
ABSTRACT
There has been growing interest in the role of B cells in antitumour immunity and potential use in adoptive cellular therapies. To date, the success of such therapies is limited. The intrinsic capacity of B cells to specifically activate tumour-specific CD4+ T cells in vivo via TCR-dependent interactions remains poorly defined. We have developed an in vivo tumour model that utilizes MHCII I-E restriction which limits antigen presentation to tumour-specific CD4 T cells to either tumour-specific B cells or host myeloid antigen presenting cells (APCs) in lymphopenic RAG-/-mice. We have previously shown that these naive tumour-specific CD4+ T cells can successfully eradicate established tumours in this model when activated by host APCs. When naïve tumour-specific B cells are the only source of I-E+ APC, very limited proliferation of naïve CD4+ T cells is observed, whereas host I-E+ APCs are potent T cell activators. B cells pre-activated with an anti-CD40 agonistic antibody in vivo support increased T cell proliferation, although far less than host APCs. CD4+ T cells that have already differentiated to an effector/central memory phenotype proliferate more readily in response to naïve B cells, although still 100-fold less than in response to host APCs. This study demonstrates that even in a significantly lymphopenic environment, myeloid APCs are the dominant primary activators of tumour-specific T cells, in contrast to the very limited capacity of tumour-specific B cells. This suggests that future anti-tumour therapies that incorporate activated B cells should also include mechanisms that activate host APCs.
GRAPHICAL ABSTRACT
Introduction
Despite the success of immune checkpoint therapy, there is an ongoing need to broaden response rates in patients with cancer. While antigen presenting cell (APC)-T cell interactions are key, the roles of professional APCs are very difficult to assess in patients and the relative contributions of dendritic cells (DCs) versus B cells are not well understood. Tumour infiltrating B cells have been observed in a growing number of cancer types and likely interact with T cells in both secondary lymphoid tissues and tumour tissue.Citation1 Recent interest has focussed on B cells as a candidate for CD4+ T cell-based cellular vaccine approaches.
Both DCs and B cells express high levels of surface MHC class II (MHCII). MHCII expression by DCs is generally accepted as the initial driver of CD4+ T cell activation, while B cell MHCII is required for receipt of antigen-specific MHCII-restricted CD4+ T cell help. However B cell MHCII expression may also play a role in primary CD4+ T cell activation in certain settings. In vitro CD40L-activated B cells have shown some promise in stimulating anti-tumour CD4+ and CD8+ T cell responses leading to reduced tumour growth in mice.Citation2 The specific mechanisms involved remain unclear. It is not known whether such tumour responses involve transfer of tumour antigen to endogenous APCs, nor whether they are driven by CD4+ T cells alone or a combination of CD8+ and CD4+ T cells. Moreover, the in vivo capacity of tumour-specific B cells to activate tumour-specific CD4+ T cell responses without prior in vitro manipulation has not been assessed.
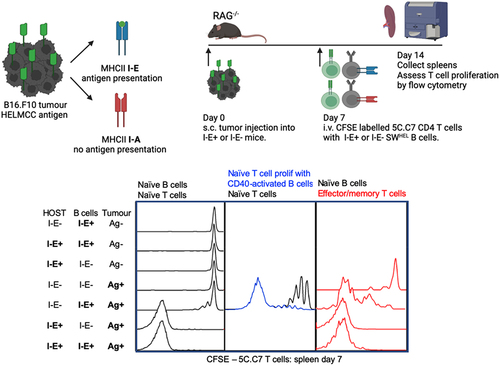
To establish an MHCII-restricted in vivo model to test the ability of tumour-specific B cells to specifically activate tumour-specific CD4+ T cells, we utilized Hen Egg Lysozyme (HEL) specific SWHEL B cellsCitation3 and Moth Cytochrome C residues 87–103 (MCC) T cell receptor (TCR) transgenic 5C.C7 CD4+ T cells in combination with a B16.F10 tumour line that expresses recombinant HELMCC antigen containing the relevant B and T cell epitopes.Citation4,Citation5 The ability of tumour-specific B cells to activate tumour-specific CD4+T cells independently of other APCs was determined. A key aspect of this model is that the 5C.C7 CD4+ T cell can recognize its cognate antigen MCC only when presented on the MHCII I-E allele. H-2b C57BL/6 (termed B6) mice lack the ability to produce functional MHCII I-E molecules, whereas H-2k B10.BR (termed BR) mice have a functional I-E gene. Through the specific interbreeding of B6 and BR mouse lines, wildtype expression levels of MHCII are maintained while antigen-presentation via MHCII I-E expression can be limited to either host APCs or adoptively transferred SWHEL B cells. As hosts of adoptively transferred T and B cells, the model utilizes lymphopenic Rag2-/- tumour-bearing mice that have been shown to support naïve T cell activation in multiple tumour models.Citation5,Citation6 In the B16.F10 HELMCC model used here, we have previously demonstrated that Rag2-/- mice support potent IFN-γ-dependent CD4+ tumour responses capable of complete tumour clearance.Citation5 The purpose of this study was to specifically ask if naïve and activated tumour specific B cells have the capacity to activate naïve tumour specific CD4 T cells in vivo without the confounders of other endogenous B cell or CD4 T cell populations being present. In addition, this model dissects this CD4 T cell: B cell interaction in the absence of CD8 T cells, allowing us to uniquely address the interactions of B cells and CD4 T cells in an MHC-restricted system.
Methods
Mice
All mice were bred and housed under SPF conditions in the Centenary Institute Animal Facility. SWHEL mice were a gift from Robert Brink.Citation3 These mice generate HyHEL10 B cell receptor (BCR)-expressing B cells and antibodies specific for the model antigen Hen Egg Lysozyme (HEL) and can switch to all antibody isotypes. SWHEL mice were maintained on a C57BL/6 Rag2−/− backgroundCitation7 and crossed with Rag2−/−mice on a B10.BR (H-2k) background when required. 5C.C7 TCR transgenic (tg) mice specific for Moth Cytochrome C residues 87–103 (MCC) plus I-EkCitation8 have been described previously. 5C.C7 TCR tg mice were maintained on a Rag1−/− B10.BR (H-2k) background and crossed with C57BL/6 Rag1−/− mice for experimental use. Host and donor mice were also bred to express various combinations of CD45.1 and CD45.2 in order to unequivocally identify adoptively transferred cells.
Cell lines and immunizations
The B16.F10 melanoma cell line was originally obtained from ATCC. B16.F10 cells were retrovirally transducedCitation9 to express HELMCC, which consists of HEL protein with residues 64–76 replaced with residues 87–103 of MCC.Citation10 To generate a membrane bound form of HELMCC, the connecting peptide, transmembrane and cytoplasmic domains of H-2KbCitation11 were fused at the C-terminus. A stable high expressing clone designated B16.mHELMCC was used for all experiments.Citation5 1 × 106 B16.mHELMCC cells were injected subcutaneously (s.c.) seven days prior to intravenous transfer of 5C.C7 CD4+ T cells and/or SWHEL B cells, when large palpable tumours were present.
For in vivo activation of B cells, SWHEL mice received a s.c. immunization with 5 × 106 live B16.mHELMCC tumour cells followed by two intraperitoneal (i.p.) injections of anti-CD40 (FGK45, 25 μg/injection) on days 3 and 6. To generate effector/memory (eff/mem) 5C.C7 T cells, TCR tg mice were immunized s.c. in both flanks and the neck scruff with a total of 10 μg of MCC peptide 87–103 emulsified in Freund’s complete adjuvant (CFA) 3 weeks before cell harvest.
Adoptive cell transfer, flow cytometry analysis and cell sorting
For adoptive transfer, SWHEL B cells were isolated from spleens and TCR Tg 5C.C7 T cells from pooled lymph nodes. B cells were co-transferred with T cells at a 5:1 ratio (5×106 B cells, 1 × 106 T cells). Eff/mem 5C.C7 T cells were FACS sorted as CD4+TCR+CD44hi CD62L±CD103− using FACSAria II or Influx BD sorters. Samples were analyzed on LSR-II, Fortessa or FACSCanto BD flow cytometers. Antibodies were obtained from BD Pharmingen or eBioscience, or produced from B cell hybridomas in-house. The following monoclonal Abs were used to stain cells: anti-CD4(RM4–5), anti-CD11b(M1/70), anti-NK1.1(PK136), anti- CD45(30-F11), anti-MHCII(M5/114.15.2), anti-Ter119(TER 119) and anti-B220(RA3-6B2) obtained from BD Biosciences (Franklin Lakes, NJ, USA); anti-CD19(6D5), anti-CD45.2(104), anti- CD45.1(A20) and anti-Gr1(RB6-8C5) obtained from BioLegend (San Diego, CA, USA). All antibodies were directly conjugated. Non-specific binding to Fc receptors blocked using anti- CD16/32 purified in house from the 2.4G2-hybdridoma.
Statistics
All tests were performed using GraphPad Prism Software. For nonparametric data, the Mann-Whitney test to compare ranks was used. For parametric data, unpaired Student t tests were used for comparisons between two populations and one-way ANOVA was used when comparing more than two groups. All data shown as mean ± SEM. (*, P < .05; **, P < .01; ***, P < .001; ****, P < .0001). There were no data point exclusions for each experiment.
Results
To compare the ability of tumour-specific B cells and host myeloid APCs to present tumour antigen to CD4+ T cells, we developed a general experimental approach of adoptively co-transferring CFSE labeled 5C.C7 T cells with either I-E positive or I-E negative (termed I-E+ and I-E−, respectively) SWHEL B cells, into I-E+ or I-E− tumour bearing hosts (). A 5:1 B:T ratio was chosen to provide ample numbers of APCs for initial activation. Spleens and lymph nodes were harvested seven days after adoptive transfer for flow cytometric analysis (). The gating strategy to identify 5C.C7 T cells and SWHEL B cells is shown in Supplementary Fig. S1A. When naïve T cells and naïve B cells were co-transferred, no T cell proliferation was observed in the spleen 7 days later, if I-E was absent from both host APCs and SWHEL B cells (). Limited T cell proliferation was observed when SWHEL B cells expressed I-E. Strong T cell proliferation occurred when host APCs expressed I-E (), with the vast majority of transferred T cells becoming CFSE-negative (having undergone > 7 cell divisions). Increased frequency () and absolute number () of T cells was seen. Divided T cells were predominantly CD62Llo CD44hi effector cells (). Spontaneous proliferation of adoptively transferred CD4+ T cells was not observed when tumour antigen was not present, even when both host APCs and SWHEL B cells expressed I-E (Sup ). T cell frequencies in tumour draining lymph nodes were similar to those obtained for spleens (Sup ).
Figure 1. Naïve tumour-specific B cells are poor activators of naïve tumour-specific CD4+ T cells in vivo despite a lymphopenic environment.

B cell expansion in the spleen was the greatest when both host myeloid APCs and transferred SWHEL B cells expressed I-E (). When only B cells expressed I-E, modest expansion was observed when compared to the I-E negative control. This was reflected in both frequency () and absolute number of transferred B cells (). Similar B cell frequencies were observed in tumour draining lymph nodes (Sup ). Interestingly, when host APCs were the only source of I-E, SWHEL B cells were rapidly deleted. This was in the context of strong CD4+ T cell proliferation in response to host APCs. Given T cell and B cell frequencies were similar between the spleen and tumour draining lymph nodes, only spleens were analysed in subsequent experiments.
In vitro stimulated B cells have been shown to activate CD4+ T cells in anti-tumour responses.Citation12 We next examined if in vivo activated B cells could stimulate naive CD4+ T cell proliferation. Activated SWHEL B cells were co-transferred with naïve CFSE labeled 5C.C7 CD4+ T cells into I-E- tumour bearing hosts (). Moderate T cell expansion was seen for activated compared to naïve B cells (), with some T cells undergoing > 7 rounds of division. This was reflected in both increased frequency () and absolute number (). There was a trend toward an increase in activated compared to naïve SWHEL B cell number but this did not reach statistical significance (). A significant increase in the MHCII I-E expression level on in vivo activated B cells () may have contributed to the observed increase in T cell activating capacity. We have previously demonstrated these in vivo activated B cells express CD19, B220, CD23, HEL+ with subtle upregulation of MHC-II and CD95.Citation4,Citation5 Importantly, no proliferation is observed when naïve 5C.C7 CD4 T cells are transferred into I-E- anti-CD40 activated SWHEL transgenic tumour bearing mice (Figure S1f). These data suggest that in vivo activated tumour-specific B cells have a modest capacity to activate naïve CD4+ T cells.
Figure 2. CD40 activated tumour-specific B cells have a limited capacity to activate naïve tumour-specific CD4 T cells.

The activation threshold of eff/mem T cells is known to be lower than that of naïve T cells,Citation13 so we tested whether naïve B cells could activate eff/mem CD4+ T cells. Similar to the naïve setting, strong proliferation was induced when host myeloid APCs could present antigen, with all adoptively transferred T cells undergoing at least 8 cell divisions (). This expansion was reflected in both increased frequency () and absolute number of T cells . When antigen presentation was limited to naïve B cells, eff/mem T cells proliferated more than naïve T cells ( compared with ). However B cells generated 100-fold fewer eff/mem T cells than host APCs.
Figure 3. Naïve tumour-specific B cells have a limited capacity to activate effector/memory tumour-specific CD4 T cells.

In the presence of eff/mem T cells, B cell expansion was the greatest when both host myeloid APCs and transferred SWHEL B cells expressed MHC I-E (), although it was lower than in the presence of naïve T cells (). There was no significant difference in B cell expansion when B cells were the only APC expressing I-E, compared to the I-E negative control. Interestingly, deletion of I-E negative SWHEL B cells was once again observed in the presence of strong host APC-induced T cell proliferation. Taken together, these data suggest that B cells have only a limited capacity to induce CD4+ T cell proliferation to tumour antigens in vivo.
Discussion
The dominant MHCII-dependent activators of both naïve and effector memory CD4+ T cells in this model were host myeloid APCs, with B cells playing only a minor role, even after prior in vivo activation of the T or B cells. The small degree of activation experienced by CD4+ T cells responding to B cell-presented antigen in our experiments is unlikely to fully support an effective anti-tumour response, even in a highly lymphopenic Rag2−/− environment. In contrast, host APC-dependent activation drives a potent CD4+-dependent anti-tumour response in this mouse model.Citation5
Similar results have been observed in a mouse model of experimental autoimmune encephalomyelitis (EAE) in which Archambault et al. used a cre-mediated conditional approach to limit antigen presentation to CD19+ cells.Citation14 They observed only limited CD4+ T cell activation in initial and secondary responses. These experiments were conducted in wild type mice with wild type B cells. Importantly, when presentation was limited to B cells, T cell activation was insufficient to induce EAE disease.
These data do not negate the involvement of B cells in optimal antigen presentation in more complex anti-tumour immune responses involving both CD4+ and CD8+ T cells. There has been growing interest in the role of B cells in anti-tumour immunity and a number of possible roles have been considered [Reviewed inCitation15]. B cell-based vaccines may influence a number of established MHCII dependent and independent functions, including antigen presentation to both CD4+ and CD8 T+ cells, antibody production and potentially suppression via regulatory B cell populations. In contrast, although DCs have generally been considered to be the more relevant APC for T cell priming in a wide range of contexts, tumour lysate vaccination and DC-based adoptive cellular therapies have shown only limited success in treating cancer [Reviewed inCitation16]. In addition, DC-based therapy faces major practical constraints, whereas B cells can be easily purified from the peripheral blood of patients.
In vitro T cell proliferation and IFNγ production has been observed in response to polyclonal B cells purified from PBMCs and cultured with CD40L and tumour cell lysates.Citation17 Similarly, CD40-activated B cells loaded with myeloma lysates were capable of activating myeloma antigen-specific T cells in vitro.Citation18 These results collectively demonstrate that polyclonal human B cells stimulated in vitro can contribute to T cell mediated anti-tumour immune responses. The ability to identify human B cells with tumour reactive specificities in vitro may also generate new therapeutic approaches.Citation18
In murine models, several lines of evidence support a role for B cells in anti-tumour responses. In the B16.F10 mouse melanoma model, in vivo depletion of murine B cells with an anti-CD20 monoclonal antibody attenuated the antigen-specific responses of CD4+ and CD8+ T cells.Citation19 Antibodies coupled to tumour proteins have been shown to induce CD8+ T cell activation via cross presentation of antibody complexes by mouse DCs.Citation20 In addition, a cancer vaccine based on coupling tumour antigens such as her-2/neu to an scFv anti-CD19 mAb has been shown to reduce tumour growth.Citation21
Our data suggest that therapies aimed at generating CD4+ dependent anti-tumour T cell responses should not target B cells alone. They also underline the importance of T cell:B cell contact for B cell survival during activation. B cell deletion was apparent when B cells lacked MHCII I-E but host APCs did not (). The deleted SWHEL B cells were still capable of binding the HEL antigen but were unable to make MHCII-specific interactions with proliferating T cells responding to host APCs. It is possible that activation with tumour-derived antigen and absence of T cell help may be inducing anergy leading to deletion of the SWHEL B cells. Landmark studies of the HEL transgenic B cell system demonstrated that in contexts where HEL was expressed as a self-antigen, B cells underwent rapid deletion within 1 week, similar to what was observed in these experiments.Citation22 Furthermore, T cells may play a role in deletion of anergic B cells through CD40- and Fas-ligands interactions.Citation23,Citation24 This phenomenon is currently under investigation. We appreciate that the use of high affinity transgenic CD4 T cell and B cells in combination with lymphopenic RAG-/- mice and a transplantable tumour line is an artificial system and does not recapitulate the complexities of the adaptive immune systems interaction with cancer. However despite this, it is striking how poor B cells were at activating the tumour-specific CD4 T cells in one of the most lymphopenic systems available to study in mice. This reiterates the importance of DCs being the most important APC in the activation of naïve CD4 T cells in vivo. Of note, maximum T cell and B cell expansion in the effector/memory experiments described in were an order of magnitude lower compared to the experiments described in . This may reflect partial homing of effector T cells directly to the tumour tissue rather than the spleen, but may also reflect a blunted ability to proliferate compared to naïve CD4 T cells, which is consistent with previous comparisons of naïve vs effector T cell proliferation.Citation25
In summary, while published studies indicate that B cells may have important role in the cooperative immune response against tumours, our use of MHC-restriction to isolate the in vivo effects of tumour-specific B cells on tumour-specific CD4+ T cells indicate that primary cognate interactions between CD4+ T cells and B cells are unlikely to serve as a major mechanism of CD4+ T cell activation.
Supplemental Material
Download MS Word (304.1 KB)Acknowledgments
We thank C. Jolly for providing HEL-Alexa647 and scientific discussion; the staff of the Centenary Institute Flow Cytometry and Animal Facilities for technical support, and members of the T cell biology and Immune Imaging Labs for scientific discussion. This work was supported by Australian National Health and Medical Research Council grants 1001020, 1012930 and 1051843 and a New South Wales Cancer Council grant RG 13-13.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Raw data were generated at the Centenary Institute. Derived data supporting the findings of this study are available from the corresponding author BF on request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2023.2290799
Additional information
Funding
References
- Wennhold K,Thelen, M, Lehmann, J, Schran, S, Preugszat, E, Garcia-Marquez, M, Lechner, A, Shimabukuro-Vornhagen, A, Ercanoglu, MS, Klein, F, et al. CD86(+) antigen-presenting B cells are increased in Cancer, localize in tertiary lymphoid structures, and induce specific T-cell responses. Cancer Immunol Res. 2021;9(9):1098–8.
- Wennhold K, Weber TM, Klein-Gonzalez N, Thelen M, Garcia-Marquez M, Chakupurakal G, Fiedler A, Schlösser HA, Fischer R, Theurich S, et al. CD40-activated B cells induce anti-tumor immunity in vivo. Oncotarget. 2017;8(17):27740–27753. doi:10.18632/oncotarget.7720.
- Phan TG, Amesbury M, Gardam S, Crosbie J, Hasbold J, Hodgkin PD, Basten A, Brink R. B cell receptor–independent stimuli trigger immunoglobulin (ig) class switch recombination and production of IgG autoantibodies by anergic self-reactive B cells. J Exp Med. 2003;197(7):845–60. doi:10.1084/jem.20022144.
- Guy TV, Terry AM, Bolton HA, Hancock DG, Zhu E, Brink R, McGuire HM, Shklovskaya E, de St Groth BF. Collaboration between tumor-specific CD4+ T cells and B cells in anti-cancer immunity. Oncotarget. 2016;7(21):30211–29. doi:10.18632/oncotarget.8797.
- Shklovskaya E, Terry AM, Guy TV, Buckley A, Bolton HA, Zhu E, Holst J, Fazekas de St Groth B. Tumour-specific CD4 T cells eradicate melanoma via indirect recognition of tumour-derived antigen. Immunol Cell Biol. 2016;94(6):593–603. doi:10.1038/icb.2016.14.
- Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EKM, Muranski P, Restifo NP, Antony PA. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010;207(3):651–67. doi:10.1084/jem.20091921.
- Shinkai Y, Lam, KP, Oltz, EM, Stewart, V, Mendelsohn, M, Charron, J, Datta, M, Young, F, Stall, AM, Alt, FW. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68(5):855–867. doi:10.1016/0092-8674(92)90029-C.
- Seder RA, Paul WE, Davis MM, Fazekas de St Groth B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J Exp Med. 1992;176(4):1091–1098. doi:10.1084/jem.176.4.1091.
- Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DAA. Generation of T-cell receptor retrogenic mice. Nat Protoc. 2006;1(1):406–17. doi:10.1038/nprot.2006.61.
- Shklovskaya E, Roediger B, Fazekas de St Groth B. Epidermal and dermal dendritic cells display differential activation and migratory behavior while sharing the ability to stimulate CD4+ T cell proliferation in vivo. J Immunol. 2008;181(1):418–30. doi:10.4049/jimmunol.181.1.418.
- Brooks A, Hartley S, Kjer-Nielsen L, Perera J, Goodnow CC, Basten A, McCluskey J. Class II-restricted presentation of an edogenously derived immunodominant T-cell determinant of hen egg lysozyme. Proc Natl Acad Sci U S A. 1991;88(8):3290–3294. doi:10.1073/pnas.88.8.3290.
- Li Q, Lao X, Pan Q, Ning N, Yet J, Xu Y, Li S, Chang AE. Adoptive transfer of tumor reactive B cells confers host T-Cell immunity and tumor regression. Clin Cancer Res. 2011;17(15):4987–4995. doi:10.1158/1078-0432.CCR-11-0207.
- Rogers PR, Dubey C, Swain SL. Qualitative changes accompany memory T cell generation: faster, more effective responses at lower doses of antigen. J Immunol. 2000;164(5):2338–2346. doi:10.4049/jimmunol.164.5.2338.
- Archambault AS, Carrero JA, Barnett LG, McGee NG, Sim J, Wright JO, Raabe T, Chen P, Ding H, Allenspach EJ, et al. Cutting edge: conditional MHC class II expression reveals a limited role for B cell antigen presentation in primary and secondary CD4 T cell responses. J Immunol. 2013;191(2):545–550. doi:10.4049/jimmunol.1201598.
- Laumont CM, Banville AC, Gilardi M, Hollern DP, Nelson BH. Tumour-infiltrating B cells: immunological mechanisms, clinical impact and therapeutic opportunities. Nat Rev Cancer. 2022;22(7):414–430. doi:10.1038/s41568-022-00466-1.
- Linette GP, Carreno BM. On the twentieth anniversary of dendritic cell vaccines - riding the next wave. Cancer Res. 2022;82(6):966–968. doi:10.1158/0008-5472.CAN-21-4440.
- Lapointe R, Bellemare-Pelletier A, Housseau F, Thibodeau J, Hwu P. CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res. 2003;63:2836–43.
- Kim SK, Nguyen Pham T-N, Nguyen Hoang TM, Kang H-K, Jin C-J, Nam J-H, Chung S-Y, Choi SJN, Yang D-H, Kim Y-K, et al. Induction of myeloma-specific cytotoxic T lymphocytes ex vivo by CD40-activated B cells loaded with myeloma tumor antigens. Ann Hematol. 2009;88(11):1113–1123. doi:10.1007/s00277-009-0721-y.
- DiLillo DJ, Yanaba K, Tedder TF. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol. 2010;184(7):4006–4016. doi:10.4049/jimmunol.0903009.
- Pham CD, Woo M-Y, Kim Y-S, Park S, Kwon M-H. An anti-nucleic acid antibody delivers antigen to the cross-presentation pathway in dendritic cells and potentiates therapeutic antitumor effects. J Immunol. 2012;189(12):5755–63. doi:10.4049/jimmunol.1200804.
- Ma Y, Xiang D, Sun J, Ding C, Liu M, Hu X, Li G, Kloecker G, Zhang H-G, Yan J, et al. Targeting of antigens to B lymphocytes via CD19 as a means for tumor vaccine development. J Immunol. 2013;190(11):5588–5599. doi:10.4049/jimmunol.1203216.
- Fulcher DA, Basten A. Reduced life span of anergic self-reactive B cells in a double-transgenic model. J Exp Med. 1994;179(1):125–134. doi:10.1084/jem.179.1.125.
- Rathmell JC, Townsend SE, Xu JC, Flavell RA, Goodnow CC. Expansion or elimination of B cells in vivo: dual roles for CD40- and fas (CD95)-ligands modulated by the B cell antigen receptor. Cell. 1996;87(2):319–29. doi:10.1016/S0092-8674(00)81349-5.
- Fulcher DA, Lyons AB, Korn SL, Cook MC, Koleda C, Parish C, Fazekas de St Groth B, Basten A. The fate of self-reactive B cells depends primarily on the degree of antigen receptor engagement and availability of T cell help. J Exp Med. 1996;183(5):2313–28. doi:10.1084/jem.183.5.2313.
- Bikah G, Pogue-Caley RR, McHeyzer-Williams LJ, McHeyzer-Williams MG. Regulating T helper cell immunity through antigen responsiveness and calcium entry. Nat Immunol. 2000;1(5):402–12. doi:10.1038/80841.