
ABSTRACT
Banana Fusarium wilt (BFW), caused by Fusarium oxysporum f. sp. cubense (Foc), poses a major challenge to the worldwide banana industry. Fungal microRNA-like small RNAs (milRNAs) play crucial roles in regulating fungal growth, conidiation, development, and pathogenicity. However, the milRNAs and their functions in the pathogenesis of Foc remain poorly understood. In this study, we employed high-throughput sequencing and bioinformatics to profile Foc sRNAs during both pure culture and early infection stages. Our analysis identified six milRNAs exhibiting significantly upregulated expression at the initial Foc infection. Of these, milR106’s biogenesis was found to be Dicer-dependent, whereas milR87, milR133, milR138, and milR148 were associated with Dicer and Argonaute proteins. Genetic manipulation and phenotype analysis confirmed that milR106 is crucial for Foc virulence by regulating conidiation, hydrogen peroxide sensitivity, and infective growth. Gene Ontology analysis of milRNA targets in the banana genome revealed enrichment in defence response to fungus and cellular response to hypoxia, implying the importance of these target genes in response to pathogen infection. In conclusion, our sRNA profiling of Foc identified several infection-induced milRNAs. The corresponding results provide valuable molecular targets for the development of an efficient strategy to control BFW.
1. Introduction
Fusarium oxysporum f. sp. cubense (Foc) is a typical soil-borne fungus that causes Fusarium wilt by infecting the roots and blocking the vascular tissues of the host banana and threatens global banana production (Viljoen et al. Citation2020). A total of four races have been reported in Foc, of which the tropical race 4 (TR4) is the most widespread. Banana Fusarium Wilt (BFW) caused by TR4 is the most devastating disease in banana-producing countries and regions (Zhang et al. Citation2019). It is well known that the most effective method to control BFW is implying strict phytosanitary measures and planting disease-resistant cultivars (Dita et al. Citation2018). However, breeding disease-resistant cultivars that simultaneously meet market standards while maintaining high yields is challenging. Additionally, these disease control measures are very difficult to enforce by smallholder growers (Viljoen et al. Citation2020). In some severely affected banana plantations, the conventional ‘Cavendish cultivar had to be abandoned for other alternative crops due to the spread of TR4 (Warman and Aitken Citation2018). Therefore, a comprehensive understanding of the pathogenesis of BFW and the development of improved control methods are urgently needed.
MicroRNA-like small RNAs (milRNAs), first found in Neurospora crassa and Botrytis cinerea, resemble plant and animal microRNAs and play crucial roles in post-transcriptional regulation of gene expression (Lee et al. Citation2010; Weiberg et al. Citation2013; Jin et al. Citation2018). These milRNAs, typically 19–25 nt in length, derive from single-stranded RNA transcripts with stem-loop structures and silence complementary transcripts through RNA-mediated interference (RNAi) (Bartel Citation2004; Nicolás and Ruiz-Vázquez Citation2013). A growing number of milRNAs have been recognised for their roles in fungal growth and development, such as those involved in sclerotia development in Sclerotinia sclerotiorum (Xia et al. Citation2020), vegetative form of mycelial or yeast phases of dimorphic fungus Penicillium marneffei (Lau et al. Citation2013), and spore morphological differences of Metarhizium acridum (Zhang et al. Citation2022). Recent studies reported that milRNAs secreted by phytopathogenic fungi through extracellular vesicles play important roles in plant-microbe interaction (Huang et al. Citation2019; He et al. Citation2023). For instance, milRNAs of B. cinerea have been identified as small RNA effectors that suppress host immunity and facilitate fungal infection through cross-kingdom RNAi (Weiberg et al. Citation2013; Wang et al. Citation2017). Similarly, the Puccinia striiformis f. sp. tritici (Pst) miRNA, Pst-milR1, promotes plant disease susceptibility by cleaving the PR2 gene (Wang et al. Citation2017). The milRNA Vm-milR37 exclusively expressed in the mycelium, was found to contribute to pathogenicity in Valsa mali (Feng et al. Citation2021).
Although the regulatory role of endogenous milRNAs in pathogenicity has been characterised in some fungi, such as B. cinerea, P. striiformis, and Va. mali (Weiberg et al. Citation2013; Wang et al. Citation2017; Feng et al. Citation2021). Little is known about the role of small RNAs produced by the soil-borne fungus Foc in pathogenicity and other biological processes. In this study, we conducted high-throughput sequencing and bioinformatics analysis to profile the sRNAs of Foc during both pure culture and early infection stages. We aimed to identify the milRNAs that were significantly induced during the initial infection process of Foc, laying the foundation for further elucidation of the pathogenic mechanism of Foc. We confirmed that one of the milRNAs is Dicer-dependent and involved in the pathogenicity of Foc. The results provide valuable molecular targets for the development of efficient strategies to control BFW and breed disease-resistant cultivars.
2. Materials and methods
2.1. Fungal strains and sample preparation for small RNA sequencing
The wild-type (WT) strain XJZ2 of F. oxysporum f. sp. cubense tropical race 4 (Li et al. Citation2013) was activated on PDA plates at 28 °C and then transferred to YPD media to prepare the conidia suspension. The triploid cultivated banana cultivar ‘Canvendish’, widely grown in production (Li et al. Citation2024), was used for infection inoculation following the previously described method (Li et al. Citation2014). Briefly, healthy banana roots were immersed in the suspension with a conidia concentration of 1 × 108 conidia/mL. After 36 hours of inoculation, banana roots with attached Foc conidia and mycelia were collected for total RNA extraction. The same concentration of conidia was inoculated in a minimal medium (MM) and cultured for 36 h to collect pure cultured conidia and mycelia as a control. Each sample was prepared in triplicate.
2.2. RNA extraction and sRNA sequencing
200 mg of banana roots or Foc mycelia from different treatments were collected and ground into powder using liquid nitrogen. Total RNA extraction was performed using a CTAB reagent (De Wever et al. Citation2020). RNA integrity was assessed and quantified with a Bioanalyzer 2100 (Agilent, Santa Clara, USA), and only qualified RNA samples with a RIN (RNA Integrity Number) value between 7 and 10 were sent to Novogene (Beijing, China) for sRNA library construction and high-throughput sequencing (Ouyang et al. Citation2021).
2.3. Sequence data analysis and infection-induced milRNA prediction
Raw sequencing data were trimmed and analysed using CLC Genomics Workbench v11 (CLC bio) following our previous description (Li et al. Citation2022). Only reads mapped to the Foc II5 (TR4 strain) genome were used for data analysis. To minimise false positive sRNAs induced by mRNA degradation only reads mapped to the UTR, intron, and intergenic regions were used for sRNA length distribution and sRNA locus identification. The RNAstructure (Version 6.3) was employed to predict the secondary structure of the sRNA precursor (Reuter and Mathews Citation2010).
To identify differentially expressed sRNAs and their loci reads from the pure culture stage and initial infection stage of Foc were compared. To assess sRNA expression, read counts of more than 10 were normalised to transcripts per million (TPM) values, and log2 ratios were calculated using the TPM value from the samples of XJZ2-36 hpi and XJZ2. Those sRNAs, that were significantly up-regulated [log2TPM (XJZ2–36 hpi/XJZ2) >1, p < 0.05] in the infection stage compared to the pure culture stage, along with their precursors exhibiting a typical secondary hairpin structure, were considered as infection-induced milRNAs (Lee et al. Citation2010; Li et al. Citation2022).
2.4. Validation of milRNA by qRT‑PCR
To validate the predicted and differentially expressed milRNAs, quantitative reverse transcription-PCR (qRT-PCR) was performed according to our previously described method (Li et al. Citation2022). The milRNA was first added to a PolyA tail, and then the cDNA was reverse-transcribed using an oligo-dT adaptor. Subsequently, the expression of milRNAs was quantified by qRT-PCR. At least three biological replicates were performed for each sample.
2.5. Generation and validation of the milRNA deletion mutants and the complemented strains
The precursor sequence (approximately 300 bp) of the milRNA was replaced and complemented through homologous recombination through PEG-mediated protoplast transformation (Li et al. Citation2014). The milRNA precursor was first introduced into the Geneticin resistance gene cassette of pKNT-G418 and then transformed after ligating it with its left and right border sequences, respectively. The milRNA deletion mutants and the complemented strains were identified by PCR using the primers provided in Table S1.
2.6. Fungal invasion assays and pathogenicity test
The ability of mycelia to penetrate cellophane and their invasive growth on tomato fruit surfaces were compared between the WT and the mutants according to the previously described method (Li et al. Citation2014). Pathogenicity on banana plantlets was assessed as described previously (Li et al. Citation2013). Each treatment involved the use of 30 banana plantlets, and the incidence and disease indexes were calculated for disease severity assessment (Li et al. Citation2013).
2.7. MilRNA target gene prediction and functional annotation
The potential target genes of the milRNAs were predicted using the psRNATarget algorithm (https://www.zhaolab.org/psRNATarget/analysis) with default parameters (Li et al. Citation2022). The Gene Ontology (GO) pathway classification of target genes from the Foc genome was implemented on the NovoMagic cloud analysis platform (Novogene, China). For annotation of target genes derived from the banana genome (Ahmad et al. Citation2020), the website of Banana Genome Hub (https://banana-genome-hub.southgreen.fr/content/go-enrichment) was utilised for GO enrichment analysis, with a highly sensitive parameter to merge similar GO terms.
2.8. Statistical analysis
In the study, a Student’s t-test was used for the significance analysis of two samples. A Duncan’s multiple range test was used to analyse the data of multiple samples at both levels (α = 0.05 and α = 0.01). And different letters on the bars indicated significant differences.
3. Results
3.1. Sequence analysis of sRNAs in Foc during pure culture and initial infection stages
In this study, the small RNAs of F. oxysporum f. sp. cubense in the pure cultured stage (Culture) and the initial infection stage (36 hours post-inoculation, 36 hpi) of banana roots were sequenced respectively. After the elimination of the 5’ adaptor, contamination, and low-quality sequences from the raw data, reads with lengths ranging from 16 to 40 nucleotides (nt) were preserved. For subsequent sRNA analysis, only reads that perfectly matched the Foc II5 genome were used. After data processing, we obtained over 16 million high-quality small RNA sequences, with 9,221,095 reads from the pure cultured stage and 6,899,403 reads from the initial infection stage (Table S2). Each sample was replicated three times, and each replicate contained more than two million reads, providing sufficient data for subsequent analysis. Less than 7% of the sequences from Foc-36 hpi could be mapped to the genome, compared to an average of 27% of the sequences from Foc-culture (Table S2). This discrepancy may be attributed to the substantial number of sequences derived from the host banana in the Foc-36 hpi data, in contrast to the pure culture data of Foc.
All sRNA reads mapped to the Foc II5 genome were aligned to the data sets of the Rfam database, including rRNA, tRNA, snRNA, snoRNA, and repeat elements, as well as transcripts, genes, and the upstream and downstream regions (1,000 bp before and after the gene sequence) in turn. Very few small RNAs without annotation were found in the two samples. In the pure cultured stage, the majority of reads (76.25%) were mapped to the Rfam database. Among them, most of the reads (70.16%) were aligned to tRNA. However, in the infection stage (36 hpi), approximately 61% of the reads were aligned to the Rfam database, with only 29.56% derived from tRNA. Notably, the reads from rRNA and snRNA/snoRNA showed a significant increase in the 36 hpi sample compared to the pure cultured sample (). The ratios of reads mapped to introns, 3’/5’ UTRs, as well as intergenic regions (upstream and downstream of genes) were significantly higher in the infection stage, compared to the pure cultured stage (). However, there was no significant difference in the proportion of reads mapped to transcripts between the two samples. After sequence alignment, reads that were mapped to the Rfam database and transcripts were excluded, and the remaining reads from introns, 3’/5’ UTRs, and intergenic regions were used for further sRNA data analysis.
Figure 1. Small RNA sequencing analysis in Fusarium oxysporum f. sp. cubense (Foc). (a) Classification and annotation of sRNAs. (b) sRNA length distribution. (c) and (d) Statistical analysis of initial bases in sRNA from XJZ2-culture (c) and XJZ2-36 hpi (d).
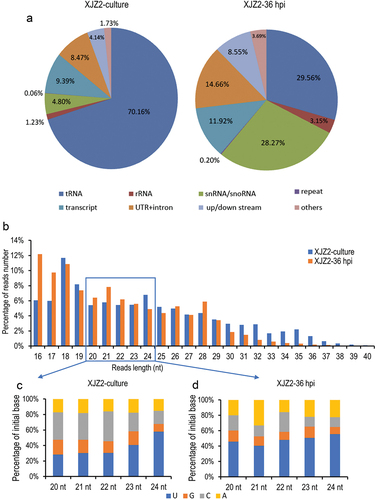
The lengths of the reads were statistically analysed, and the results showed that approximately 58% of the sRNA reads were 18–26 nt in length in both samples (). Among these, the most frequently sequenced reads were 18 nt in length, followed by 19 nt and 24 nt in the pure cultured stage. In the infection stage of 36 hpi, 16-nt reads were the most abundant, followed by 18-nt and 21-nt reads.
Considering that the reported size of miRNA in Eukaryotes is typically between 20 and 24 nt (Bartel Citation2004; Lee et al. Citation2010), reads with lengths ranging from 20 to 24 nt were used to analyse the bias in the initial nucleotide. In both samples, the majority of sRNAs started with U. Reads starting with U averagely accounted for 37.5%, followed by reads starting with C and A, with average proportions of 29.82% and 16.87%, respectively, while sRNAs starting with G were the least abundant in the pure cultured Foc (). In the infection stage of 36 hpi, sRNAs starting with U remained the most abundant, accounting for an average of 48.15%, followed by sRNAs starting with A and C, accounting for 22.72% and 17.01%, respectively, while those starting with G were the least abundant (). Compared to the pure cultured Foc, the reads starting with U and A were significantly increased upon inoculation of the host plant, indicating that sRNAs with a bias towards U and A as the initial nucleotides may play important roles in infection processes.
3.2. Screening and identification of induced milRNAs in Foc during the initial infection stage
To identify milRNAs in Foc, unannotated reads were blasted against the miRbase (Release 21.0). Only a few reads were mapped to known miRNAs in the database, and no difference in the expression of miRNAs was detected between the two samples. Consequently, we failed to predict any known milRNA in Foc, which is consistent with the absence of miRNA orthologous between filamentous fungi and other eukaryotic species (Chen et al. Citation2014).
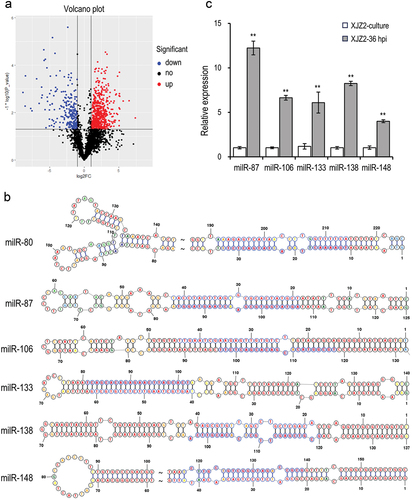
To identify infection-induced milRNAs in Foc, we determined the sRNAs-producing sites and screened the precursors with hairpin structures. Based on normalization by calculating the transcripts per million (TPM) value of each sRNA-producing locus, the expression levels of sRNAs were compared between the two samples. A total of 3,439 sRNAs-producing loci were captured. Among them, 953 and 327 sRNAs showed significant up- and down-regulated expression, respectively, in the infection stage compared to the pure-cultured stage (). After predicting the secondary structure of sRNA precursors and verifying their expression, we identified six loci of infection-induced milRNAs with hairpin structures (). These milRNAs were primarily derived from intergenic regions, except for milR106, which was from an intron. The results of qRT-PCR confirmed that the expression of the milRNAs was consistent with sequencing results when using small nuclear RNA U4 of Foc as the internal control (). However, one milRNA (milR-80) did not show specific products for its low GC content.
Figure 2. Identification of infection-induced milRNAs in Fusarium oxysporum f. sp. cubense. (a) Differential expression analysis of sRNAs of Foc in the pure culture stage and the initial infection stage. (b) Precursors’ secondary structure and sequences of infection-induced milRNAs in Foc. The mature milRNA sequences were marked in light blue. (c) Expression of the milRNAs in pure culture stage and initial infection stage by qRT-PCR. A Student’s t-test was used for significant analysis. **, p < 0.01. Error bars indicate S.D. (n = 3).
To validate the identified milRNA sequences in Foc, we amplified the milRNAs using small RNA reverse transcription products as templates. Subsequently, the PCR products were cloned into a T vector for sequencing. The resequencing results of fungal milRNA clones (Data not shown) confirmed that the mature milRNA sequences were consistent with the original high-throughput sequencing results and with a length ranging from 21 to 24 nt (). These findings provide additional evidence that the six small RNAs obtained through our screening process met the criteria for fungal milRNAs and were recognised as Foc_milRNAs induced by infection.
3.3. Identification of infection-induced milRNAs biosynthetic pathways in Foc
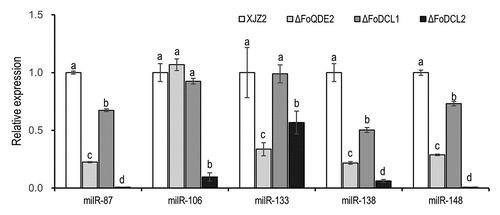
Argonaute (AGO) and Dicer proteins are known to play key roles in milRNA production in fungi (Lee et al. Citation2010). The expression of infection-induced milRNAs was detected by qRT-PCR in previously screened ΔFoQDE2, ΔFoDCL1, and ΔFoDCL2 mutants (Li et al. Citation2022), as well as WT strain. And the biosynthetic pathways of the milRNAs were analysed according to their expression levels. The results showed that the expression levels of milR87, milR133, milR138, and milR148 were significantly decreased in both ΔFoQDE2 and ΔFoDCL2 mutants compared to the WT, indicating the biosynthesis of these four milRNAs is dependent on the FoDCL2 and FoQDE2 genes. On the other hand, milR106 expression was significantly decreased only in ΔFoDCL2 mutants, suggesting that its biosynthesis is solely dependent on the FoDCL2 gene ().
Figure 3. Expression of the milRNAs in different gene deletion mutants involved in the sRNA biogenesis pathway in Fusarium oxysporum f. sp. cubense. Transcriptional levels of the infection-induced milRNAs were detected by qRT-PCR. A Duncan’s multiple range test was used for significant analysis. Error bars indicate S. D. (n = 3). Different letters on the bars indicate significant differences at the level of α = 0.01.
3.4. Deletion and complementation of milR106 in Foc
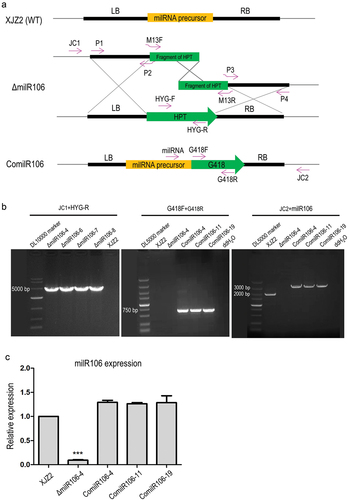
Among the six infection-induced milRNAs identified, milR106 is exclusively dependent on the FoDCL2 gene for its biogenesis. These unique biogenesis characteristics and the expression pattern of upregulation during the early infection stages prompted us to select milR106 for comprehensive functional investigation. To investigate the function of milR106 in Foc, we generated the milRNA precursor deletion mutants from the WT strain XJZ2 via homologous recombination. Additionally, we created complementation transformants based on one of the deletion mutants ΔmilR106-4 (). Four milRNA-deleted mutants were confirmed by PCR with the primer JC1 from the 5’ flanking region and primer HYG-R located in the hygromycin resistance gene (HYG) (). Using the primer pair G418F/G418R designed from the geneticin resistance gene, an amplicon was observed in the complemented transformants (ComilR106-4/-11/-19), but not in the WT strain, ΔmilR106 mutant or the negative control (ddH2O) (). The three complemented transformants also showed an amplicon when PCR amplification was performed with primer milR106 combined with primer JC2 from 3’ flanking region as shown in , indicating successful complementation of milR106 in the ΔmilR106 deletion mutant. This result was further confirmed by qRT-PCR ().
Figure 4. Verification of the milR106 deletion and complementation mutants by PCR and qRT-PCR. (a) Schematic diagram for deletion and complementation of milR106 in Foc. (b) PCR identification of the milR106 deletion mutants and complemented strains. (c) Transcriptional level of milR106 in different mutants of Foc was detected by qRT-PCR. To assess the statistical significance between the wild-type strain XJZ2 and the ΔmilR106-4 mutant, a Student’s t-test was used. ***, p < 0.001. Error bars indicate S.D. (n = 3).
3.5. MilR106 regulates tolerance to hydrogen peroxide and is involved in the virulence of Foc
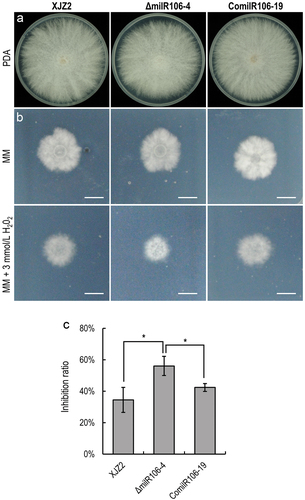
To decipher the biological functions of milR106 in Foc, we assessed the colony growth and conidiation in the WT strain and the mutants. The WT and the milR106 complemented strains exhibited normal growth on PDA () and minimal medium (MM) (). The ΔmilR106 mutant showed a slight attenuated growth on PDA, although statistical analysis revealed no significant difference compared to the WT strain (). The ΔmilR106 mutant produced much fewer microconidia than the WT and the complemented transformants after 7 days of culture on PDA (Figure S1). Moreover, the ΔmilR106 mutant displayed hypersensitive to oxidative stress when cultured on MM supplemented with 3 mmol/L H2O2 (). In contrast, the growth of the milR106 complemented strains was unaffected by oxidative stress (). These results indicate that milR106 positively regulates tolerance to oxidative stress in Foc.
Figure 5. Characterization of milR106-deleted mutant and complemented strains in Fusarium oxysporum f. sp. cubense. (a) Colony morphology of the WT strain XJZ2, the ΔmilR106 mutant (ΔmilR106-4), and milR106 complemented strain ComilR106-19 on PDA plates. (b) Mycelial sensitivity to H2O2. Mycelial growth of the above strains was measured at MM and MM with 3 mmol/L H2O2. Bar=1 cm. (c) Percentage of mycelial growth inhibition by H2O2. A Student’s t-test was used for significant analysis. *, p < 0.05. Error bars indicate S.D. (n = 3).
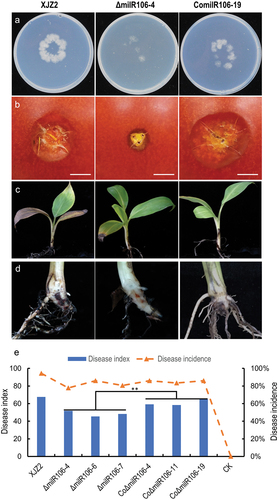
Pathogenicity tests showed that the ΔmilR106 mutant exhibited a compromised ability to penetrate the cellophane membrane () and caused smaller necrosis areas on the surface of tomato fruits than the WT (). Conversely, the complemented transformant showed the opposite phenotype (). In the banana seedling infection assay, ΔmilR106 showed significantly reduced virulence compared to the WT (). However, the virulence of the complemented transformants was significantly enhanced, as evidenced by the presence of obvious internal disease symptoms of brown discoloration and higher disease indices (). These findings suggest that milR106 contributes to the infective growth and virulence of Foc.
Figure 6. Assessment of invasive growth and Pathogenicity. (a) Cellophane penetration assay comparing the invasive growth of the WT strain XJZ2, ΔmilR106-4 mutant, and the complemented strain ComilR106-19. Conidia suspensions (100 μL per strain) with the same concentration of 1 × 105 spores/mL were put on cellophane-covered PDA plates and incubated at 28 °C for 4 days, then the cellophane sheets were removed, and samples were incubated for an additional 3 days and the colony size indicated mycelial growth were photographed. (b) Invasive growth on tomato fruits. The surfaces of tomato fruits were inoculated with discs of the different strains. The symptoms of necrosis on tomato fruits were recorded at 5 days post-inoculation. Bar =1 cm. (c) and (d) Symptoms of pathogenicity on the leaves (c) and corms (d) of the banana plantlets. (e) Disease index and disease incidence of banana plantlets after inoculation with the different strains. A Student’s t-test was used for significant analysis. **, p < 0.01.
3.6. Prediction and GO enrichment of target genes of infection-induced milRNAs in Foc
To investigate the significance of milRNAs in the Foc infection process, we predicted the target genes of milRNAs that expression was significantly induced in the initial infection stage. Using psRNATarget online software, we predicted a total of 296 unigenes from the Foc II5 genome and 1,256 unigenes from the banana genome (M. acuminata DH Pahang v4.3) as targets of the infection-induced milRNAs. In the banana genome, some Foc-milRNAs could target the same gene, such as milR106 and milR87, they both targeted one gene of Macma4_06_g01500. Additionally, some milRNAs targeted a single gene but had multiple target sites, such as milR133, milR148, and milR80. Multiple target sites were more common in the Foc genome.
GO enrichment analysis of biological processes demonstrated that GO terms such as defence response to fungus (GO:0050832) and cellular response to hypoxia (GO:0071456) were exclusively enriched among the target genes from the banana genome (), suggesting that these infection-induced milRNAs are more likely to target resistant genes in host plants. Furthermore, a lot of genes involved in proteolysis (GO:0006508) and response to oxidative stress (GO:0006979) were also enriched (). The most prevalent GO terms for molecular functions were ATPase activity (GO:0016887), nucleotide binding (GO:0000166), and transporter activity (GO:0005215). As for cellular components, the GO term early endosome (GO: 0005769) was uniquely enriched ().
Figure 7. Go enrichment of target genes of six infection-induced milRNAs in Foc. (a) Target genes from the host banana genome. (b) Target genes from the Foc II5 genome.
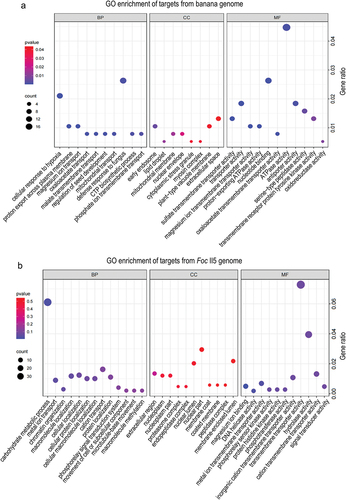
GO term analysis of target genes in Foc showed significant enrichment in biological processes such as methylation (GO:0032259), carbohydrate metabolic process (GO:0005975), and metal ion transport (GO:0030001) (). For molecular functions, a large number of genes engaged in transmembrane transporter activity (GO:0022857) and hydrolase activity (GO:0004553) were enriched. However, there was no significant GO terms enrichment for cellular components (). The diverse functions of these target genes indicate that milRNAs have crucial roles in regulating gene expression in a wide range of biological processes, particularly in plant defence responses to pathogen infections.
In this study, knockout of milR106 in Foc results in increased sensitivity to hydrogen peroxide and decreased virulence, suggesting a potential involvement of the milRNA in hydrogen peroxide clearance, reactive oxygen species (ROS) homoeostasis regulation, antioxidant defence, and response to oxidative stress. To elucidate the mechanism underlying the phenotype, we checked the target genes of milR106 in both pathogen and host plants. Three of the target genes (Macma4_04_g05490, Macma4_06_g02360, and Macma4_02_g06450) encoding peroxidases in banana were enriched in the response to oxidative stress (GO:0006979) (Table S3). The target genes in Foc were not enriched in any GO terms related to oxidative stress (Table S4).
4. Discussion
milRNAs involved in plant-pathogen interaction have been discovered in some pathogenic fungi by the application of high-throughput sequencing techniques and bioinformatics analysis (Wang et al. Citation2017; Jin et al. Citation2019; Feng et al. Citation2021). In this study, we identified six infection-induced milRNAs by profiling the sRNAs of Foc during pure culture and early infection stages. These milRNAs, like other milRNAs identified in N. crossa (Lee et al. Citation2010) and F. oxysporum (Chen et al. Citation2014), lack sequence conservation. Their producing sites are mainly located in the intergenic and untranslated regions (3’/5’ UTR and intron). The findings contrast with those of sRNAs discovered in B. cinerea and S. sclerotiorum, where the sRNAs are primarily derived from transposable elements (Weiberg et al. Citation2013; Derbyshire et al. Citation2019). This discrepancy may be attributed to our sequencing data analysis of the elimination of reads mapped to repetitive elements.
In fungi, at least five types of milRNA biogenesis pathways have been reported (Lee et al. Citation2010; Jin et al. Citation2019). Among them, N. crossa milR-3 biogenesis is similar to that of plants, which require only Dicer to produce mature milRNA (Jones-Rhoades et al. Citation2006; Lee et al. Citation2010). The milR-4 is derived from tRNA, and its production is partially dependent on Dicer. The biogenesis of milR-2 requires AGO protein QDE2 and its catalytic activity but is completely independent of Dicer. For milR-1, the most abundant milRNAs in N. crossa, its production requires Dicer, QDE2, QIP, and MRPL3 (Lee et al. Citation2010; Chang et al. Citation2012). Our data showed that the transcript level of milR106 was considerably lower in the ∆FoDCL2 mutant but not in the ΔFoQDE2 and ∆FoDCL1 mutants when compared to the WT. The result indicates that milR106 is a Dicer-dependent milRNA of the milR-3 type. Whereas the transcript levels of the other three milRNAs, milR133, milR138, and milR148, were significantly decreased in both the ΔFoQDE2 and ∆FoDCL2 mutants, like the expression pattern of milR87, which has been identified as a milRNA of the milR-1 type (Li et al. Citation2022).
Increasing evidence indicates that milRNAs produced by plant pathogenic fungi play a crucial regulatory role in developmental processes and pathogenicity (Wang et al. Citation2015; Huang et al. Citation2019). For instance, a novel milRNA (VdmilR1) was identified to accumulate during the late stage of V. dahliae infection and was found to regulate pathogenicity by targeting a virulence gene from the fungus itself (Jin et al. Citation2019). In V. mali, the expression of Vm-milR37 was observed in the mycelium, but not in the stage of the infection process. Overexpression of this milRNA did not affect vegetative growth, but significantly decreased pathogenicity (Feng et al. Citation2021). Here, we identified six milRNAs that were dramatically induced in the early stage of Foc infection (36 hpi). Through deletion and complementation of the precursor gene, we confirmed that milR106 is involved in the infective growth and virulence of Foc. Our previous study also found milR87 contributes to Foc virulence during the early infection stage by silencing a GH79 glycosyl hydrolase, which could activate the general host defence response (Li et al. Citation2022). These investigations on the function of milRNAs in various fungi reveal that milRNAs expressed at different stages of infection and vegetative growth employ diverse mechanisms to regulate pathogen pathogenicity.
Recently, small RNAs were reported to function by trafficking between the host plants and the fungal phytopathogens and play significant role in the nature of the infection (Weiberg and Jin Citation2015; Hua et al. Citation2018; Mahanty et al. Citation2023). In this study, milR106 and milR87 both targeted Macma4_06_g01500, a gene encoding a pentatricopeptide repeat-containing (PPR) protein. PPR proteins have been demonstrated to participate not only in plant growth and development regulation but also in coping with various biotic and abiotic stresses (Liu et al. Citation2010; Xing et al. Citation2018). For instance, PPR proteins found in potatoes are significantly induced in response to the infection of Ralstonia solanacearum, a bacterial wilt pathogen, suggesting their involvement in disease resistance (Park et al. Citation2016).
GO enrichment analysis of target genes from the host plants and the pathogens will help to annotate the function of infection-induced milRNAs. In our study, GO terms of defence response to fungus and cellular response to hypoxia were enriched. Hypoxia stress can be caused by submergence or pathogen infection. These two stresses often occur sequentially or at the same time in nature. Therefore, plants have evolved economical and efficient strategies to deal with the stresses, such as responding to both stresses via a single signalling pathway (Tang and Liu Citation2021; Valeri et al. Citation2021). Although no significant enrichment of GO terms related to oxidative stress was observed among the target genes of milR106 in the pathogen, three peroxidase-encoding genes in the host banana were enriched in the GO term “response to oxidative stress” (GO:0006979). Peroxidases, as integral components of antioxidant defence systems, play a crucial regulatory role in maintaining ROS homoeostasis and supporting cellular functions, such as antioxidant defence and plant defence responses, by modulating ROS levels (Daudi et al. Citation2012). Our finding revealed that milR106 targets several genes encoding peroxidases. Therefore, it is not difficult to understand that infection-induced milR106 may target and suppress plant peroxidase expression, inhibiting plant defence responses such as hypersensitive reaction response and ROS burst, thereby facilitating pathogen infection. In contrast, the loss of milR106 makes the pathogen more sensitive to peroxide accumulation, compromising its ability to counteract the plant’s defence. These findings imply that the target genes associated with these enriched GO terms are important in response to pathogen infection, and the milRNAs that inhibit their target gene expression may play a role in preventing the activation of plant resistance or recognition. However, the functions of these target genes need to be verified further.
Our research about the infection-induced milRNAs in Foc provides a scientific theoretical foundation for exploring the function of this kind of milRNAs in fungi. It also supplies potential targets for the development of an efficient BFW control strategy, as well as the breeding of banana-resistant cultivars.
Supplemental Material
Download JPEG Image (178.3 KB){kind=link}
Supplemental Material
Download MS Excel (52.8 KB)Acknowledgments
We are grateful to Prof. Yingzi Yun, College of Plant Protection, Fujian Agriculture and Forestry University for providing a vector of pKNT-G418 for fungal genetic transformation.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21501203.2024.2345917
Additional information
Funding
References
- Ahmad F, Martawi NM, Poerba YS, De Jong H, Schouten H, Kema GHJ. 2020. Genetic mapping of Fusarium wilt resistance in a wild banana Musa acuminata ssp malaccensis accession. Theor Appl Genet. 133(12):3409–3418. doi: 10.1007/s00122-020-03677-y.
- Bartel DP. 2004. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5.
- Chang S, Zhang Z, Liu Y. 2012. RNA interference pathways in fungi: Mechanisms and functions. Annu Rev Microbiol. 66(1):305–323. doi: 10.1146/annurev-micro-092611-150138.
- Chen R, Jiang N, Jiang Q, Sun X, Wang Y, Zhang H, Hu Z, Han KH. 2014. Exploring microRNA-like small RNAs in the filamentous fungus Fusarium oxysporum. PLoS One. 9(8):e104956. doi: 10.1371/journal.pone.0104956.
- Daudi A, Cheng Z, O’Brien JA, Mammarella N, Khan S, Ausubel FM, Bolwell GP. 2012. The apoplastic oxidative burst peroxidase in Arabidopsis is a major component of pattern-triggered immunity. The Plant Cell. 24(1):275–287. doi: 10.1105/tpc.111.093039.
- De Wever J, Tulkens D, Verwaeren J, Everaert H, Rottiers H, Dewettinck K, Lefever S, Messens K. 2020. A combined RNA preservation and extraction protocol for gene expression studies in cacao beans. Front Plant Sci. 11:992. doi: 10.3389/fpls.2020.00992.
- Derbyshire M, Mbengue M, Barascud M, Navaud O, Raffaele S. 2019. Small RNAs from the plant pathogenic fungus Sclerotinia sclerotiorum highlight host candidate genes associated with quantitative disease resistance. Mol Plant Pathol. 20(9):1279–1297. doi: 10.1111/mpp.12841.
- Dita M, Barquero M, Heck D, Mizubuti ESG, Staver CP. 2018. Fusarium wilt of banana: Current knowledge on epidemiology and research needs toward sustainable disease management. Front Plant Sci. 9:1468. doi: 10.3389/fpls.2018.01468.
- Feng H, Xu M, Gao Y, Liang J, Guo F, Guo Y, Huang L. 2021. Vm-milR37 contributes to pathogenicity by regulating glutathione peroxidase gene VmGP in Valsa mali. Mol Plant Pathol. 22(2):243–254. doi: 10.1111/mpp.13023.
- He B, Wang H, Liu G, Chen A, Calvo A, Cai Q, Jin H. 2023. Fungal small RNAs ride in extracellular vesicles to enter plant cells through clathrin-mediated endocytosis. Nat Commun. 14(1):4383. doi: 10.1038/s41467-023-40093-4.
- Hua C, Zhao J, Guo H. 2018. Trans-kingdom RNA silencing in plant-fungal pathogen interactions. Mol Plant. 11(2):235–244. doi: 10.1016/j.molp.2017.12.001.
- Huang C, Wang H, Hu P, Hamby R, Jin H. 2019. Small RNAs – big players in plant-microbe interactions. Cell Host Microbe. 26(2):173–182. doi: 10.1016/j.chom.2019.07.021.
- Jin Y, Zhao P, Fang Y, Gao F, Guo H, Zhao J. 2018. Genome-wide profiling of sRnas in the Verticillium dahliae-infected Arabidopsis roots. Mycology. 9(3):155–165. doi: 10.1080/21501203.2018.1426062.
- Jin Y, Zhao J, Zhao P, Zhang T, Wang S, Guo H. 2019. A fungal milRNA mediates epigenetic repression of a virulence gene in Verticillium dahliae. Philos Trans R Soc B-Biol Sci. 374(1767):20180309. doi: 10.1098/rstb.2018.0309.
- Jones-Rhoades MW, Bartel DP, Bartel B. 2006. MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol. 57:19–53. doi: 10.1146/annurev.arplant.57.032905.105218.
- Lau SK, Chow WN, Wong AY, Yeung JM, Bao J, Zhang N, Lok S, Woo PC, Yuen KY, Vinetz JM. 2013. Identification of microRNA-like RNAs in mycelial and yeast phases of the thermal dimorphic fungus Penicillium marneffei. PLoS Negl Trop Dis. 7(8):e2398. doi: 10.1371/journal.pntd.0002398.
- Lee HC, Li L, Gu W, Xue Z, Crosthwaite SK, Pertsemlidis A, Lewis ZA, Freitag M, Selker EU, Mello CC, et al. 2010. Diverse pathways generate microRNA-like RNAs and Dicer-independent small interfering RNAs in fungi. Mol Cell. 38(6): 803–814. doi: 10.1016/j.molcel.2010.04.005.
- Li M, Shi J, Xie X, Leng Y, Wang H, Xi P, Zhou J, Zhong S, Jiang Z. 2013. Identification and application of a unique genetic locus in diagnosis of Fusarium oxysporum f. sp. cubense tropical race 4. Can J Plant Pathol. 35(4):482–493. doi: 10.1080/07060661.2013.828321.
- Li M, Xie L, Wang M, Lin Y, Zhong J, Zhang Y, Zeng J, Kong G, Xi P, Li H, et al. 2022. FoQDE2-dependent milRNA promotes Fusarium oxysporum f. sp. cubense virulence by silencing a glycosyl hydrolase coding gene expression. PLoS Pathog. 18(5):e1010157. doi: 10.1371/journal.ppat.1010157.
- Li M, Xie X, Lin X, Shi J, Ding Z, Ling J, Xi P, Zhou J, Leng Y, Zhong S, et al. 2014. Functional characterization of the gene FoOCH1 encoding a putative α-1, 6-mannosyltransferase in Fusarium oxysporum f. sp. cubense. Fungal Genet Biol. 65:1–13. doi: 10.1016/j.fgb.2014.01.005.
- Li X, Yu S, Cheng Z, Chang X, Yun Y, Jiang M, Chen X, Wen X, Li H, Zhu W, et al. 2024. Origin and evolution of the triploid cultivated banana genome. Nature Genet. 56(1):136–142. doi:10.1038/s41588-023-01589-3.
- Liu Y, He J, Chen Z, Ren X, Hong X, Gong Z. 2010. ABA overly-sensitive 5 (ABO5), encoding a pentatricopeptide repeat protein required for cis-splicing of mitochondrial nad2 intron 3, is involved in the abscisic acid response in Arabidopsis. Plant. 63:749–765. doi: 10.1111/j.1365-313X.2010.04280.x.
- Mahanty B, Mishra R, Joshi RK. 2023. Cross-kingdom small RNA communication between plants and fungal phytopathogens-recent updates and prospects for future agriculture. RNA Biol. 20(1):109–119. doi: 10.1080/15476286.2023.2195731.
- Nicolás FE, Ruiz-Vázquez RM. 2013. Functional diversity of RNAi-associated sRnas in fungi. Int J Mol Sci. 14(8):15348–15360. doi: 10.3390/ijms140815348.
- Ouyang S, Park G, Ji H, Borkovich KA. 2021. Small RNA isolation and library construction for expression profiling of small RNAs from Neurospora crassa and Fusarium oxysporum and analysis of small RNAs in Fusarium oxysporum-infected plant root tissue. Methods Mol Biol. 2170:199–212. doi: 10.1007/978-1-0716-0743-5_14.
- Park S, Gupta R, Krishna R, Kim ST, Lee DY, Hwang DJ, Bae SC, Ahn IP. 2016. Proteome analysis of disease resistance against Ralstonia solanacearum in potato cultivar CT206-10. Plant Pathol J. 32(1):25–32. doi: 10.5423/PPJ.OA.05.2015.0076.
- Reuter JS, Mathews DH. 2010. Rnastructure: Software for RNA secondary structure prediction and analysis. BMC Bioinf. 11(1):129. doi: 10.1186/1471-2105-11-129.
- Tang H, Liu H. 2021. Roles of single gene in plant hypoxia and pathogen responses. Plant Signal Behav. 16(10):1934295. doi: 10.1080/15592324.2021.1934295.
- Valeri MC, Novi G, Weits DA, Mensuali A, Perata P, Loreti E. 2021. Botrytis cinerea induces local hypoxia in Arabidopsis leaves. New Phytol. 229(1):173–185. doi: 10.1111/nph.16513.
- Viljoen A, Ma L, Molina AB. 2020. Fusarium Wilt (Panama disease) and monoculture in banana production: Resurgence of a century-old disease. In: Emerging Plant Diseases and Global Food Security. St Paul: APS Press; p. 159–184.
- Wang B, Sun Y, Song N, Zhao M, Liu R, Feng H, Wang X, Kang Z. 2017. Puccinia striiformis f. sp. tritici microRNA-like RNA 1 (Pst-milR1), an important pathogenicity factor of Pst, impairs wheat resistance to Pst by suppressing the wheat pathogenesis-related 2 gene. New Phytol. 215(1):338–350. doi: 10.1111/nph.14577.
- Wang M, Weiberg A, Dellota EJ, Yamane D, Jin H. 2017. Botrytis small RNA Bc-siR37 suppresses plant defense genes by cross-kingdom RNAi. RNA Biol. 14(4):421–428. doi: 10.1080/15476286.2017.1291112.
- Wang M, Weiberg A, Jin H. 2015. Pathogen small RNAs: A new class of effectors for pathogen attacks. Mol Plant Pathol. 16(3):219–223. doi: 10.1111/mpp.12233.
- Warman NM, Aitken EAB. 2018. The movement of Fusarium oxysporum f. sp. cubense (sub-tropical race 4) in susceptible cultivars of banana. Front Plant Sci. 9:1748. doi: 10.3389/fpls.2018.01748.
- Weiberg A, Jin H. 2015. Small RNAs-the secret agents in the plant-pathogen interactions. Curr Opin Plant Biol. 26:87–94. doi: 10.1016/j.pbi.2015.05.033.
- Weiberg A, Wang M, Lin F, Zhao H, Zhang Z, Kaloshian I, Huang H, Jin H. 2013. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science. 342(6154):118–123. doi: 10.1126/science.1239705.
- Xia Z, Wang Z, Kav NNV, Ding C, Liang Y. 2020. Characterization of microRNA-like RNAs associated with sclerotial development in Sclerotinia sclerotiorum. Fungal Genet Biol. 144:103471. doi: 10.1016/j.fgb.2020.103471.
- Xing H, Fu X, Yang C, Tang X, Guo L, Li C, Xu C, Luo K. 2018. Genome-wide investigation of pentatricopeptide repeat gene family in poplar and their expression analysis in response to biotic and abiotic stresses. Sci Rep. 8(1):2817. doi: 10.1038/s41598-018-21269-1.
- Zhang E, Zhang J, Zhao R, Lu Y, Yin X, Lan X, Luo Z. 2022. Role of microRNA-like RNAs in the regulation of spore morphological differences in the entomopathogenic fungus Metarhizium acridum. Pol J Microbiol. 71(3):309–324. doi: 10.33073/pjm-2022-028.
- Zhang L, Cenci A, Rouard M, Zhang D, Wang Y, Tang W, Zheng S. 2019. Transcriptomic analysis of resistant and susceptible banana corms in response to infection by Fusarium oxysporum f. sp. cubense tropical race 4. Sci Rep. 9(1):8199. doi: 10.1038/s41598-019-44637-x.