
ABSTRACT
Huntington's Disease is caused by inheritance of a single disease-length allele harboring an expanded CAG repeat, which continues to expand in somatic tissues with age. Whether somatic expansion contributed to toxicity was unknown. From extensive work from multiple laboratories, it has been made clear that toxicity depended on length of the inherited allele, but whether preventing or delaying somatic repeat expansion in vivo would be beneficial was unknown, since the inherited disease allele was still expressed. In Budworth et al., we provided definitive evidence that suppressing the somatic expansion in mice substantially delays disease onset in littermates that inherit the same disease-length allele. This key discovery opens the door for therapeutic approaches targeted at stopping or shortening the CAG tract during life. The analysis was difficult and, at times, non-standard. Here, we take the opportunity to discuss the challenges, the analytical solutions, and to address some controversial issues with respect to expansion biology.
Introduction
Over the years, the proteins that contributed to the mutation underlying HD were deduced by crossing HD mice with mice lacking key DNA repair enzymes.Citation1,2 Specifically, the proteins whose loss in mice attenuated or reduced expansion were implicated in causing the mutation. However, no one had taken the next step to determine whether somatic expansion had effects on pathophysiology. It was difficult to take that step because loss of DNA repair often comes with pathology of its own. The most notable example of this was the R6/1 Citation3-7 or HdhQ111 Citation8,9 crosses with the Msh2(−/−) mouse. Indeed, loss of Msh2(−/−) attenuated expansions of CAG tracts in all of the tested HD crosses, but the relationship to toxicity was difficult to establish. Msh2(−/−) animals are characterized by methylation tolerance, hyper-recombination, and lymphomas at early ages (peak at 8 weeks of age), as well as global instability in repetitive elements throughout the genome.Citation10 Although many laboratories had generated HD/Msh2(−/−) animals over the years, so many other sites expanded, contracted, or recombined that it was impossible to link pathophysiology to expansion in the disease allele.
Loss of 7,8-dihydro-8-oxo-guanine (8-oxo-G) glycosylase (ogg1) in HD animals, on the other hand, suppressed CAG expansion without widespread pathology.Citation11-13 Thus, for the first time, we were able to construct a line in which all littermates inherited the disease allele, but only some (those with ogg1) could expand it somatically. We generated mice by breeding the HD mouse knock-in line Hdh(Q150/Q150) of 150 CAG repeats with the ogg1(−/−) mouse to generate Hdh(Q150/Q150)/ogg1(−/−) or Hdh(Q150/Q150)/ogg1(+/+) genotypes. To determine whether somatic instability contributed to disease, all we needed to do was to compare the onset of pathophysiology in the two lines, i.e., compare the onset of toxicity in Hdh(Q150/Q150)/ogg1(+/+) (somatic expansion) and Hdh(Q150/Q150)/ogg1(−/−) animals (no somatic expansion). However, the analysis was difficult. The methods we used were key to resolve the effects of the inherited and somatic repeats, but did not follow the most conventional paradigms. Here, we discuss and clarify, in detail, the methodology we used to draw conclusions, and why particular analysis approaches were chosen over others. We deal with three relevant issues: (1) how to distinguish the significance of small expansions, (2) the variability in animal testing, and (3) the basis for the experimental design of animal testing. These methods may be useful to others. Finally, we comment on two issues of relevance to expansion toxicity, the contribution of large versus small expansion, and whether some brain regions contribute more than others to toxicity.
Results and discussion
An approach for measuring small variable changes in repeat tracts that occur at the onset of behavioral phenotypes
The first goal was to establish that loss of ogg1 reduced somatic expansion in a large group of animals. Expansion is easily and robustly measured during age in mouse models, particularly in mid-age or in older animals. However, our goal in this analysis was to measure the impact of expansion size at the beginning of onset. It goes without saying that we expected the changes in repeat tract size to be small, and the analysis would be challenging. Was there a mathematical basis to evaluate the impact of somatic changes at early stages in disease? Could we discriminate differences in somatic expansion between Hdh(Q150/Q150)/ogg1(+/+) and Hdh(Q150/Q150)/ogg1(−/−) animals if the repeat tract changes were small and non-Gaussian?
Indeed, the key to evaluating small changes is a large n. To deal with the small sizes, we created distributions comprising the sum of all the tract changes in length that occurred in 1200 animals between 5 weeks to 40 weeks.Citation12 For any one animal, the size of repeat tracts at a particular age comprises hundreds of lengths (see , each panel is a brain region from a single animal), many of which will change as the animal ages. Considering all animals and summing all changes in tract lengths in each one, the probability that loss of ogg1 reduces the somatic expansion size is based on hundreds of repeat tract changes in any particular age group, and thousands of repeat tract changes if ages between 5–40 weeks are pooled. The pooling provided the large n that was needed for robust statistics. Indeed, linear regression confirmed with high probability that loss of ogg1 reduced the somatic expansion size early in their life. To ensure that the repeat tracts were analyzed in an unbiased manner, the repeat tract length for all samples was commercially analyzed (ACGT inc., Illinois USA), and these data directly forwarded for statistical analysis of repeat changes. What we learned from the analysis was that the majority of small individual changes were spread throughout the distribution of expansion size from the Hdh(Q150/Q150)/ogg1(+/+) and Hdh(Q150/Q150)/ogg1(−/−) animals. Thus, one could not always see differences in the size distributions by visual inspection, but the pooled changes could be calculated for statistical significance. Although both lines inherited the same disease allele, the probability that somatic expansion was higher in Hdh(Q150/Q150)/ogg1(+/+) line was significant.
Figure 1. The mutation length distribution within tissues of Hdh(CAG)150 mice at 11 months. The histograms show a representative sample of CAG repeat lengths within the striatum, cortex, cerebellum and liver of an 11-month-old mouse. These data are compiled from small pool PCR analyses of reactions containing only 1–4 Alifiable mutant molecules in a few cells of one animal. The median CAG repeat length and number of mutant alleles (n) examined in each tissue is indicated. STR is striatum, CTX is cortex, CBL is cerebellum. Summing the change in tract length over many animals provides a large n at this particular age group.
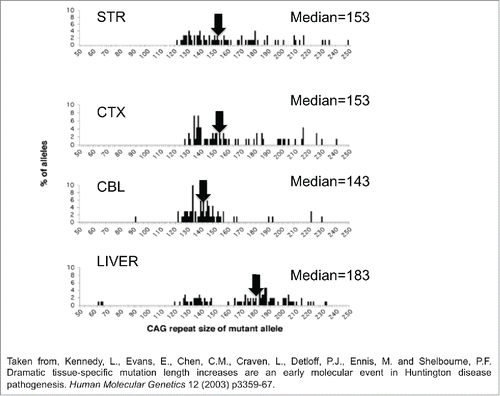
The spread of tract sizes in the distribution in Hdh(Q150/Q150)/ogg1(+/+) line ranged from 90-120 repeats. Thus, a second complication to be faced was how to link a large number of tract sizes in any one age to predict a behavioral response, which was also variable. One solution was to use an average value for repeat tract length and correlate it with the average performance at each age. This approach had significant shortcomings. Although it is easiest to think of measurements in terms of averages (and often very useful to do so), the average is not meaningful in a highly variable situation, and it was unlikely that behavior could be described by the average repeat tract sizes. Peggy Shelbourne nicely illustrates the point in her seminal analysis ().Citation14,15 As she observes in the Hdh(Q150/Q150) line, the repeat distribution among brain regions is intrinsically variable () and spans 100-200 repeats (). The average or median does not reflect well the size distribution (). For example, the median mutation length in the cerebellum (143rpts) is significantly lower than the corresponding measure in the striatum (P < 0 .01) (153rpts) and cortex (P < 0 .001) (153rpts) (). However, most of the sizes in all three tissues cluster around 130-155rpts, i.e., most of the repeats were of similar size. A smaller population of longer repeats skews the striatal and cortical median toward a higher value. The median in the liver is 184rpts, but the distribution is distinctly bi-modal (). Indeed, the average value for the liver repeat tract length does not correlate well with either of the two modes (). Thus, linking performance to the average tract length is not an accurate description of the relationship. Similarly, the average performance of the Hdh(Q150/Q150)/ogg1(+/+) and Hdh(Q150/Q150)/ogg1(−/−) animals is also not a good measure of distribution of performances, as we illustrate in Figure 4A (in Budworth et al., 2015,Citation12).
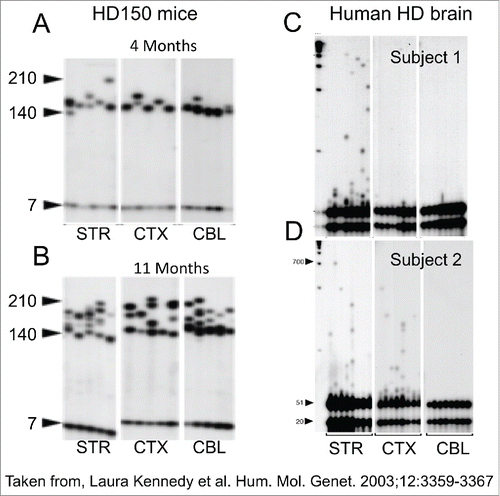
Figure 2. CAG repeat copy number of the progenitor mutation influences tissue-specific mutation length profiles. (A) Representative data from the SP–PCR mutation length profiles within striatum (St), cortex (Cx), cerebellum (Cb) and liver at 4 and 11 months of age in Hdh(Qwt/150) mice. Each lane contains the products from 1 to 10 cells worth of DNA from the tissue indicated. The numbers on the left hand side of the panels indicate the number of CAG repeats carried by the Hdh alleles amplified (mo=months). (B) Dramatic mutation length increases in human HD striata prior to pathological cell loss. Tissue from subject 1 shows mutation length variability in the striatum but not cortex (Brodmann's area 7) or hypothalamus. SP–PCR data reveal that while the median mutation length within tissues is ˜41 CAG repeats, some cells within the striatum contain mutations >1000 CAG repeats in length. Each lane contains the amplification products from ˜50 cells. (C,D) Tissue from subject 2 has mutation length variability in the striatum and cortex (Brodmann's area 7) but not cerebellum. SP–PCR data indicate the median mutation length within both tissues is ˜51 CAG repeats but some striatal cells contain mutations >700 CAG repeats in length. Each lane contains the amplification products from 40 to 50 cells. The numbers on the left side of each panel indicate the number of CAG repeats carried by the HD alleles amplified. The lanes marked M contain the 1Kb plus size marker.
To more accurately link the distribution of repeat tract sizes to behavior, we took a two-part analysis. First, as described above, we established, by linear regression, that the somatic expansion was greater in Hdh(Q150/Q150)/ogg1(+/+) animals relative to Hdh(Q150/Q150)/ogg1(−/−) animals. We subdivided the expansion size according to age groups differing by 10 or 20 week intervals, and the entire distribution for the Hdh(Q150/Q150)/ogg1(+/+) and Hdh(Q150/Q150)/ogg1(−/−) genotypes was compared at each age. For example, if the repeats between 10-20 weeks in Hdh(Q150/Q150)/ogg1(+/+) were larger than the Hdh(Q150/Q150)/ogg1(−/−) as judged by linear regression, then this age group was assigned a “yes,” and if the repeats in Hdh(Q150/Q150)/ogg1(+/+) animals were smaller than in the Hdh(Q150/Q150)/ogg1(−/−), the age group was assigned a “no.,” This analysis considered all of the repeats in the distributions, and transformed the entire set into a single value (yes or no at each age) for each genotype. Indeed, repeat tracts in Hdh(Q150/Q150)/ogg1(+/+) were larger than the Hdh(Q150/Q150)/ogg1(−/−) in all age groups in the first 40 weeks. We then considered whether the distribution of behavioral performances was better in each age group. Box plots, a Tukey analysis, evaluated the probability that the entire performance distribution in an age group from a particular genotype is distinct from another. If suppression of somatic expansion accompanied a delay in motor decline, then we could establish a direct relationship between the two. Indeed, somatic expansion (in Hdh(Q150/Q150)/ogg1(+/+) animals) had a negative effect on performance, and suppression of somatic expansion in Hdh(Q150/Q150)/ogg1(−/−) animals provided an improvement in motor function within the age window where somatic expansion was suppressed (between 5 and 40 weeks).
Based on probabilities, differences in performance were observed even when the sizes of the repeat tracts were very small (p=0.04 at 11 weeks and p=.0001 at 31 weeks). Although the performance distributions were variable, the majority of the Hdh(Q150/Q150)/ogg1(−/−) mice significantly outperformed their Hdh(Q150/Q150)/ogg1(+/+) littermates at any age tested below 40 weeks. The loss of somatic expansion in the homozygous Hdh(Q150/Q150)/ogg1(−/−) crosses led to a remarkable 7–10 month delay in the onset of motor decline relative to Hdh(Q150/Q150)/ogg1(+/+) littermates, who had inherited the same disease-length allele.
In the second approach, both the repeat distributions and the behavioral distributions were evaluated by regression analysis to establish whether behavior was a linear function of the repeat length. Indeed, there was a quantitative relationship between performance and somatic length at the time of onset: the smaller repeat tracts corresponded to better performance, and the larger repeat tracts corresponded to poorer performance. Collectively, our findings demonstrate, for the first time, that somatic expansion contributes to toxicity at the age of onset, when the changes were small. The implications are enormous. Drug companies are reluctant to invest in compound development without a basis. The demonstration that suppressing expansion of the repeat tracts delays onset establishes a viable therapeutic option for treating these deadly diseases.
Study design
This leaves us with the final consideration - the study design. In the conventional testing design for mouse models, a group of animals is tested continuously with age to evaluate a developing motor phenotype. While this approach is extremely useful and cost-effective, this type of analysis fails to identify whether the changes in one group of animals describes all animals. Expansion occurs in most animals, each of which will harbor a distinct range of repeat tract lengths.Citation16,17 Therefore, the relationship between repeat tract length and behavior is averaged over many animals, each of which has its own tract length spread. Furthermore, the effects of learning bias on the motor performance over long periods of testing cannot be quantified (in our case 100 weeks of testing).
To deal with this situation, we created random populations in each age group. Each animal group was allowed to reach a defined age, the performance was tested only at that age, and the animal was immediately sacrificed for repeat tract analysis. The value in this approach is power. Each age group independently validated the relationship between behavior and repeat tract length since each age group contained different mice, i.e., the animals at 10–20 weeks were different than the ones at 20–30 weeks. Four age groups rather than one validated the results.
Furthermore, the ancillary measurements such as histology and motor analysis could be performed on the same set of animals.
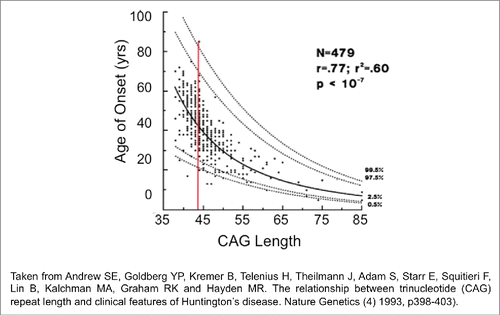
The study design is reminiscent of clinical testing in human patients. All individuals are tested for repeat length and performance to create a population. The same set of HD subjects is not necessarily measured with time. If they were, then we would know only that a handful of patients develop disease at some rate as they aged. We would know nothing about how those humans fit into the general population. Indeed, the problems that can arise from testing only one group are obvious. In humans, the length of the inherited CAG repeat predicts the average age of onset, but any two unrelated HD patients of the same repeat length differ in onset from 20 to 80-years ().Citation18 Therefore, following only a subset of patients may not reflect the population, if only the late onset group happened to be selected for testing. Thus, human tract changes are both small and variable, as we see in our mouse analysis, but there are robust methods to deal with the variability. Statistical evaluation in a population-based analysis is capable of drawing firm conclusions as to whether the onset of disease is generally related to the inherited repeat length.
Figure 3. The wide distribution of behavioral responses for the same CAG repeat length in HD patients. The regression curve was calculated on log transformed data. Confidence intervals for predicted age given the size of the CAG repeat. Outer curves delineate the 99% confidence interval while inner curves show the 95% confidence interval.
Do expansions in all regions contribute equally to toxicity?
Our quantitative analysis provides some insights into two long-standing questions relevant to the impact of expansion on pathophysiology: whether a large population of small repeats has more of an impact than a larger population of small repeats, and the related question, do regions with the longest repeats contribute the most to pathology.Citation19 While in HD, for example, the striatum is the most sensitive area targeted for death, and has the longest alleles [], is it really the striatum that contributes the most to disease onset?
It is widely observed that the striatum has the longest expansion sizes, and Vonsattel classification clearly identifies the striatum as the earliest affected tissue.Citation20 Thus, it makes sense that the striatum would be most sensitive if the somatic repeats are longest in that region and contribute to toxicity. However, in addressing this question, again the averages are not accurate measures to judge the relationship between repeat length and toxicity. The median value of cerebellum (143rpts) and the cortex (153rpts) is different by only 10 repeats, yet by Vonsattel criteria, the onset of toxicity for the striatum (stage I) and cerebellum (stage III) are years apart.
The benefit in linear regression analysis is that the onset of performance decline can be evaluated as a linear function of repeat tract sizes that were in the population. At the time of onset, the short repeats were linearly related to better motor performance, and all four brain regions contribute equally to toxicity.Citation12 That is because all four-brain regions had similar populations of shorter lengths. At the upper end of the distribution in the striatum, the longer tracts were linearly related to poor outcome, but the distribution of lengths differed among brain regions. The striatum had the longest allele lengths, overall, but there were fewer of them. The longest allele sizes in the hippocampus and cerebellum were shorter relative to the striatum, but there were more of them. Thus, we find, using linear regression, that the hippocampus and cerebellum contributed more to the poor performance than did the striatum. Our analysis supports a model in which a larger group of small mutations has more impact on toxicity than does a smaller group of long expansions [see ]. It is important to note that all of the repeat tract changes measured between 5 and 40 weeks in the mouse preceded neuronal death, supporting the hypothesis that some of the clinical features of HD are caused by pathological processes that precede gross neuronal cell death, and are likely to be effects of the somatic changes in the CAG tract.
With the exception of the tissue dependence of toxicity, our results measured at onset in the mouse are consistent with the seminal work of Wheeler and colleagues in human postmortem brains.Citation21 Both analyses agree that the pathology is more significant in individuals with longer repeats, the mouse data and the human predictions differ in the importance of the tissue contribution. This may be because the mouse data are collected directly at the time of onset. Because somatic expansion changes with age, the repeat tracts measured at the end of life in humans must be used to predict the sizes at the time of onset, which occurs decades earlier. Thus, statistically, it is possible that the striatum appears to have a more significant contribution to toxicity than the other regions in human brain, because the toxicity is extrapolated from repeat lengths that occur at the end of life. More analyses will need to be conducted before this question is resolved. On this point, however, it is interesting to note that mutation sizes late in the disease course parallel the aggregate load in human HD brain tissue, which is generally higher in cortex than in the striatum.Citation22 Although the striatum is the most affected region in HD, only 1-4% of striatal neurons in all grades of HD have nuclear aggregates, even though the cortical changes in repeat length were smaller.Citation22
Conclusions
Huntington's Disease (HD) is caused by inheritance of a single disease-length allele harboring an expanded CAG repeat, which continues to expand in somatic tissues with age. We demonstrate in Budworth, et al., 2015.Citation12 that suppression of somatic expansion delays the onset of disease. Our analysis suggests that the increased mutation lengths in somatic cells are likely to have a primary role in affecting pathological events, and that all of the brain regions contribute to disease at onset. Collectively, these results are consistent with the notion that a large group of small changes contribute more to early pathology than do a smaller group of large repeats.
The significance of these findings is substantial. Gene replacement therapy for hereditary disease is a long way off. However, if somatic expansion contributes to disease, then a therapeutic approach is possible by inhibiting the somatic expansions that occur in the brain during life. If we do nothing, a hereditary disease such as HD is a death sentence. However, if suppressing somatic expansion substantially delays the onset of disease, treatment is possible during life. The window widens for future therapeutics designed to shorten the repeat.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Institutes of Health grants ES020766 (to CTM), NS060115 (to CTM), and CA092584 (to CTM).
References
- McMurray CT. Mechanisms of Trinucleotide Repeat Instability During Human Development. Nat Rev Genet 2010; 11(11):786-99; PMID:20953213; http://dx.doi.org/10.1038/nrg2828
- Mirkin SM. Expandable DNA repeats and human disease. Nature 2007 Jun 21; 447(7147):932-40; PMID:17581576; http://dx.doi.org/10.1038/nature05977
- Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genetics 1999; 23(4):p. 471-3; http://dx.doi.org/10.1038/70598
- Owen BA, Yang Z, Lai M, Gajec M, Badger II JD, Hayes JJ, Edelman W Kucherlapati R, Wilson TM, McMurray CT. (CAG)(n)-hairpin DNA Binds to Msh2-Msh3 and Changes Properties of Mismatch Recognition. Nat Struct Mol Biol 2005; 12(8):663-70; PMID:16025128; http://dx.doi.org/10.1038/nsmb965
- Savouret C, Brisson E, Essers J, Kanaar R, Pastink A, te Riele H, Junien C, Gourdon G. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J 2003; 22:2264-73; PMID:12727892; http://dx.doi.org/10.1093/emboj/cdg202
- van den Broek WJ, Nelen MR, Wansink DG, Coerwinkel MM, te Riele H, Groenen PJ, Wieringa B. Somatic expansion behavior of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet 2002; 11:191-8; PMID:11809728; http://dx.doi.org/10.1093/hmg/11.2.191
- Mangiarini L, Sathasivam K, Mahal A, Mott R, Seller M, Bates GP. Instability of highly expanded CAG repeats in mice transgenic for the Huntington's disease mutation. Nat Genetics 1997; 15(2):p. 197-200; http://dx.doi.org/10.1038/ng0297-197
- Dragileva E, Hendricks A, Teed A, Gillis T, Lopez ET, Friedberg EC, Kucherlapati R, Edelmann W, Lunetta KL, MacDonald ME, Wheeler VC. Intergenerational and striatal CAG repeat instability in Huntington's disease knock-in mice involve different DNA repair genes. Neuro Biol Dis 2009; 33(1):p. 37-47; PMID:18930147; http://dx.doi.org/10.1016/j.nbd.2008.09.014
- Wheeler VC, Lebel LA, Vrbanac V, Teed A, te Riele H, MacDonald ME. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum Mol Genet 2003; 12(3):p. 273-81; PMID:12554681; http://dx.doi.org/10.1093/hmg/ddg056
- Bak ST, Sakellariou D, Pena-Diaz J. The dual nature of mismatch repair as antimutator and mutator: for better or for worse. Front Genet 2014; 5: p. 287; http://dx.doi.org/10.3389/fgene.2014.00287
- Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007; 447(7143):p. 447-52; PMID:17450122; http://dx.doi.org/10.1038/nature05778
- Budworth H, Harris FR, Williams P, Lee do Y, Holt A, Pahnke J, Szczesny B, Acevedo-Torres K, Ayala-Pena S, McMurray CT. Suppression of Somatic Expansion Delays the Onset of Pathophysiology in a Mouse Model of Huntington's Disease. PLoS Genetics 2015; 11(8):p. e1005267; PMID:26247199; http://dx.doi.org/10.1371/journal.pgen.1005267
- Mollersen L, Rowe AD, Larsen E, Rognes T, Klungland A. Continuous and periodic expansion of CAG repeats in Huntington's disease R6/1 mice. PLoS Genetics 2010; 6(12):p. e1001242; PMID:21170307; http://dx.doi.org/10.1371/journal.pgen.1001242
- Kennedy L, Evans E, Chen CM, Craven L, Detloff PJ, Ennis M, Shelbourne PF. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet 2003; 12(24):p. 3359-67; PMID:14570710; http://dx.doi.org/10.1093/hmg/ddg352
- Shelbourne PF, Keller-McGandy C, Bi WL, Yoon SR, Dubeau L, Veitch NJ, Vonsattel JP, Wexler NS, Arnheim N, Augood SJ. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet 2007; 16(10):p. 1133-42; PMID:17409200; http://dx.doi.org/10.1093/hmg/ddm054
- Wheeler VC, Auerbach W, White JK, Srinidhi J, Auerbach A, Ryan A, Duyao MP, Vrbanac V, Weaver M, Gusella JF, et al. Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum Mol Genet 1999; 8(1):p. 115-22; PMID:9887339; http://dx.doi.org/10.1093/hmg/8.1.115
- Lee JM, Zhang J, Su AI, Walker JR, Wiltshire T, Kang K, Dragileva E, Gillis T, Lopez ET, Boily MJ, Cyr M, Kohane I, Gusella JF, MacDonald ME, Wheeler VC. A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst Biol 2010; 4:p. 29; PMID:20302627; http://dx.doi.org/10.1186/1752-0509-4-29
- Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, Starr E, Squitieri F, Lin B, Kalchman MA, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genetics 1993; 4(4):p. 398-403; http://dx.doi.org/10.1038/ng0893-398
- Kennedy L, Shelbourne PF. Dramatic mutation instabilityin HD mouse striatum: does polyglutamine load contribute to cell-specific vulnerability in Huntington's disease? Hum. Mol. Genet 2000; 9:2539-44
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP, Jr. Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol 1985; 44:559-77
- Swami M, Hendricks AE, Gillis T, Massood T, Mysore J, Myers RH, Wheeler VC. Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet 2009; 18(16):p. 3039-47; PMID:19465745; http://dx.doi.org/10.1093/hmg/ddp242
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci 1999; 19:2522-34; PMID:10087066