the reticulophagy upon reduction of USP20 levels (, quantification in 2C). We further employed the ssRFP-GFP-KDEL system to evaluate the involvement of USP20 in regulating reticulophagy [Citation23] (). Under normal conditions, we did not observe a significant amount of red KDEL signal. Prolonged treatment with specific ER stress inducers, such as dithiothreitol (DTT), thapsigargin, or tunicamycin, led to marginal induction of reticulophagy (Figure S3A). However, upon treatment with EBSS, a noticeable increase in red (RFP+ GFP−) KDEL puncta was observed, indicating successful induction of reticulophagy (). Conversely, when USP20 was knocked down, we observed a significant decrease in the number of red (RFP+ GFP−) KDEL puncta and a reduction in the levels of cleaved RFP under starvation conditions, compared to the control (, quantification in 2F, and ). Consistently, overexpression of USP20 resulted in an increased number of red (RFP+ GFP−) KDEL puncta under starvation conditions (Figure S3B, quantification in S3C). These observations strongly support the notion that USP20 positively regulates reticulophagy.
Figure 2. USP20 is involved in the process of reticulophagy. (A) U-2 OS cells were transfected with either the empty vector or FLAG-USP20, along with RFP-SEC61B. After 24 h of transfection, cells were fixed and immunostained with antibody against FLAG (green). Scale bar: 5 μm. (B) U-2 OS cells were transfected with scramble shRNA or USP20 shRNA, along with GFP-SEC61B. After 72 h of transfection, cells were lysed with RIPA buffer, and the lysates were subjected to immunoblotting using the indicated antibodies. ACTB was used as a loading control. (C) The relative fold changes of the ratio of GFP to GFP-SEC61B as shown in (B) are presented as means of three replicate experiments ± SEM. The significance levels are indicated as *p < 0.05 and **p < 0.01 (one-way ANOVA with Tukey’s test). (D) The dual fluorescent ssRFP-EGFP-KDEL was constructed by fusing the ER signal sequence with RFP-EGFP, followed by the ER retention signal KDEL. This construct enables the characterization of different stages of reticulophagy. ER fragments trapped in autophagosomes are represented in yellow (RFP+ GFP+), while those trapped in autolysosomes appear in red (RFP+ GFP−) due to the quenching of GFP under acidic conditions in the lysosome. (E) HeLa cells stably expressing Tet-on ssRFP-GFP-KDEL were transfected with either scramble shRNA or USP20 shRNA. After 48 h, doxycycline (DOX) was added to induce the expression of ssRFP-GFP-KDEL. After 24 h of induction, cells were treated with DMEM or EBSS for 9 h. Cells were then fixed for fluorescence detection. Scale bar: 5 μm. (F) The number of red (RFP+ GFP−) KDEL puncta per cell was quantified from (E). Statistical analyses were performed on data from three independent experiments, with counts of more than 100 cells. Error bars represent SEM. The significance levels are indicated as n.s., not significant, and ****p < 0.0001 (one-way ANOVA with Tukey’s test). (G) Samples from (E) were subject to immunoblotting with the indicated antibodies to assess the cleavage of ssRFP-GFP-KDEL, resulting in the generation of RFP. ACTB was used as a loading control. LE: long exposure; SE: short exposure.
USP20 specifically mediates the deubiquitination of the reticulophagy receptor RETREG1
To investigate the regulatory mechanism of USP20 in reticulophagy, we performed a screening of currently identified reticulophagy receptors to identify specific substrates undergoing deubiquitination by USP20. A denaturing immunoprecipitation assay was conducted to examine changes in the ubiquitination levels of six known ER-localized reticulophagy receptors (RETREG1, ATL3, RTN3L, SEC62, CCPG1 and TEX264) upon overexpression of USP20. The results indicated that RETREG1 is specifically deubiquitinated by USP20, as the ubiquitination level of RETREG1 significantly decreased under conditions of USP20 overexpression (Figure S4A). In contrast, the ubiquitination levels of the other reticulophagy receptors did not exhibit significant changes under the same conditions (Figure S4B-F). This suggests that USP20 specifically targets RETREG1 for deubiquitination, highlighting its selective role in regulating the ubiquitination status of this particular reticulophagy receptor.
Furthermore, the catalytically inactive mutant of USP20, USP20C154S,H643Q, did not result in a decrease in the ubiquitination level of RETREG1 (). Additionally, treatment with the specific USP20 inhibitor GSK2643943A [Citation43] led to elevated levels of RETREG1 ubiquitination compared to the control (). Employing a stable cell line expressing FLAG-GFP-RETREG1 and utilizing FLAG for pulldown of the conjugated ubiquitin signal, we observed an increase in the conjugated ubiquitin signal on RETREG1 upon USP20 knockdown (), aligning with the data obtained from treating cells with the USP20 inhibitor GSK2643943A. Moreover, the immunoprecipitation assay demonstrated an interaction between MYC-USP20 and FLAG-RETREG1, indicating their potential binding capability (). In addition, the endogenous interaction between USP20 and RETREG1 was confirmed through immunoprecipitation experiments using antibodies against RETREG1 (). Therefore, these results suggest that RETREG1 is a substrate of USP20, and that the DUB activity is essential for regulating the ubiquitination of RETREG1.
Figure 3. USP20 interacts with RETREG1 and mediates the deubiquitination of RETREG1. (A) HEK293FT cells were transfected with either the empty vector, MYC-USP20, or MYC-USP20C154S,H643Q, along with FLAG-RETREG1 and HA-UB. After 24 h of transfection, FLAG affinity isolation was performed under denaturing conditions using anti-FLAG magnetic beads, and the immunoprecipitated samples were subjected to immunoblotting analysis using the specific antibodies. The numbers indicate the relative amount of FLAG-RETREG1 signal compared to the control condition. (B) RETREG1 is deubiquitinated by USP20. HEK293FT cells were transfected with FLAG-RETREG1 and HA-UB. The cells were then treated with different concentration of USP20 inhibitor GSK2643943A for 12 h. FLAG affinity isolation was performed under denaturing conditions using anti-FLAG magnetic beads, followed by immunoblotting analysis using the indicated antibodies. (C) USP20 knockdown leads to the accumulation of RETREG1-conjugated ubiquitin signal. HeLa cells stably expressing FLAG-GFP-RETREG1 were established through lentivirus-mediated transfection. Subsequently, the cells were transfected with either scramble shRNA or USP20 shRNA, and FLAG affinity isolation was performed under denaturing conditions. The analysis was conducted by immunoblotting with the indicated antibody. (D) USP20 interacts with RETREG1 in cells. HEK293FT cells were transfected with either the empty vector or MYC-USP20, along with FLAG-RETREG1. MYC affinity isolation was performed using anti-MYC magnetic beads, and the samples were subjected to immunoblotting analysis using the specific antibodies. (E) The endogenous interaction between USP20 and RETREG1. Cell lysates from HEK293FT cells were subjected to immunoprecipitation using antibodies against RETREG1. The immunoprecipitated samples were then analyzed by immunoblotting to detect the presence of USP20. (F) USP20 cleaves both K48- and K63-linked ubiquitin chains from RETREG1. HEK293FT cells were transfected with either the empty vector or MYC-USP20, together with HA-UB or HA-UB[K48 only] or HA-UB[K63 only], along with FLAG-RETREG1. FLAG affinity isolation was performed under denaturing conditions using anti-FLAG magnetic beads, and the immunoprecipitated samples were subjected to immunoblotting analysis using the specific antibodies. (G and I) cycloheximide (CHX) chase analysis of FLAG-RETREG1 under control or USP20 overexpression conditions (G), or under scramble shRNA or USP20 shRNA knockdown conditions (I). HEK293FT cells were treated with CHX at a concentration of 100 μg/ml for 0, 4, or 8 h to block protein synthesis. The degradation rate of FLAG-RETREG1 was monitored over time by immunoblotting. (H and J) statistical analysis of the degradation rate of FLAG-RETREG1 in (G and I). Data are mean ± SEM from three independent experiments. (K) The degradation of FLAG-RETREG1 protein is dependent on the proteasome. HEK293FT cells expressing FLAG-RETREG1 were subjected to various treatments including control, MG132 (10 μM)or BafA1 (100 nM) for 12 h. Protein synthesis was blocked using CHX at a concentration of 100 μg/ml for 0, 6, or 12 h. The degradation kinetics of FLAG-RETREG1 were monitored over time using immunoblotting. ACTB was used as the loading control for all immunoblotting analyses. The numbers indicate the relative amount of FLAG-RETREG1 signal compared to the initial time point (0 h), normalized to ACTB.
![Figure 3. USP20 interacts with RETREG1 and mediates the deubiquitination of RETREG1. (A) HEK293FT cells were transfected with either the empty vector, MYC-USP20, or MYC-USP20C154S,H643Q, along with FLAG-RETREG1 and HA-UB. After 24 h of transfection, FLAG affinity isolation was performed under denaturing conditions using anti-FLAG magnetic beads, and the immunoprecipitated samples were subjected to immunoblotting analysis using the specific antibodies. The numbers indicate the relative amount of FLAG-RETREG1 signal compared to the control condition. (B) RETREG1 is deubiquitinated by USP20. HEK293FT cells were transfected with FLAG-RETREG1 and HA-UB. The cells were then treated with different concentration of USP20 inhibitor GSK2643943A for 12 h. FLAG affinity isolation was performed under denaturing conditions using anti-FLAG magnetic beads, followed by immunoblotting analysis using the indicated antibodies. (C) USP20 knockdown leads to the accumulation of RETREG1-conjugated ubiquitin signal. HeLa cells stably expressing FLAG-GFP-RETREG1 were established through lentivirus-mediated transfection. Subsequently, the cells were transfected with either scramble shRNA or USP20 shRNA, and FLAG affinity isolation was performed under denaturing conditions. The analysis was conducted by immunoblotting with the indicated antibody. (D) USP20 interacts with RETREG1 in cells. HEK293FT cells were transfected with either the empty vector or MYC-USP20, along with FLAG-RETREG1. MYC affinity isolation was performed using anti-MYC magnetic beads, and the samples were subjected to immunoblotting analysis using the specific antibodies. (E) The endogenous interaction between USP20 and RETREG1. Cell lysates from HEK293FT cells were subjected to immunoprecipitation using antibodies against RETREG1. The immunoprecipitated samples were then analyzed by immunoblotting to detect the presence of USP20. (F) USP20 cleaves both K48- and K63-linked ubiquitin chains from RETREG1. HEK293FT cells were transfected with either the empty vector or MYC-USP20, together with HA-UB or HA-UB[K48 only] or HA-UB[K63 only], along with FLAG-RETREG1. FLAG affinity isolation was performed under denaturing conditions using anti-FLAG magnetic beads, and the immunoprecipitated samples were subjected to immunoblotting analysis using the specific antibodies. (G and I) cycloheximide (CHX) chase analysis of FLAG-RETREG1 under control or USP20 overexpression conditions (G), or under scramble shRNA or USP20 shRNA knockdown conditions (I). HEK293FT cells were treated with CHX at a concentration of 100 μg/ml for 0, 4, or 8 h to block protein synthesis. The degradation rate of FLAG-RETREG1 was monitored over time by immunoblotting. (H and J) statistical analysis of the degradation rate of FLAG-RETREG1 in (G and I). Data are mean ± SEM from three independent experiments. (K) The degradation of FLAG-RETREG1 protein is dependent on the proteasome. HEK293FT cells expressing FLAG-RETREG1 were subjected to various treatments including control, MG132 (10 μM)or BafA1 (100 nM) for 12 h. Protein synthesis was blocked using CHX at a concentration of 100 μg/ml for 0, 6, or 12 h. The degradation kinetics of FLAG-RETREG1 were monitored over time using immunoblotting. ACTB was used as the loading control for all immunoblotting analyses. The numbers indicate the relative amount of FLAG-RETREG1 signal compared to the initial time point (0 h), normalized to ACTB.](/cms/asset/6eee70c5-60da-4d9d-900c-bb09df676092/kaup_a_2347103_f0003_oc.jpg)
USP20-mediated deubiquitination of RETREG1 facilitates the stabilization of RETREG1
Previous studies have shown that USP20 preferentially hydrolyzes K48- and K63-linked ubiquitin chain and reduces the ubiquitination of HMGCR (3-hydroxy-3-methylglutaryl-CoA reductase), resulting in substrate stabilization, activation and subsequent influence on cholesterol biogenesis within the liver after feeding [Citation43]. To investigate the deubiquitination patterns of USP20 on RETREG1, we utilized ubiquitin variants that specifically form K48 or K63 polyubiquitin linkages (referred to as UB[K48 only] and UB[K63 only], respectively) in the ubiquitination assay. Denaturing immunoprecipitation experiments confirmed that USP20 can remove both K48- and K63-linked ubiquitin chains from RETREG1 (). Subsequent investigations were conducted to explore the involvement of USP20 in the regulation of RETREG1 degradation. Our findings showed that overexpression of USP20 led to a slight increase in RETREG1 levels compared to the control cells, which was dependent on the DUB activity of USP20 (). Since RETREG1 is a protein with high stability [Citation23], its degradation is not easily detectable at the steady-state level. We therefore conducted cycloheximide chase experiments to assess its degradation kinetics. Remarkably, our results indicated that overexpression of USP20 significantly enhanced the stability of RETREG1, whereas knockdown of USP20 led to reduced RETREG1 protein stability (). Treatment with the proteasome inhibitor MG132, but not the lysosome inhibitor BafA1, effectively blocked the degradation of RETREG1, as demonstrated by the accumulation of RETREG1 protein levels compared to untreated cells (). These findings indicate that USP20 participates in the regulation of RETREG1 degradation through the proteasome pathway.
Next, we sought to identify the specific sites where USP20 deubiquitinates RETREG1. RETREG1 has been previously reported to undergo ubiquitination primarily at K160 and K247 [Citation44,Citation45]. We therefore investigated whether mutations in these two lysine residues led to decreased ubiquitination levels and impaired cleavage by USP20. Surprisingly, we observed that mutating K160 and K247 did not result in a significant reduction in RETREG1 ubiquitination levels (Figure S5A-B). Furthermore, USP20 was still able to effectively deubiquitinate RETREG1 even in the presence of these mutations. These findings suggest that there might be other lysine residues or alternative modifications contributing to the ubiquitination and subsequent regulation of RETREG1.
The RETREG1 protein sequence contains 24 lysine residues distributed in the transmembrane RHD domain and the C terminus (Figure S5A). We generated a series of RETREG1 truncation mutants, specifically deleting either the RHD or the C-terminal domain (Figure S5C). Mapping results showed that USP20 was capable of interacting with all the truncation mutants of RETREG1 (Figure S5D). Additionally, denaturing immunoprecipitation experiments provided evidence that USP20 could still deubiquitinate RETREG1 even when its RHD domain or the C-terminal domain was deleted (Figure S5E). These results suggest that RETREG1 has multiple ubiquitination sites, and USP20 exerts comprehensive deubiquitination regulation on RETREG1.
USP20 promotes autophagy by facilitating the interaction between RETREG1 and LC3B
RETREG1 serves as an essential receptor for reticulophagy, playing a critical role in the process by interacting with LC3. Next, we sought to determine whether the increased expression of USP20 affects the interaction between RETREG1 and LC3B. Our results showed a significant increase in the number of LC3B puncta colocalized with RETREG1 in USP20-overexpressing cells compared to control cells, both under normal conditions and EBSS plus BafA1 treatment (, quantification in 4B). In addition, our immunoprecipitation results demonstrated a notable enhancement of the interaction between RETREG1 and LC3B upon overexpression of USP20, while the catalytically inactive mutant USP20C154S,H643Q showed no such effect (). Furthermore, introducing mutations in the two previously identified crucial lysine residues for the ubiquitination of RETREG1 to arginine (RETREG1K160R,K247R) enhanced its binding with LC3B compared to wild-type RETREG1. While the overexpression of USP20 also increased the interaction between RETREG1K160R,K247R and LC3B, the enhancement was not as significant as observed with wild-type RETREG1 (Figure S5F). These data support the idea that the interaction between RETREG1 and LC3B depends on USP20’s deubiquitination activity on RETREG1. Deubiquitination of RETREG1 by USP20 promotes its binding with LC3B and consequently enhances reticulophagy.
Figure 4. USP20 promotes the interaction between RETREG1 and LC3B. (A) HeLa cells were transfected with either the empty vector or FLAG-USP20, along with GFP-RETREG1. Following transfection, the cells were treated with either DMEM or EBSS+BafA1 (100 nM) for 9 h before fixation. Subsequently, cells were immunostained with FLAG (cyan) and LC3B (red), and nuclei were labeled with DAPI (blue). Scale bar: 10 μm. Insets indicate the enlarged area. Scale bar: 2 μm. (B) The number of colocalized LC3B with RETREG1 per cell was quantified. Statistical analyses were performed on data from three independent experiments, with counts of more than 100 cells. Error bars represent SEM. The significance levels are indicated as *p < 0.05 and **p < 0.01 (one-way ANOVA with Tukey’s test). (C) RETREG1 exhibits increased interaction with LC3B upon overexpression of USP20, but not with USP20C154S,H643Q. HEK293FT cells were co-transfected with either the control vector or FLAG-RETREG1, along with the vector, MYC-USP20 or USP20C154S,H643Q. FLAG affinity isolation was performed using anti-FLAG magnetic beads, and the samples were immunoblotted with the indicated antibody. ACTB was used as a loading control. The numbers indicate the ratio of LC3B-II to FLAG. (D) Increased colocalization of USP20 with RETREG1 during starvation. HeLa cells were transfected with FLAG-USP20 and GFP-RETREG1. After transfection, the cells were treated with either DMEM or EBSS for 9 h before fixation. Immunostaining was performed using antibodies against FLAG (red). Z-stack projection of representative images showing the signals of FLAG-USP20 and GFP-RETREG1 was acquired by SIM. Scale bar: 10 μm. Insets indicate magnified orthogonal sectioning views of regions within the boxes. Scale bar: 2 μm. The areas within box I and II are rendered in 3D and displayed from different angles. Scale bar: 0.5 μm.
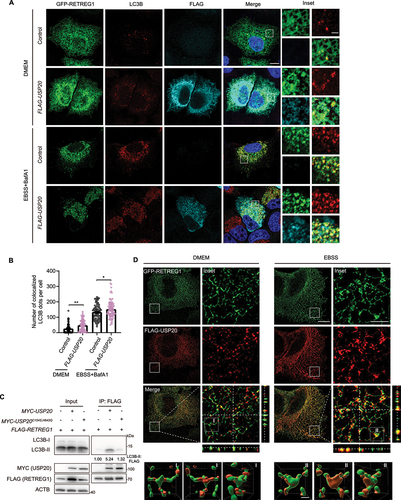
To gain further insights into the localization pattern of RETREG1 and USP20, we employed super-resolution structured illumination microscopy (SIM) to resolve their distribution on the ER. Our observations revealed that both RETREG1 and USP20 exhibit a non-uniform distribution on the ER membrane (). Under normal conditions, USP20 was found to colocalize with RETREG1 specifically in areas where RETREG1 formed punctate structures (). Remarkably, under starvation conditions, we observed an increased colocalization between USP20 and RETREG1, accompanied by a greater number of RETREG1 puncta (). These findings suggest that USP20 plays a role in stabilizing RETREG1 and facilitating the recruitment of LC3B to RETREG1, thereby promoting reticulophagy in response to nutrient deprivation.
VAPs mediate the localization of USP20 to the ER
Since USP20 lacks a transmembrane domain but localizes to the ER, we were intrigued by whether it is anchored to the ER by specific ER proteins. Remarkably, USP20 contains two FFAT motifs, typical motifs known to interact with ER-transmembrane proteins VAPA and VAPB () [Citation46]. We initially generated a USP20-FLAG-GFP knock-in cell line by inserting FLAG-GFP before the stop codon of the USP20 open reading frame via CRISPR-Cas9-mediated homologous recombination (). Immunostaining results indicated its colocalization with VAPB on the ER (). To gain insight into this mechanism, we investigated the interaction between USP20 and VAPs. VAPs have been reported to interact with proteins containing FFAT motifs via their MSP domain, leading to their localization to the ER [Citation47]. Immunoprecipitation experiments provided evidence that the FFAT motifs of USP20 play a central role in mediating its interaction with VAPs (). To further confirm the FFAT-dependent localization of USP20 to the ER, we conducted a microsome fractionation experiment. The results demonstrated that wild-type USP20 predominantly localized to the microsomes derived from the ER, whereas the FFAT1/2-deleted mutant of USP20 was primarily found in the cytosol (). Furthermore, immunofluorescence imaging analysis revealed that upon loss of the FFAT1/2 motifs, USP20 exhibited cytoplasmic localization (). The mutants VAPAK94D,M96D and VAPBK87D,M89D, which lost the interaction with the FFAT motif [Citation12], exhibited decreased interaction with USP20 compared to their wild-type counterparts (). Additionally, in VAPA and VAPB double-knockout cells, USP20 exhibited a higher distribution in the cytosol fraction compared to the control cells (). These findings suggest that VAPs play a crucial role in mediating the ER localization of USP20.
Figure 5. VAPs mediate the localization of USP20 to the ER. (A) The protein sequence of USP20 contains multiple domains, including a zinc finger (ZF) domain, a catalytic USP domain, and two DUSP domains. Two FFAT motifs, located in the USP domain, are conserved among different organisms. Multiple sequence alignment of USP20 proteins from various species was performed using the clustal omega in the European Bioinformatics Institute (EMBL-EBI) (https://www.ebi.ac.uk/Tools/msa/clustalo/). Conserved regions are shaded in grey. (B) The construction of USP20-FLAG-GFP knock-in cell line enables the characterization of endogenous USP20 level. The FLAG-GFP fragment was inserted into the C terminus of USP20 via CRISPR-Cas9-mediated knock-in. (C) Endogenous USP20 colocalized with VAPB. The USP20-FLAG-GFP knock-in cells were fixed and immunostained with antibody against VAPB (red). Scale bar: 10 μm. (D) The FFAT motifs of USP20 are responsible for its interaction with VAPA and VAPB. HEK293FT were transfected with FLAG-USP20 wild-type or its mutants, including FLAG-USP20[ΔFFAT1], FLAG-USP20[ΔFFAT2] or FLAG-USP20[ΔFFAT1/2]. Subsequently, FLAG affinity isolation was performed using anti-FLAG magnetic beads, and the samples were analyzed by immunoblotting with the indicated antibodies. ACTB was used as a loading control. (E) Deleting the FFAT motifs of USP20 results in reduced enrichment in the ER. HEK293FT cells were transfected with either FLAG-USP20 or FLAG-USP20[ΔFFAT1/2]. A microsome fractionation assay was performed to illustrate the localization of USP20 in the ER. The ER marker VAPB was used to indicate the ER fraction, while ACTB was used to indicate the cytosol fraction. (F) FLAG-USP20[ΔFFAT1/2] does not localize to the ER. HeLa cells were transfected with either FLAG-USP20 or FLAG-USP20[ΔFFAT1/2]. After 24 h of transfection, the cells were fixed and immunostained with anti-FLAG antibody (green), and the nuclei were labeled with DAPI (blue). Scale bar: 10 μm. (G) VAPA and VAPB exhibit reduced interaction with USP20 when their FFAT-binding motifs are mutated. HeLa cells were transfected with GFP-VAPA, GFP-VAPAK94,M96D, GFP-VAPB or GFP-VAPBK87D,M89D. After 24 h of transfection, a GFP immunoprecipitation assay was performed, and the samples were analyzed by immunoblotting using the specific antibodies. ACTB was used as a loading control. (H) USP20 exhibits reduced localization to the ER in VAPA and VAPB double-knockout (DKO) cells. A microsome fractionation assay was performed using both HEK293FT wild-type cells and VAPA and VAPB double-knockout cells to evaluate the enrichment of USP20 in the ER. The effectiveness of VAPA and VAPB knockout was confirmed by immunoblotting with VAPA and VAPB antibodies, while ACTB was used as a marker for the cytosol fraction. (I) Localization of USP20 and VAPB on the ER. HeLa cells were transfected with FLAG-USP20. Following transfection, the cells were subjected to DMEM or EBSS treatment for 9 h prior to fixation. Subsequently, cells were immunostained with FLAG (red) and VAPB (green). Z-stack projection of representative images showing the signals of FLAG-USP20 and VAPB was acquired by SIM. Scale bar: 10 μm. Insets indicate magnified orthogonal sectioning views of regions within the boxes. Scale bar: 1 μm.
![Figure 5. VAPs mediate the localization of USP20 to the ER. (A) The protein sequence of USP20 contains multiple domains, including a zinc finger (ZF) domain, a catalytic USP domain, and two DUSP domains. Two FFAT motifs, located in the USP domain, are conserved among different organisms. Multiple sequence alignment of USP20 proteins from various species was performed using the clustal omega in the European Bioinformatics Institute (EMBL-EBI) (https://www.ebi.ac.uk/Tools/msa/clustalo/). Conserved regions are shaded in grey. (B) The construction of USP20-FLAG-GFP knock-in cell line enables the characterization of endogenous USP20 level. The FLAG-GFP fragment was inserted into the C terminus of USP20 via CRISPR-Cas9-mediated knock-in. (C) Endogenous USP20 colocalized with VAPB. The USP20-FLAG-GFP knock-in cells were fixed and immunostained with antibody against VAPB (red). Scale bar: 10 μm. (D) The FFAT motifs of USP20 are responsible for its interaction with VAPA and VAPB. HEK293FT were transfected with FLAG-USP20 wild-type or its mutants, including FLAG-USP20[ΔFFAT1], FLAG-USP20[ΔFFAT2] or FLAG-USP20[ΔFFAT1/2]. Subsequently, FLAG affinity isolation was performed using anti-FLAG magnetic beads, and the samples were analyzed by immunoblotting with the indicated antibodies. ACTB was used as a loading control. (E) Deleting the FFAT motifs of USP20 results in reduced enrichment in the ER. HEK293FT cells were transfected with either FLAG-USP20 or FLAG-USP20[ΔFFAT1/2]. A microsome fractionation assay was performed to illustrate the localization of USP20 in the ER. The ER marker VAPB was used to indicate the ER fraction, while ACTB was used to indicate the cytosol fraction. (F) FLAG-USP20[ΔFFAT1/2] does not localize to the ER. HeLa cells were transfected with either FLAG-USP20 or FLAG-USP20[ΔFFAT1/2]. After 24 h of transfection, the cells were fixed and immunostained with anti-FLAG antibody (green), and the nuclei were labeled with DAPI (blue). Scale bar: 10 μm. (G) VAPA and VAPB exhibit reduced interaction with USP20 when their FFAT-binding motifs are mutated. HeLa cells were transfected with GFP-VAPA, GFP-VAPAK94,M96D, GFP-VAPB or GFP-VAPBK87D,M89D. After 24 h of transfection, a GFP immunoprecipitation assay was performed, and the samples were analyzed by immunoblotting using the specific antibodies. ACTB was used as a loading control. (H) USP20 exhibits reduced localization to the ER in VAPA and VAPB double-knockout (DKO) cells. A microsome fractionation assay was performed using both HEK293FT wild-type cells and VAPA and VAPB double-knockout cells to evaluate the enrichment of USP20 in the ER. The effectiveness of VAPA and VAPB knockout was confirmed by immunoblotting with VAPA and VAPB antibodies, while ACTB was used as a marker for the cytosol fraction. (I) Localization of USP20 and VAPB on the ER. HeLa cells were transfected with FLAG-USP20. Following transfection, the cells were subjected to DMEM or EBSS treatment for 9 h prior to fixation. Subsequently, cells were immunostained with FLAG (red) and VAPB (green). Z-stack projection of representative images showing the signals of FLAG-USP20 and VAPB was acquired by SIM. Scale bar: 10 μm. Insets indicate magnified orthogonal sectioning views of regions within the boxes. Scale bar: 1 μm.](/cms/asset/1464324e-6104-4d0a-a3b2-2338a52bafb5/kaup_a_2347103_f0005_oc.jpg)
We then used SIM to investigate the localization of USP20 and VAPB on the ER membrane. The two proteins displayed an intercalated localization pattern with overlapping signals to some extent under normal conditions, indicating their proximity on the ER membrane (). Upon treatment with EBSS, we observed a specific colocalization of USP20 and VAPB at the sites where USP20 formed puncta, suggesting their close association within the ER (). These findings imply a potential functional interaction between USP20 and VAPB in the regulation of reticulophagy, particularly under starvation conditions.
USP20 facilitates RETREG1-VAPB association and enhances early autophagy protein recruitment for reticulophagy
We used SIM to study the localization of RETREG1 and VAPB on the ER membrane and observed their adjacent localization with partially overlapping signals under normal conditions (). After EBSS treatment, VAPB showed specific colocalization with RETREG1 at the sites where RETREG1 formed puncta (). We then investigated the potential role of VAPs in mediating the deubiquitination of RETREG1 by USP20. Surprisingly, even in the absence of the USP20 FFAT motifs, we observed that USP20 was still able to bind to and deubiquitinate RETREG1 (Figure S6A-B). These findings suggest that while VAPs may play a role in mediating the localization of USP20 to the ER, they may have additional functions beyond facilitating USP20-mediated deubiquitination of RETREG1.
Figure 6. USP20 enhances the interaction between RETREG1 and VAPB on the ER. (A) Localization of RETREG1 and VAPB on the ER. HeLa cells were transfected with GFP-RETREG1. Following transfection, the cells were subjected to DMEM or EBSS treatment for 9 h prior to fixation. Subsequently, cells were immunostained with VAPB (red). Z-stack projection of representative images showing the signals of GFP-RETREG1 and VAPB was acquired by SIM. Scale bar: 10 μm. Insets indicate magnified orthogonal sectioning views of regions within the boxes. Scale bar: 2 μm. (B) USP20 promotes the interaction of RETREG1 with VAPB. HEK293FT cells were co-transfected with either the empty vector or MYC-USP20, along with the empty vector or FLAG-RETREG1. FLAG affinity isolation was performed using anti-FLAG magnetic beads, and the immunoprecipitated samples were subjected to immunoblotting analysis using the specific antibodies. ACTB was used as a loading control. (C) The USP20 FFAT1/2-deleted mutant does not enhance the interaction between RETREG1 and VAPB. HEK293FT cells were co-transfected with either the empty vector or MYC-USP20 or MYC-USP20[ΔFFAT1/2], along with FLAG-RETREG1. FLAG affinity isolation was performed using anti-FLAG magnetic beads, and the immunoprecipitated samples were subjected to immunoblotting analysis using the specific antibodies. ACTB was used as a loading control. (D) Overexpression of USP20 leads to an increase in the colocalization of WIPI2 with RETREG1 under both normal and starvation conditions. HeLa cells were transfected with the control vector or FLAG-USP20, along with GFP-RETREG1. Following transfection, the cells were subjected to DMEM or EBSS treatment for 9 h prior to fixation. Subsequently, cells were immunostained with WIPI2 (red) and FLAG (cyan). Scale bar: 10 μm. Insets indicate the enlarged area. Scale bar: 2 μm. (E and F) the number of WIPI2 dots per cell (E) and the percentage of RETREG1-colocalized WIPI2 (F) from (D) was quantified.
![Figure 6. USP20 enhances the interaction between RETREG1 and VAPB on the ER. (A) Localization of RETREG1 and VAPB on the ER. HeLa cells were transfected with GFP-RETREG1. Following transfection, the cells were subjected to DMEM or EBSS treatment for 9 h prior to fixation. Subsequently, cells were immunostained with VAPB (red). Z-stack projection of representative images showing the signals of GFP-RETREG1 and VAPB was acquired by SIM. Scale bar: 10 μm. Insets indicate magnified orthogonal sectioning views of regions within the boxes. Scale bar: 2 μm. (B) USP20 promotes the interaction of RETREG1 with VAPB. HEK293FT cells were co-transfected with either the empty vector or MYC-USP20, along with the empty vector or FLAG-RETREG1. FLAG affinity isolation was performed using anti-FLAG magnetic beads, and the immunoprecipitated samples were subjected to immunoblotting analysis using the specific antibodies. ACTB was used as a loading control. (C) The USP20 FFAT1/2-deleted mutant does not enhance the interaction between RETREG1 and VAPB. HEK293FT cells were co-transfected with either the empty vector or MYC-USP20 or MYC-USP20[ΔFFAT1/2], along with FLAG-RETREG1. FLAG affinity isolation was performed using anti-FLAG magnetic beads, and the immunoprecipitated samples were subjected to immunoblotting analysis using the specific antibodies. ACTB was used as a loading control. (D) Overexpression of USP20 leads to an increase in the colocalization of WIPI2 with RETREG1 under both normal and starvation conditions. HeLa cells were transfected with the control vector or FLAG-USP20, along with GFP-RETREG1. Following transfection, the cells were subjected to DMEM or EBSS treatment for 9 h prior to fixation. Subsequently, cells were immunostained with WIPI2 (red) and FLAG (cyan). Scale bar: 10 μm. Insets indicate the enlarged area. Scale bar: 2 μm. (E and F) the number of WIPI2 dots per cell (E) and the percentage of RETREG1-colocalized WIPI2 (F) from (D) was quantified.](/cms/asset/3e5d4c32-34aa-44a1-9db0-906eecd42954/kaup_a_2347103_f0006_oc.jpg)
Previous studies have demonstrated that VAPs promote autophagy by facilitating ER-phagophore contact through binding with WIPI2. This interaction enables the recruitment of diverse autophagy proteins, facilitating the formation of the autophagy initiation complex at the ER [Citation12,Citation48]. In our study, we observed an enhanced interaction between RETREG1 and VAPB following the overexpression of USP20 (). Conversely, their interaction was notably diminished in cells expressing the mutant USP20[ΔFFAT1/2], compared to those with wild-type USP20 (). Concurrently, we observed a reduced binding of RETREG1 to LC3B when USP20 was unable to localize to the ER (). Additionally, there was a notable increase in the colocalization of RETREG1 with the early autophagy protein WIPI2 upon USP20 overexpression under normal conditions. While a mild increase in colocalization was observed under starvation conditions, the p-value did not achieve significance (, quantification in 6E-F). These findings suggest that USP20 may enhance the association between RETREG1 and VAPB, thereby facilitating reticulophagy by recruiting more early autophagy proteins to the initiation sites.
We further explored the regulatory implications of USP20-mediated deubiquitination of RETREG1 in the context of reticulophagy. Employing ssRFP-GFP-KDEL as a reporter, we conducted experiments involving USP20 knockdown and subsequent rescue. The results demonstrated that in USP20 knockdown cells, the expression of wild-type USP20 or RETREG1 fully restored reticulophagy function, whereas expression of the catalytically inactive mutant USP20C154S,H643Q or the FFAT1/2-deleted mutant USP20[ΔFFAT1/2] failed to initiate reticulophagy (). These findings underscore the pivotal role of the deubiquitination activity of USP20 in regulating reticulophagy.
Figure 7. The inhibition of reticulophagy caused by USP20 knockdown can be rescued by the expression of RETREG1 or wild-type USP20, but not by the USP20 mutants. (A) The expression of RETREG1 or wild-type USP20, but not the catalytically inactive mutant USP20C154S,H643Q or FFAT1/2-deleted mutant USP20[ΔFFAT1/2], fully rescued reticulophagy function. HeLa cells stably expressing Tet-on ssRFP-GFP-KDEL were transfected with either scramble shRNA or USP20 shRNA, together with the control, FLAG-RETREG1, FLAG-USP20, FLAG-USP20C154S,H643Q or FLAG-USP20[ΔFFAT1/2], respectively. After 48 h, DOX was added to induce the expression of ssRFP-GFP-KDEL. Following 24 h of induction, cells were treated with EBSS for 9 h, and then fixed for fluorescence detection. Scale bar: 5 μm. (B) The number of red (RFP+ GFP−) KDEL puncta per cell was quantified from (A). Statistical analyses were performed on data from three independent experiments, with counts of more than 100 cells. Error bars represent SEM. The significance levels are indicated as n.s., not significant, and ****p < 0.0001 (one-way ANOVA with Tukey’s test). (C) A proposed model elucidating the role of USP20 in regulating RETREG1 deubiquitination and facilitating reticulophagy. USP20 deubiquitinates and stabilizes RETREG1, while also being recruited to the ER by VAPs. Under starvation conditions, USP20 and RETREG1 form puncta, and USP20 further recruits VAPs to these loci. This concerted action leads to the recruitment of early autophagy proteins, including WIPI2, thereby promoting reticulophagy.
![Figure 7. The inhibition of reticulophagy caused by USP20 knockdown can be rescued by the expression of RETREG1 or wild-type USP20, but not by the USP20 mutants. (A) The expression of RETREG1 or wild-type USP20, but not the catalytically inactive mutant USP20C154S,H643Q or FFAT1/2-deleted mutant USP20[ΔFFAT1/2], fully rescued reticulophagy function. HeLa cells stably expressing Tet-on ssRFP-GFP-KDEL were transfected with either scramble shRNA or USP20 shRNA, together with the control, FLAG-RETREG1, FLAG-USP20, FLAG-USP20C154S,H643Q or FLAG-USP20[ΔFFAT1/2], respectively. After 48 h, DOX was added to induce the expression of ssRFP-GFP-KDEL. Following 24 h of induction, cells were treated with EBSS for 9 h, and then fixed for fluorescence detection. Scale bar: 5 μm. (B) The number of red (RFP+ GFP−) KDEL puncta per cell was quantified from (A). Statistical analyses were performed on data from three independent experiments, with counts of more than 100 cells. Error bars represent SEM. The significance levels are indicated as n.s., not significant, and ****p < 0.0001 (one-way ANOVA with Tukey’s test). (C) A proposed model elucidating the role of USP20 in regulating RETREG1 deubiquitination and facilitating reticulophagy. USP20 deubiquitinates and stabilizes RETREG1, while also being recruited to the ER by VAPs. Under starvation conditions, USP20 and RETREG1 form puncta, and USP20 further recruits VAPs to these loci. This concerted action leads to the recruitment of early autophagy proteins, including WIPI2, thereby promoting reticulophagy.](/cms/asset/986afccf-052e-4bc1-875a-b4c8652bd7c1/kaup_a_2347103_f0007_oc.jpg)
Discussion
Our study demonstrates that USP20 deubiquitinates and stabilizes the reticulophagy receptor RETREG1 to facilitate reticulophagy. USP20 has previously been shown to regulate autophagy by deubiquitinating and stabilizing ULK1, thus promoting autophagy [Citation36]. Given the localization of USP20 to the ER, we investigated whether USP20 regulates the deubiquitination of ER-targeted proteins. In this study, we elucidated the recruitment of USP20 to the ER by VAP proteins, its role in deubiquitinating the reticulophagy receptor RETREG1, and its function in stabilizing RETREG1 to promote reticulophagy under starvation conditions ().
Previous studies on USP20 have focused on its activity in metabolic diseases, antiviral immunity and cancer pathogenesis [Citation43,Citation49,Citation50]. For example, the phosphorylated USP20 binds to ER-localized AMFR/gp78 (autocrine motility factor receptor) and stabilizes HMGCR by cleaving K48- and K63-linked ubiquitin chains, resulting in increased cholesterol biosynthesis within the liver after feeding [Citation43]. Furthermore, USP20 plays a pivotal role in cellular antiviral responses by promoting deconjugation of K48-linked ubiquitin chains from STING1/MITA (stimulator of interferon response cGAMP interactor 1), leading to enhanced stability of STING1 and increased expression of type I interferons and proinflammatory cytokines following DNA virus infection [Citation49,Citation50]. USP20 reduces atherogenic signaling in smooth muscle cells by deubiquitinating RIPK1 (receptor interacting serine/threonine kinase 1) in response to TNF (tumor necrosis factor) and IL1B (interleukin 1 beta) stimulation [Citation51]. Moreover, a study reported that USP20 promotes breast cancer metastasis by stabilizing the metastasis-promoting transcription factor SNAI2 (snail family transcriptional repressor 2) [Citation52]. The research findings also revealed a correlation between higher expression of USP20 and worse metastasis-free survival, consistent with the observations made in mice, highlighting the role of USP20 in promoting metastasis [Citation52]. Additional investigations have unveiled USP20’s involvement in the deubiquitination and stabilization of diverse substrates, including ADRB2 (adrenoceptor beta 2) and SQSTM1 [Citation53,Citation54]. In this study, we revealed the novel role of USP20 in cleaving K48 and K63-linked ubiquitin chains on RETREG1, ultimately stabilizing RETREG1 and facilitating the process of reticulophagy. Thus, our findings provided compelling evidence for USP20’s multifaceted engagement in diverse pathways, including reticulophagy, where it exerts its deubiquitinating and substrate-stabilizing effects.
Although USP20 lacks a predicted transmembrane domain, it is localized to the ER. In this study, we identified the pivotal role of the FFAT motifs in mediating USP20’s ER localization. Our experiments revealed that mutations in the FFAT motifs of USP20 disrupted its localization to the ER, underscoring the critical importance of these domains in this process (). Surprisingly, even with the FFAT mutations, USP20 retained its ability to interact with ER-localized RETREG1 and carried out deubiquitination of RETREG1 (Figure S6A-B). The mechanism underlying this unique binding and deubiquitination ability of the USP20 FFAT mutants to ER-localized RETREG1 remains to be elucidated. Furthermore, our investigation has shed light on the interplay between the FFAT motifs of USP20 and VAPs, which serves as a key mechanism for recruiting VAPs to specific ER subdomains (). Through this orchestrated recruitment, USP20 facilitated the subsequent assembly of autophagy initiation proteins at these sites, culminating in the promotion of efficient reticulophagy (). This intricate interplay among USP20, VAPs, and the autophagy machinery emerges as a crucial determinant in maintaining ER homeostasis, unraveling novel insights into the regulation of reticulophagy in response to starvation stress.
Reticulophagy relies on ER-localized receptors that typically present LIR motifs to interact with LC3 and initiate the autophagic process. However, the mechanism of how the phagophore is initiated and extended during reticulophagy has not been previously elucidated. Previous studies have demonstrated that certain ER proteins interact with autophagy initiation proteins, contributing to the regulation of autophagosome biogenesis [Citation12,Citation48]. In the current study, we provide evidence indicating that reticulophagy necessitates a membrane recruitment event, which subsequently provides lipidated LC3 for interaction with the reticulophagy receptor RETREG1. This interaction is essential for the efficient execution of reticulophagy.
During our study, two reports from the Dikic and Hubner labs claimed that RETREG1 ubiquitination promotes clustering, driving ER membrane remodeling, and facilitating reticulophagy flux [Citation55,Citation56]. They identified AMFR as the E3 ubiquitin ligase responsible for ubiquitinating RETREG1. However, the type of ubiquitination chain was not defined. In our study, we found that USP20 deubiquitinates K48- and K63-linked ubiquitin chains on RETREG1 and promotes its stabilization. Furthermore, we observed an enhanced interaction between RETREG1 and LC3B upon USP20 overexpression, whereas the catalytically inactive mutant, USP20C154S,H643Q, did not exhibit the same effect (). All of these data verified the importance of USP20 in promoting reticulophagy through the deubiquitination of RETREG1. Notably, previous studies have shown that USP20 collaborates with USP33 in the efficient removal of K48-linked ubiquitin chains from tail-anchored proteins, following their insertion into the ER. This process rescues membrane proteins that have been ubiquitinated by protein quality control in the cytosolic site, highlighting the essential role of USP20 in maintaining ER homeostasis [Citation42,Citation57]. Based on these findings, it is possible that RETREG1 undergoes ubiquitination/deubiquitination regulation at the early stage of reticulophagy. During this stage, USP20 deubiquitinates RETREG1, stabilizing it and facilitating the recruitment of VAPs, along with early autophagy-related proteins, to the ER subdomain for the initiation of autophagy. Subsequently, after the assembly of the autophagy initiation complex, RETREG1 may experience another round of ubiquitination to promote its clustering. Considering that AMFR is a classical E3 ubiquitin ligase responsible for ER-associated degradation (ERAD), it would be interesting to understand the nature of the ubiquitin chains formed on RETREG1 by AMFR for clustering and driving reticulophagy.
In summary, our findings demonstrate that USP20 plays a crucial role in deubiquitinating and stabilizing the reticulophagy receptor RETREG1 at specific ER subdomains. Additionally, USP20’s interaction with VAPs further enhances its role in promoting efficient reticulophagy.
Materials and methods
Cell culture
HEK293FT (PTA-5077), HeLa (CRM-CCL-2) and U-2 OS (HTB-96) cells obtained from ATCC were cultured in DMEM (Thermo Fisher Scientific 12,800,017) supplemented with 10% fetal bovine serum (Lonsera, S711-001S) and 100 U/ml penicillin G and 100 μg/ml streptomycin (Thermo Fisher Scientific 15,140,148) at 37°C under 5% CO2. For autophagy and reticulophagy induction experiments, cells were subject to Earle’s Balanced Salt Solution (EBSS; Sigma-Aldrich, E2888) for 4 h and 9 h, respectively.
Generating knockout and knock-in cell lines
The knockout cell lines were generated using the CRISPR-Cas9 system. Guide RNAs sequences were designed using the website http://chopchop.cbu.uib.no/. The guide RNAs sequences were inserted into the pGL3-U6-sgRNA-PGK-puromycin plasmid (Addgene 51,133; Xingxu Huang). U-2 OS, HeLa or HEK293FT cells were co-transfected with the sgRNA plasmids and pST1374-NLS-flag-linker-Cas9 (Addgene 44,758; Xingxu Huang). After 24 h, cells were selected with 5 μg/ml puromycin (Sigma-Aldrich 540,411) and 10 μg/ml blasticidin (Thermo Fisher Scientific, R21001) for 7 days. The knockout of the target gene was confirmed by immunoblotting. Single-cell colonies were then sorted by flow cytometry into 96-well plates, and each colony was screened by immunoblotting. The target sequences used are listed in Table S1.
The USP20-FLAG-GFP knock-in cell lines were generated using the CRISPR-Cas9 system. Guide RNAs sequences were designed near the stop codon. The guide RNAs sequences were inserted into the pGL3-U6-sgRNA-PGK-puromycin plasmid. HeLa cells were co-transfected with the sgRNA plasmids, pST1374-NLS-flag-linker-Cas9, and the pC3.1-USP20-linker-FLAG-linker-GFP donor plasmid. After 24 h, cells were selected with 5 μg/ml puromycin and 10 μg/ml blasticidin for 7 days. The knock-in of the target gene was confirmed by immunoblotting. Single-cell colonies were then sorted by flow cytometry into 96-well plates, and each colony was screened by immunoblotting and sequencing. The target sequences used are listed in Table S1.
Constructing stable cell lines
HeLa cells stably expressing inducible ssRFP-GFP-KDEL were established via lentivirus-mediated transfection. To generate lentiviral particles, HEK293FT cells were co-transfected with the Tet-on ssRFP-GFP-KDEL plasmid (Addgene 128,257; Noboru Mizushima) and the lentivirus packaging plasmids psPAX2 (Addgene 12,260; Didier Trono) and pMD2.G (Addgene 12,259; Didier Trono) using Lipofectamine® 2000 (Thermo Fisher Scientific 11,668,019). After 48 h of transfection, lentiviral particles were harvested and centrifuged to remove cell debris. HeLa cells were then plated in 6-well plates and infected with the viral particles for 24 h. Subsequently, the virus particle-containing medium were replaced with fresh medium. After 48 h of infection, cells were selected with 2 μg/ml puromycin. Single-cell colonies were then sorted by flow cytometry into 96-well plates, and each colony was screened by immunoblotting. Doxycycline treatment was applied for 24 h to induce the expression of ssRFP-GFP-KDEL. The same method was used to generate HeLa cells stably expressing FLAG-GFP-RETREG1.
Plasmids
FLAG-USP20 and FLAG-RETREG1 were generated by cloning the corresponding cDNA into the pRK5-FLAG vector [Citation58]. MYC-USP20 and MYC-RETREG1 were generated by inserting the corresponding cDNA into the pRK5-MYC vector. FLAG-USP20[ΔFFAT1], FLAG-USP20[ΔFFAT2], FLAG-USP20[ΔFFAT1/2] and MYC-USP20C154S,H643Q were generated by performing overlap extension of PCR-mediated deletions on pRK5-FLAG-USP20 and pRK5-MYC-USP20. FLAG-RETREG1[ΔRHD], FLAG-RETREG1[ΔC], MYC-RETREG1[ΔRHD] and MYC-RETREG1[ΔC] were generated by overlap extension of PCR-mediated deletions on pRK5-FLAG-RETREG1 and pRK5-MYC-RETREG1. EGFP-SEC61B was generated by cloning the corresponding cDNA into the pEGFP.C1 vector (Clontech, 6084–1). The plasmid pRK5-HA-UB was described previously [Citation59]. HA-UB[K48 only] and HA-UB[K63 only] were generated through PCR-mediated site-directed mutagenesis on pRK5-HA-UB. All mutations were confirmed by DNA sequencing. The plasmids encoding GFP-VAPA, GFP-VAPAK94D,M96D, GFP-VAPB and GFP-VAPBK87D,M89D were kindly provided by Dr. Hong Zhang (Institute of Biophysics, Chinese Academy of Sciences). PC3.1-USP20-linker-FLAG-linker-GFP was generated by cloning the corresponding genomic fragment and linker-FLAG-linker-GFP into the PC3.1-puro vector. PCDH-FLAG-linker-GFP-linker-RETREG1 was generated by cloning the corresponding cDNA and FLAG-linker-GFP-linker into pCDH-CMV-MCS-EF1-Puro (System Biosciences, CD510B–1).
Antibodies and reagents
Mouse anti-MYC (2276), rabbit anti-HA (3724) and rabbit anti-RETREG1 (83414S, for immunoprecipitation) were purchased from Cell Signaling Technology. Rabbit anti-FLAG (F3165), mouse anti-FLAG (F7425) and rabbit anti-LC3B (L7543, for immunoblotting) were purchased from Sigma-Aldrich. Rabbit anti-RETREG1 (21537–1-AP, for immunoblotting), rabbit anti-VAPA (15275–1-AP) and rabbit anti-VAPB (14477–1-AP) were purchased from Proteintech. Rabbit anti-UB (A19686), rabbit anti-SQSTM1 (A11247), rabbit anti-MYC (AE070) and mouse anti-HA (AE008) were purchased from ABclonal, Inc. Mouse anti-MYC (sc-166,247) was purchased from Santa Cruz Biotechnology. Rabbit anti-WIPI2 (ab105459) was purchased from Abcam. Rabbit anti-USP20 (A301-189A) was purchased from Bethyl Laboratories. The rabbit polyclonal antibody against GFP was generated using a recombinant GFP protein derived from Aequorea victoria. Mouse anti-LC3B (M152–3, for immunofluorescence), Anti-ACTB (HRP-DirecT; PM053–7) were purchased from Medical & Biological Laboratories Co., LTD. The secondary antibodies goat anti-mouse IgG (H+L), HRP (111-035-146), goat anti-rabbit IgG (H+L), HRP (111-035-144), and Cy5-AffiniPure donkey anti-rabbit IgG (H+L) (711-175-152) were purchased from Jackson ImmunoResearch Inc. The secondary antibodies goat anti-rabbit IgG (H+L), Alexa Fluor 488 (A-11034), goat anti-mouse IgG (H+L), Alexa Fluor 488 (A-11029), goat anti-rabbit IgG (H+L), Alexa Fluor 568 (A-11036), goat anti-mouse IgG (H+L), Alexa Fluor 568 (A-11031), and goat anti-mouse IgG (H+L), Alexa Fluor 633 (A-21052) were purchased from Invitrogen. MG132 (S2619) was purchased from Selleck. Chloroquine (C6628) was purchased from Sigma-Aldrich. BafA1 (sc-201550A) was purchased from Santa Cruz Biotechnology. Doxycycline (60204ES03) was purchased from Yeasen. GSK2643943A (HY-111458) was purchased from MedChemExpress (MCE).
Immunoprecipitation and immunoblotting
For immunoprecipitation, HEK293FT cells were transfected and lysed 24 h post-transfection in NP40 lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.5% NP40 [Sangon Biotech, A600385–0100]) containing EDTA-free protease inhibitor cocktail (Bimake, B14002) for 30 min at 4°C. The lysates were centrifuged at 17,000 × g for 10 min at 4°C, and the soluble supernatant fractions were subjected to immunoprecipitation. Immunoprecipitation was performed using protein A beads (Bimake, B23202) pre-bound with RETREG1 or GFP antibody, FLAG (Bimake, B26102), HA (Bimake, B26201), or MYC (Bimake, B26302) magnetic beads. After incubation for 2 h at 4°C, the beads were washed three times with NP40 wash buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.1% NP40), and the samples were immunoblotted with the indicated antibodies.
For immunoprecipitation under denaturing condition, harvested cells were lysed in a buffer with 1% SDS (Sinopharm Chemical Reagent Co., 30166480) and 5 mM DTT (MDBio, Inc., D023) in PBS (Sangon Biotech, B548117–0500). The samples were heated at 65°C for 15 min and diluted with NP40 lysis buffer. The soluble supernatant fractions were harvested and subjected to immunoprecipitation as described above.
Immunoblotting was performed using polyvinylidene fluoride membrane (Bio-Rad 1,620,177) and the indicated antibodies. The protein signals were detected using ECL western blotting detection reagents (PerkinElemer, NEL105001EA). The chemiluminescence bands were imaged under Amersham Imager 680 (GE Healthcare Life Sciences, USA). The bands were adjusted within the linear range and quantified using ImageJ2 (v2.3.0, NIH).
Immunofluorescence microscopy
Cells grown on cover glasses were fixed with 4% paraformaldehyde (Thermo Fisher Scientific 43,368) in PBS for 15 min at room temperature. Subsequently, cells were permeabilized by 0.1% NP40 in PBS for 10 min at room temperature. After permeabilization, the cells were incubated with blocking buffer (2% BSA [Yeasen, 36104ES25] in cell staining buffer [4A Biotech, FXP005]) for 1 h at room temperature. Next, the cells were incubated with the indicated primary antibodies diluted in blocking buffer for 1 h at room temperature, and then incubated with fluorescent dye-conjugated secondary antibodies and DAPI in blocking buffer for 30 min at room temperature. Finally, the cells were mounted with Anti-Fade Fluorescence Mounting Medium (Abcam, ab104135). Images were acquired using a Zeiss LSM800 microscope (Zeiss, Germany) with a 63 × 1.4 NA oil objective. The same acquisition parameters were used for a specific set of experiments. Z-series images were displayed as maximum Z-projections and orthogonal views processed with ImageJ2. The ratio of colocalization was determined by analyzing a total of 100 cells from three independent experiments. SIM images were obtained using the Zeiss Lattice SIM microscope (Zeiss, Germany) with a Plan-Apochromat oil objective lens 63×, 1.4 NA. Images were captured using PCO edge sCMOS camera (Acquisition pixel size, 63 nm at 63× objective) and G6 gride (23 μm). Data reconstruction was performed using the default 3D method in Zen Black software.
GFP cleavage assay
U-2 OS cells were transiently co-transfected with GFP-SEC61B, along with either scramble shRNA or USP20 shRNA. After transfection, the cells were lysed using RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% Triton X-100 [Sangon Biotech, A110694–0100]), 0.5% sodium pyrophosphate [Sangon Biotech, A500883–0500], containing EDTA-free protease inhibitor cocktail [Bimake, B14002]) for 30 min at 4°C. The cell lysates were subjected to immunoblotting using antibodies against GFP, USP20, and ACTB.
Cycloheximide chase
Cycloheximide chase experiments were performed by incubating the cells in DMEM containing 100 µg/ml cycloheximide (Selleck, S7418) at 37°C. Equal number of cells was taken at different time points as indicated in the figures for immunoblotting analysis. Cells were lysed with the lysis buffer.
Microsome fraction assay
HEK293FT cells were washed three times with PBS buffer and then collected by centrifugation at 200 × g for 5 min. The cells were resuspended in three volumes of ice-cold sucrose buffer (10 mM HEPES, pH 7.4, 250 mM sucrose [Sangon Biotech, A100335–0250], 2 mM MgCl2) containing EDTA-free protease inhibitor cocktail. The cells were mechanically lysed by passing through a 26-gauge needle, and the resulting cell lysate was centrifuged at 3,800 × g at 4°C for 30 min. The supernatant was collected and subjected to a second centrifugation under the same conditions to remove any remaining debris. The resulting post-nuclear supernatant was centrifuged at 75,000 × g for 1 h at 4°C using a TLA-55 rotor. The resulting microsome pellet was resuspended in microsome buffer (10 mM HEPES, pH 7.4, 250 mM sucrose, 1 mM MgCl2, 0.5 mM DTT). Both the microsome and cytosolic fractions were subjected to immunoblotting using specific antibodies.
Quantification and statistical analysis
All experiments were independently repeated at least three times. Image analysis and quantification were performed using ImageJ2. The image processing steps included background subtraction, smoothing, and conversion to binary masks through thresholding. The numbers in different groups were measured using the “analyze particle” function with fixed particle size and circularity parameters. Data were pooled from three independent experiments, with at least 100 cells counted. Error bars represent the standard error of the mean (SEM). Statistical analysis was performed using Prism 8 (GraphPad). The significance between two groups was determined using the Student’s t-test. The significance among multiple groups was determined using one-way ANOVA followed by Tukey’s multiple comparisons test. n.s., not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Supplemental Material
Download MS Word (3.6 MB)Acknowledgements
We thank Dr. Hong Zhang (Institute of Biophysics, Chinese Academy of Sciences), Professor Qiming Sun (Zhejiang University, China), Professor Qing Zhong (Shanghai Jiao Tong University, China), Professor Zongping Xia (Zhengzhou University, China) and Professor Yangyang Li (ShanghaiTech University, China) for providing the various constructs and reagents. We thank Dr. Hong Zhang for providing valuable advice and critical reading of the manuscript. We thank the Image Core Facility and the Molecular and Cell Biology Core Facility in the School of Life Science and Technology at ShanghaiTech University for assisting with confocal microscopy and technical support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2024.2347103
Additional information
Funding
References
- Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20(3):233–242. doi: 10.1038/s41556-018-0037-z
- Morishita H, Mizushima N. Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol. 2019;35(1):453–475. doi: 10.1146/annurev-cellbio-100818-125300
- Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016;85(1):685–713. doi: 10.1146/annurev-biochem-060815-014556
- Mizushima N, Levine B, Longo DL. Autophagy in human diseases. N Engl J Med. 2020;383(16):1564–1576. doi: 10.1056/NEJMra2022774
- Yang Y, Klionsky DJ. Autophagy and disease: unanswered questions. Cell Death Differ. 2020;27(3):858–871. doi: 10.1038/s41418-019-0480-9
- Ganley IG, Lam du H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–12305. doi: 10.1074/jbc.M900573200
- Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. doi: 10.1091/mbc.e08-12-1248
- Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. doi: 10.1091/mbc.e08-12-1249
- Chen S, Novick P, Ferro-Novick S. ER structure and function. Curr Opin Cell Biol. 2013;25(4):428–433. doi: 10.1016/j.ceb.2013.02.006
- Borgese N, Francolini M, Snapp E. Endoplasmic reticulum architecture: structures in flux. Curr Opin Cell Biol. 2006;18(4):358–364. doi: 10.1016/j.ceb.2006.06.008
- Zhao YG, Zhang H. Autophagosome maturation: an epic journey from the ER to lysosomes. J Cell Bio. 2019;218(3):757–770. doi: 10.1083/jcb.201810099
- Zhao YG, Liu N, Miao G, et al. The ER contact proteins VAPA/B interact with multiple autophagy proteins to modulate autophagosome biogenesis. Curr Biol. 2018;28(8):1234–1245 e1234. doi: 10.1016/j.cub.2018.03.002
- Chino H, Mizushima N. ER-Phagy: quality control and turnover of endoplasmic reticulum. Trends Cell Biol. 2020;30(5):384–398. doi: 10.1016/j.tcb.2020.02.001
- Mochida K, Nakatogawa H. ER-phagy: selective autophagy of the endoplasmic reticulum. EMBO Rep. 2022;23(8):e55192. doi: 10.15252/embr.202255192
- Gubas A, Dikic I. ER remodeling via ER-phagy. Mol Cell. 2022;82(8):1492–1500. doi: 10.1016/j.molcel.2022.02.018
- Ferro-Novick S, Reggiori F, Brodsky JL. ER-Phagy, ER homeostasis, and ER quality control: implications for disease. Trends Biochem Sci. 2021;46(8):630–639. doi: 10.1016/j.tibs.2020.12.013
- Hubner CA, Dikic I. ER-phagy and human diseases. Cell Death Differ. 2020;27(3):833–842. doi: 10.1038/s41418-019-0444-0
- Khaminets A, Heinrich T, Mari M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522(7556):354–358. doi: 10.1038/nature14498
- Grumati P, Morozzi G, Holper S, et al. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife. 2017;6:e25555. doi: 10.7554/eLife.25555
- Chen Q, Xiao Y, Chai P, et al. ATL3 is a tubular ER-Phagy receptor for GABARAP-Mediated selective autophagy. Curr Biol. 2019;29(5):846–855 e846. doi: 10.1016/j.cub.2019.01.041
- Fumagalli F, Noack J, Bergmann TJ, et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol. 2016;18(11):1173–1184. doi: 10.1038/ncb3423
- Smith MD, Harley ME, Kemp AJ, et al. CCPG1 is a non-canonical autophagy cargo receptor essential for ER-Phagy and pancreatic ER proteostasis. Dev Cell. 2018;44(2):217–232 e211. doi: 10.1016/j.devcel.2017.11.024
- Chino H, Hatta T, Natsume T, et al. Intrinsically disordered protein TEX264 mediates ER-phagy. Mol Cell. 2019;74(5):909–921 e906. doi: 10.1016/j.molcel.2019.03.033
- An H, Ordureau A, Paulo JA, et al. TEX264 is an endoplasmic reticulum-resident ATG8-interacting protein critical for ER remodeling during nutrient stress. Mol Cell. 2019;74(5):891–908 e810. doi: 10.1016/j.molcel.2019.03.034
- Nthiga TM, Kumar Shrestha B, Sjottem E, et al. CALCOCO1 acts with VAMP-associated proteins to mediate ER-phagy. Embo J. 2020;39(15):e103649. doi: 10.15252/embj.2019103649
- Bhaskara RM, Grumati P, Garcia-Pardo J, et al. Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat Commun. 2019;10(1):2370. doi: 10.1038/s41467-019-10345-3
- Jiang X, Wang X, Ding X, et al. FAM134B oligomerization drives endoplasmic reticulum membrane scission for ER-phagy. Embo J. 2020;39(5):e102608. doi: 10.15252/embj.2019102608
- Wang X, Jiang X, Li B, et al. A regulatory circuit comprising the CBP and SIRT7 regulates FAM134B-mediated ER-phagy. J Cell Bio. 2023;222(5):e202201068. doi: 10.1083/jcb.202201068
- Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81(1):203–229. doi: 10.1146/annurev-biochem-060310-170328
- Kwon YT, Ciechanover A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem Sci. 2017;42(11):873–886. doi: 10.1016/j.tibs.2017.09.002
- Nijman SM, Luna-Vargas MP, Velds A, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773–786. doi: 10.1016/j.cell.2005.11.007
- Chu Y, Kang Y, Yan C, et al. LUBAC and OTULIN regulate autophagy initiation and maturation by mediating the linear ubiquitination and the stabilization of ATG13. Autophagy. 2021;17(7):1684–1699. doi: 10.1080/15548627.2020.1781393
- Liu CC, Lin YC, Chen YH, et al. Cul3-KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol Cell. 2016;61(1):84–97. doi: 10.1016/j.molcel.2015.11.001
- Chen YH, Huang TY, Lin YT, et al. VPS34 K29/K48 branched ubiquitination governed by UBE3C and TRABID regulates autophagy, proteostasis and liver metabolism. Nat Commun. 2021;12(1):1322. doi: 10.1038/s41467-021-21715-1
- Chen YH, Chen HH, Wang WJ, et al. TRABID inhibition activates cGAS/STING-mediated anti-tumor immunity through mitosis and autophagy dysregulation. Nat Commun. 2023;14(1):3050. doi: 10.1038/s41467-023-38784-z
- Kim JH, Seo D, Kim SJ, et al. The deubiquitinating enzyme USP20 stabilizes ULK1 and promotes autophagy initiation. EMBO Rep. 2018;19(4):e44378. doi: 10.15252/embr.201744378
- Xie W, Jin S, Wu Y, et al. Auto-ubiquitination of NEDD4-1 recruits USP13 to facilitate autophagy through deubiquitinating VPS34. Cell Rep. 2020;30(8):2807–2819 e2804. doi: 10.1016/j.celrep.2020.01.088
- Liu J, Li M, Li L, et al. Ubiquitination of the PI3-kinase VPS-34 promotes VPS-34 stability and phagosome maturation. J Cell Bio. 2018;217(1):347–360. doi: 10.1083/jcb.201705116
- Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147(1):223–234. doi: 10.1016/j.cell.2011.08.037
- Xu D, Shan B, Sun H, et al. USP14 regulates autophagy by suppressing K63 ubiquitination of beclin 1. Genes Dev. 2016;30(15):1718–1730. doi: 10.1101/gad.285122.116
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(5):452–460. doi: 10.4161/auto.4451
- Culver JA, Mariappan M. Deubiquitinases USP20/33 promote the biogenesis of tail-anchored membrane proteins. J Cell Bio. 2021;220(5):e202004086. doi: 10.1083/jcb.202004086
- Lu XY, Shi XJ, Hu A, et al. Feeding induces cholesterol biosynthesis via the mTORC1-USP20-HMGCR axis. Nature. 2020;588(7838):479–484. doi: 10.1038/s41586-020-2928-y
- Udeshi ND, Svinkina T, Mertins P, et al. Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics. 2013;12(3):825–831. doi: 10.1074/mcp.O112.027094
- Akimov V, Barrio-Hernandez I, Hansen SVF, et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat Struct Mol Biol. 2018;25(7):631–640. doi: 10.1038/s41594-018-0084-y
- Mikitova V, Levine TP, Zhang Z. Analysis of the key elements of FFAT-like motifs identifies new proteins that potentially bind VAP on the ER, including two AKAPs and FAPP2. PLOS ONE. 2012;7(1):e30455. doi: 10.1371/journal.pone.0030455
- James C, Kehlenbach RH. The interactome of the VAP family of proteins: an overview. Cells. 2021;10(7):1780. doi: 10.3390/cells10071780
- Liu N, Zhao H, Zhao YG, et al. Atlastin 2/3 regulate ER targeting of the ULK1 complex to initiate autophagy. J Cell Bio. 2021;220(7):e202012091. doi: 10.1083/jcb.202012091
- Zhang M, Zhang MX, Zhang Q, et al. USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res. 2016;26(12):1302–1319. doi: 10.1038/cr.2016.125
- Zhang MX, Cai Z, Zhang M, et al. USP20 promotes cellular antiviral responses via deconjugating K48-linked ubiquitination of MITA. J Immunol. 2019;202(8):2397–2406. doi: 10.4049/jimmunol.1801447
- Jean-Charles PY, Wu JH, Zhang L, et al. USP20 (Ubiquitin-Specific Protease 20) inhibits TNF (Tumor Necrosis Factor)-triggered smooth muscle cell inflammation and attenuates atherosclerosis. Arterioscler Thromb Vasc Biol. 2018;38(10):2295–2305. doi: 10.1161/ATVBAHA.118.311071
- Li W, Shen M, Jiang YZ, et al. Deubiquitinase USP20 promotes breast cancer metastasis by stabilizing SNAI2. Genes Dev. 2020;34(19–20):1310–1315. doi: 10.1101/gad.339804.120
- Berthouze M, Venkataramanan V, Li Y, et al. The deubiquitinases USP33 and USP20 coordinate beta2 adrenergic receptor recycling and resensitization. Embo J. 2009;28(12):1684–1696. doi: 10.1038/emboj.2009.128
- Ha J, Kim M, Seo D, et al. The deubiquitinating enzyme USP20 regulates the TNFalpha-induced NF-kappaB Signaling Pathway through stabilization of p62. Int J Mol Sci. 2020;21(9):3116. doi: 10.3390/ijms21093116
- González A, Covarrubias-Pinto A, Bhaskara RM, et al. Ubiquitination regulates ER-phagy and remodelling of endoplasmic reticulum. Nature. 2023;618(7964):394–401. doi: 10.1038/s41586-023-06089-2
- Foronda H, Fu YX, Covarrubias-Pinto A, et al. Heteromeric clusters of ubiquitinated ER-shaping proteins drive ER-phagy. Nature. 2023;618(7964):402–410. doi: 10.1038/s41586-023-06090-9
- Culver JA, Li X, Jordan M, et al. A second chance for protein targeting/folding: ubiquitination and deubiquitination of nascent proteins. BioEssays. 2022;44(6):e2200014. doi: 10.1002/bies.202200014
- Liu Y, Soetandyo N, Lee JG, et al. USP13 antagonizes gp78 to maintain functionality of a chaperone in ER-associated degradation. Elife. 2014;3:e01369. doi: 10.7554/eLife.01369
- Zhao Y, Feng Z, Zou Y, et al. The E3 ubiquitin ligase SYVN1 ubiquitinates atlastins to remodel the endoplasmic reticulum network. iScience. 2020;23(9):101494. doi: 10.1016/j.isci.2020.101494