
ABSTRACT
In order to utilize seaweed waste as a feedstock in biorefinery, it is essential to characterize the microbial communities and their metabolic processes in seaweed decomposition. In this study, the phylogeny and functionality of the prokaryotic and fungal microbiome of Sargassum seaweed waste that has inundated the Barbados coast were assessed for the first time by targeting the 16S rRNA gene and internal transcribed spacer (ITS) region of ribosomal RNA, respectively, using the cultivation-independent Illumina next-generation sequencing approach. The resulting 67 032 and 91 085 high-quality paired-end reads of 16S rRNA gene and ITS were assigned to 734 and 179 amplicon sequence variants (ASVs), respectively. The predominant bacterial genera recorded were Muricauda, Aminobacterium, Mesorhizobium, Marinobacter, Reichenbachiella, Hyphomonas, Simiduia, Aquamicrobium, Oceanicola and Alcanivorax. The major genera of Archaea belonged to Methanosarcina, Methanogenium and Methanosaeta, and that of fungi to Sigmoidea, Tylopilus and Mucor. The metabolic functions of the Sargassum metagenome, analysed via PICRUSt, revealed the predominant metabolic pathways related to the biosynthesis of terpenoids and polyketides, the biosynthesis of other secondary metabolites, the metabolism of cofactors and vitamins, and the xenobiotic biodegradation. Furthermore, the prevalence of diverse alginate lyase-producing bacteria suggests that Sargassum waste is an ideal niche material for the isolation of potent microbial biocatalysts to sustainably depolymerize alginate and thereby produce third-generation biofuels and bioactive alginate oligosaccharides with novel industrial applications.
Introduction
The input of nutrients via anthropogenic sources, including agricultural runoff and wastewater discharge, and the impact of coastal upwelling, hurricane and precipitation have been identified as the causal agents of coastal eutrophication and the triggering of algal blooms (Gower & King, Citation2019; Mohapatra, Schambeau, Pannell, & Kiene, Citation2017; Smetacek & Zingone, Citation2013). Global climate and land-use changes have also been identified as the environmental stressors for the induction of seaweed blooms (Morand & Merceron, Citation2005; Oviatt, Huizenga, Rogers, & Miller, Citation2019). Seaweed blooms disrupt the coastal ecosystem functionality via alteration of the biogeochemical cycles (Aires, Muyzer, Serrão, & Engelen, Citation2019; Hervé, Lambourdière, René-Trouillefou, Devault, & Lopez, Citation2021; Smetacek & Zingone, Citation2013). The bloom-forming seaweed species are classified into Chlorophyta, Phaeophyceae (Ochrophyta) and Rhodophyta. Recently, two pelagic species of the brown seaweed Sargassum (S. fluitans and S. natans) have caused massive blooms in the subtropical and tropical Atlantic (Gower & King, Citation2019; Langin, Citation2018). These blooms spanned a distance of 8850 km from the African Gulf of Guinea to the Greater Caribbean Sea and the Gulf of Mexico in the year 2018, with an estimated weight of >20 million tons (Wang et al., Citation2019). The recurring bloom of the holopelagic species of Sargassum has been adversely affecting the regional economy because of the inundation of putrefying biomass (Sargassum waste) onto the pristine beaches, which consequently deters the tourists (Chávez et al., Citation2020). Additionally, the Sargassum inundation is a threat to the public and environmental health since it fosters production of toxic and corrosive hydrogen sulphide and attracts skin-irritating chironomid insects (Merle et al., Citation2021; Resiere et al., Citation2018).
Sargassum seaweed waste is a rich source of proteins, minerals, carbohydrates and vitamins and has been used conventionally as a fertilizer, agricultural biostimulant and animal feed (Fernando, Kim, Kim, & Jeon, Citation2019; Miranda et al., Citation2021; Smetacek & Zingone, Citation2013). Recent studies have documented the concentrations of inorganic arsenic in the biomass of pelagic Sargassum species that are above the acceptable levels recommended by the US Environmental Protection Agency (Davis et al., Citation2021; Rodríguez-Martínez et al., Citation2020). Consequently, biotreatment of Sargassum biomass is recommended prior to its utilization as animal feed (Carrillo-Domínguez, Rodríguez-Martínez, Díaz-Martínez, Magaña-Gallegos, & Cuchillo-Hilario, Citation2023). The major carbohydrate components of Sargassum include alginate (ca. 40%), cellulose (ca. 28%), mannitol (ca. 7%), fucoidans (ca. 6%), mannan (ca. 2%) and laminarin (ca. 0.3%) (Ai et al., Citation2022; Davis, Volesky, & Mucci, Citation2003; Del Río et al., Citation2019; Laeliocattleya, Suloi, Gayatri, Putri, & Anggraeni, Citation2020; Milledge, Nielsen, & Bailey, Citation2016). It is expected that the complex chemical structures of Sargassum waste favour a unique ecological niche for the prevalence of diverse microbial populations (Mohapatra, Citation2017, Citation2018, Citation2022). These microbes play a vital role in facilitating the decomposition of Sargassum waste and thereby influence the surrounding ecosystems via contributing nutrients and other metabolites (Chen et al., Citation2022; Cleary & Huang, Citation2020; Hervé, Lambourdière, René-Trouillefou, Devault, & Lopez, Citation2021; Mohapatra, Citation2021a, Citation2021c). In recent years, there has been a growing interest in valorizing the seaweed waste, especially to depolymerize alginate, via utilizing the decomposed biomass as a feedstock for sustainable generation of bioenergy and value-added products with potential to be applied in agriculture, biorefinery, food processing and pharmaceutical industries (Kostas, Adams, Ruiz, Duran-Jimenez, & Lye, Citation2021; Miranda et al., Citation2021; Mohapatra, Citation2021b). The increased interest in the utilization of seaweed waste in industrial processes, especially in biorefinery, gives impetus to the characterization of the microbiomes present in the waste.
In this paper, the phylogenetic diversity of the prokaryotic and fungal communities associated with the Sargassum waste piled up at two different beaches off Barbados coast were examined via the cultivation-independent Illumina sequencing approach by targeting the 16S rRNA gene and the internal transcribed spacer (ITS) region of ribosomal RNA, respectively. Additionally, the metabolic functions of the Sargassum waste metagenome were predicted based on 16S rRNA marker gene sequences’ data (Langille et al., Citation2013). The alginate-depolymerizing efficacy of the identified prokaryotic species was also assessed via a carbohydrate-active enzymes (CAZy) database (Drula et al., Citation2022). This molecular analysis provides insight, not only into the phylogeny of microorganisms associated with the Sargassum waste piles but also their predicted functional profiles. This appears to be the first report characterizing the phylogeny and functionality of the microbiome of pelagic Sargassum seaweed waste inundating Barbados’ coast. Knowledge of the microbiome composition and the metabolic functions of its components in Sargassum waste piles are essential in evaluating the microbiome’s impact on coastal ecosystems and on efficient valorization of these renewable biomasses. Additionally, this information will assist in the isolation and identification of novel microbial strains for use in the fermentation of seaweed waste and production of bioactive compounds.
Materials and methods
Collection of Sargassum waste
The samples were collected in quadruplicates from Sargassum waste inundating two beaches [Harrismith (HS; 13.12ºN, 59.42ºW) and Long Beach (LB; 13.06ºN, 59.49ºW)], off the coast of Barbados. The sample collection sites at both beaches were located away from the splash zone. The Sargassum waste samples were comprised of the decomposed biomasses of two pelagic species of Sargassum, S. fluitans and S. natans (). Due to the decomposition of Sargassum biomasses, it was not possible to identify their morphotypes. After collection, samples were stored in sterile containers and transported to the laboratory on ice. Upon arrival, the samples were gently rinsed with autoclaved seawater to remove the dirt and were stored at −80ºC until further processing.
Figure 1. An image showing the Sargassum waste used in this study.

DNA extraction
One gram of frozen Sargassum biomass from each sample was homogenized with a sterile mortar and pestle. Then, 0.25 g of the homogenate was used for extraction of DNA via the SurePrepTM soil DNA isolation kit (Thermo Fisher Scientific, Fair Lawn, New Jersey, USA) according to the manufacturer’s instructions. The extracted DNA was resuspended in the supplied elution buffer and stored at −80ºC. The DNA concentrations were quantified using QuantiFluorTM ONE dsDNA System (Promega, Madison, Wisconsin, USA) and Quantus™ Fluorometer (Promega, Madison, Wisconsin, USA).
Illumina amplicon sequencing
The DNA extracted from quadruplicate Sargassum samples from each beach was pooled into one representative sample, then sent in dry ice to the ZymoBIOMICS® targeted sequencing service (Zymo Research Service, Irvine, California, USA) for sequencing. The sequencing libraries for prokaryotic (Bacteria and Archaea) and fungal communities were prepared using Quick NGS Library Preparation Kit (Zymo Research, Irvine, California, USA) by targeting the V3-V4 region of the 16S rRNA gene and the ITS2 region of rRNA, respectively. The libraries were prepared using qPCR to minimize the chimera formation. The cleaning of the libraries was performed using the Select-a-Size DNA Clean and Concentrator™ (Zymo Research Service, Irvine, California, USA) and quantified using TapeStaion™ (Agilent Technologies, Santa Clara, California, USA) and Qubit™ (Thermo Fisher Scientific, Waltham, Washington, USA). The sequencing of the libraries was conducted by using an Illumina MiSeq platform with a v3 reagent kit (600 cycles) and a 10% PhiX spike-in.
Bioinformatics
The unique amplicon sequence variants (ASVs) were curated from the raw reads of each beach sample after removal of the potential sequencing errors and chimeric sequences via the DADA2 (v.1.26) pipeline (Callahan et al., Citation2016). Taxonomy assignments of unique ASVs were conducted using the UCLUST program (v. 1.2.22) integrated in QIIME v.1.9.1 (Caporaso et al., Citation2010) and Zymo Research’s internally curated 16S database for prokaryotes (Zymo Research Service, Irvine, California, USA) and the UNITE database (Nilsson et al., Citation2018) for fungi as the references. Alpha- and beta-diversity of the microbial communities were assessed via QIIME v.1.9.1 (Caporaso et al., Citation2011).
Functional predictions of the metagenome
The putative functions of the metagenome of Sargassum waste samples from each beach were predicted based on the 16S rRNA gene sequences dataset via the EzBiocloud 16S-based MTP pipeline with XOR algorithm and PICRUSt analysis (ChunLab, Inc., Seoul, South Korea). Functional classifications of the metagenome were performed according to the Kyoto Encyclopedia of Gene and Genome (KEGG) modules (i.e., functional units of gene sets in KEGG metabolic pathways).
Results
Sequence information
A total of 168 634 and 186 862 paired-end raw sequences were acquired through the targeting of the V3-V4 region of the 16S rRNA gene and ITS2 region of the rRNA, respectively, from the Sargassum waste samples collected from two different beaches, Long Beach (LB: 78 552 and 82 540) and Harrismith Beach (HS: 90 082 and 104 322). After quality filtering, including trimming, paired-end reads merging, removal of the chimeric sequences and size separation, a total of 67 032 and 91 085 sequences (LB: 27 899 and 40 354; HS: 39 133 and 50 731) with an average size of 414 bp and 320 bp for V3-V4 and ITS2 regions, respectively, were retained. The retained sequences of V3-V4 and ITS2 regions were assigned to 734 and 179 amplicon sequence variants (ASVs), respectively, via the DADA2 (v. 1.26) pipeline.
Prokaryotic and fungal diversity in Sargassum seaweed waste
The rarefaction curves of the reads that were used to estimate the alpha diversity (richness: Sobs; diversity: Shannon and Simpson; evenness: Pielou’s evenness) indices displayed a plateau for the prokaryotes (Supplementary fig. S1) and fungi (Supplementary fig. S2) in both the sites (LB and HS) indicating that the sequencing depths were adequate for characterizing the prokaryotic and fungal communities with respect to taxonomy and predictive functions. For prokaryotes, the values of the indices, Sobs, Shannon, Simpson and Pielou’s evenness were recorded as 482, 4.88, 0.98 and 0.79, respectively, for LB and 359, 4.03, 0.97 and 0.68, respectively, for HS. The Sobs, Shannon, Simpson and Pielou’s evenness values of fungal communities were estimated as 80, 3.48, 0.75 and 0.79, respectively, for LB and 101, 1.93, 0.49 and 0.42, respectively, for HS. The beta diversity of the prokaryotic and fungal communities of LB (0.62 and 0.06) and HS (0.62 and 0.06) sites indicated that there was no significant difference.
Taxonomic composition of prokaryotic and fungal communities in Sargassum seaweed waste
Prokaryotes
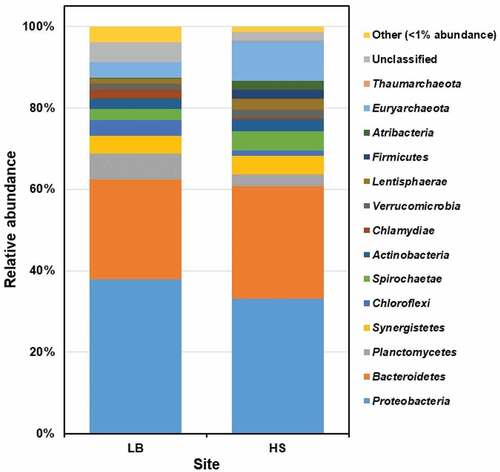
More than 91% of the acquired amplicon sequence variants (ASVs) of Sargassum waste collected from LB (91.2%) and HS (96.6%) beaches were categorized into 12 bacterial and two archaeal phyla. The relative abundances (%) of the bacterial phyla were Proteobacteria (LB: 37.8 and HS: 33.2), Bacteroidetes (LB: 24.6 and HS: 27.6), Planctomycetes (LB: 6.33 and HS: 2.93), Synergistetes (LB: 4.51 and HS: 4.58), Chloroflexi (LB: 3.83 and HS: 1.33), Spirochaetae (LB: 2.73 and HS: 4.65), Actinobacteria (LB: 2.48 and HS: 2.99), Chlamydiae (LB: 2.23 and HS: 0.32), Verrucomicrobia (LB: 1.44 and HS: 1.92), Lentisphaerae (LB: 1.99 and HS: 2.88), Firmicutes (LB: 0.12 and HS: 2.18) and Atribacteria (LB: 0.11 and HS: 2.11); and that of the archaeal phyla Euryarchaeota (LB: 3.8 and HS: 9.94) and Thaumarchaeota (LB: 0 and HS: 0.06) (). The other bacterial phyla with low relative abundances (LB: 3.98% and HS: 1.44%) were identified as Acidobacteria, Saccharibacteria, Latescibacteria, Chlorobi, Gemmatimonadetes, Hydrogenedentes, Parcubacteria, Gracilibacteria, Cyanobacteria, Deferribacteres, Armatimonadetes and Tenericutes. About 6.82% of the ASVs (LB: 4.86 and HS: 1.96) were unclassified.
Figure 2. The prokaryotic community composition of Sargassum waste at phylum level. LB, Long Beach; HS, Harrismith beach. The DNA extracted from quadruplicate Sargassum samples from each beach was pooled into one representative sample, and then used to prepare the Illumina sequencing libraries targeting the V3-V4 region of the 16S rRNA gene.
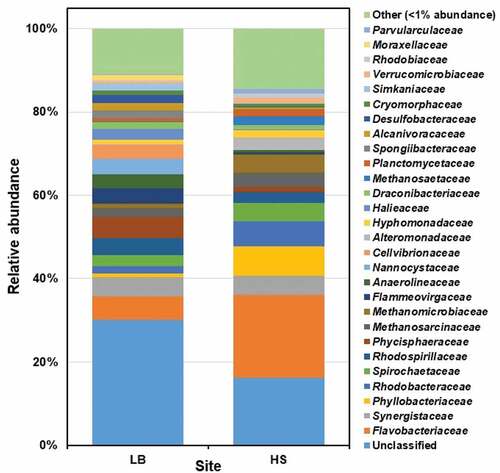
Twenty-eight prokaryotic families were detected at >1% relative abundance of ASVs in at least one sample (). The relative abundance of the unclassified prokaryotic populations at the family level was recorded as 30.1% and 16.2% for LB and HS, respectively. The dominant bacterial families were Flavobacteriaceae (LB: 5.65% and HS: 20%), Synergistaceae (LB: 4.51% and HS: 4.58%), Rhodobacteraceae (LB: 1.78% and HS: 6.04%), Spirochaetaceae (LB: 2.68% and HS: 4.4%), Rhodospirillaceae (LB: 4% and HS: 2.63%), Phycisphaeraceae (LB: 5.13% and HS: 1.18%) and Hyphomonadaceae (LB: 1.03% and HS: 1.89%). The archaeal families were Methanosarcinaceae (LB: 2.18% and HS: 3.4%) and Methanomicrobiaceae (LB: 1.09% and HS: 4.36%).
Figure 3. The prokaryotic community composition of Sargassum waste at family level. LB, Long Beach; HS, Harrismith beach. The DNA extracted from quadruplicate Sargassum samples from each beach was pooled into one representative sample, and then used to prepare the Illumina sequencing libraries targeting the V3-V4 region of the 16S rRNA gene.
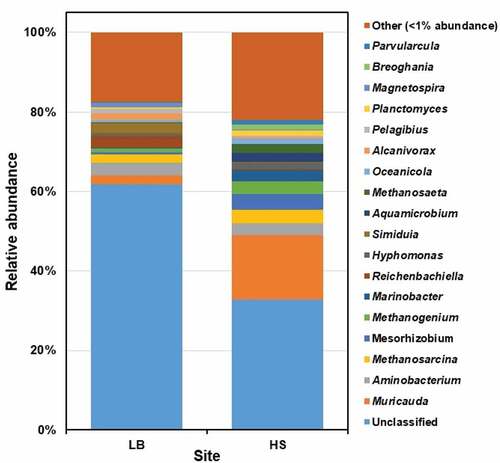
The prokaryotic communities at the genus level (>1% relative abundance of ASVs in at least one sample) were classified into 18 genera, which accounts for 32.9% of the total number of the ASVs (). About 47% of the ASVs were not classified at the genus level, indicating the presence of novel prokaryotic lineages in Sargassum waste. The 10 most abundant bacterial genera were Muricauda, Aminobacterium, Mesorhizobium, Marinobacter, Reichenbachiella, Hyphomonas, Simiduia, Aquamicrobium, Oceanicola and Alcanivorax, with the relative abundance ranging from 1% to 16.3%. The three most abundant archaeal genera were identified as Methanosarcina, Methanogenium and Methanosaeta with the relative abundance in the range 1–3%.
Figure 4. The prokaryotic community composition of Sargassum waste at genus level. LB, Long Beach; HS, Harrismith beach. The DNA extracted from quadruplicate Sargassum samples from each beach was pooled into one representative sample, and then used to prepare the Illumina sequencing libraries targeting the V3-V4 region of the 16S rRNA gene.
Fungi
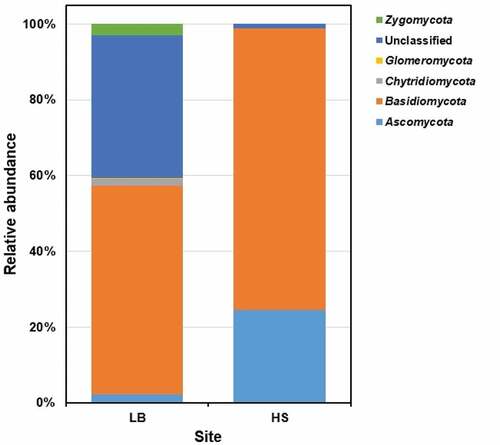
Out of the 179 ASVs acquired via sequencing the ITS2 biomarker of Sargassum waste samples from LB and HS beaches, 44 (24.6%) were not classified beyond the Fungi domain, with UNITE database indicating the presence of novel lineages. The remaining 135 ASVs (75.4%) were assigned to five phyla. The most dominant fungal phylum that was recorded in both the beaches was Basidiomycota (LB: 55.1%; HS: 74.3%), followed by Ascomycota (LB: 2.33%; HS: 24.5%), Zygomycota (LB: 3.01%; HS: 0%), Chytridiomycota (LB: 1.83%; HS: 0%) and Glomeromycota (LB: 0.03%; HS: 0%) (). At the class level, Agaricomycetes (LB: 54.5%; HS: 74.3%) of phylum Basidiomycota and Sordariomycetes (LB: 1.47%; HS: 24.3%) of phylum Ascomycota were the dominant. In Ascomycota, the highest relative abundance (LB: 1.2%; HS: 24.2%) was noted for the order Microascales and the family Halosphaeriaceae. Within Basidiomycota, the highest relative abundance (LB: 47.9%; HS: 74.3%) was recorded for the order Sebacinales and the family Sebacinaceae.
Figure 5. The fungal community composition of Sargassum waste at phylum level. LB, Long Beach; HS, Harrismith beach. The DNA extracted from quadruplicate Sargassum samples from each beach was pooled into one representative sample and then used to prepare the Illumina sequencing libraries targeting the ITS2 region of the rRNA.
The relative abundance (>1% in at least one sample) of assignable fungal ASVs was dominated by the genera Sigmoidea (24.2%), Tylopilus (2.55%) and Mucor (2.54%). The other low abundance genera (<1% of ASVs in at least one sample) belonged to Amantia, Aspergillus, Coltricia, Cortinarius, Derxomyces, Mortierella, Ophiocordyceps, Ophiostoma, Penicillium, Pulchromyces, Rhodotorula and Trechispora. The relative abundance of fungal taxa at the level of class, order, family and genus are displayed in Supplementary figs S3, S4, S5 and S6, respectively.
Functional predictions of Sargassum waste metagenome
The categorization of the predicted metagenome profiles at the levels of functional units of gene sets resulted in nine KEGG metabolic pathway modules (Suplementary table S1). Furthermore, these nine metabolic pathway modules consisted of 61 KEGG modules (Supplementary table S1). The metabolic pathways related to the biosynthesis of terpenoids and polyketides were predicted to be the most abundant (36.9%) in Sargassum waste. The other predicted metabolic pathways were related to amino acid metabolism, the biosynthesis of other secondary metabolites, the energy metabolisms, the metabolism of cofactors and vitamins, the xenobiotic biodegradation, the nucleotide metabolisms, the carbohydrate metabolism and the lipid metabolism with an average abundance of 16.6%, 16.3%, 11.9%, 11.5%, 5.71%, 0.7%, 0.35% and 0.11%, respectively.
Alginate-depolymerizing potential of prokaryotic communities
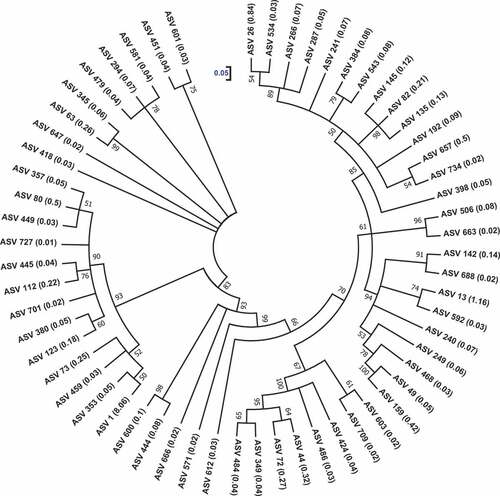
The prokaryotic communities of Sargassum waste were capable of depolymerizing alginate (10 177 of 67 032 reads: ca. 15%) by producing alginate lyases of seven polysaccharide lyase families – PL5, PL6, PL7, PL8, PL14, PL15 and PL17. Of all the ASV reads (10 177) of identified prokaryotic species analysed via CAZy, the PL7 family was recorded as the most abundant (69.5%), followed by PL17 (12.1%), PL5 (9.16%), PL14 (5.3%), PL15 (3.1%), PL8 (0.6%) and PL6 (0.19%). These alginate lyase-producing prokaryotic communities were classified into 60 ASVs. The phylogenetic relatedness of these ASVs and their relative abundances (%) are depicted in . These 60 alginate lyase-producing ASVs were classified into 35 genera of five phyla, such as Bacteroides, Proteobacteria, Planctomycetes, Verrucomicrobia and Chlorobi with a relative abundance of 10.2%, 4.36%, 0.73%, 0.04% and 0.02%, respectively (Supplementary table S2). At the genus level (>0.5% relative abundance), the predominant producers were Muricauda (8.1%; ASV1), followed by Hyphomonas (1.2%; ASV13), Alcanivorax (1%; ASVs 26, 266, 287, 534), Planctomyces (0.73%; ASVs 44, 72, 349, 424, 484, 486), Owenweeksia (0.59%; ASVs 63, 294, 345, 418, 451, 479, 581, 601, 647), Tamlana (0.55%; ASVs 80, 357, 701), Labrenzia (0.5%; ASVs 49, 159) and Microbulbifer (0.5%; ASV657).
Figure 6. The phylogenetic relatedness of alginate lyases-producing prokaryotic communities associated with Sargassum waste. ASV, amplicon sequence variant. The cladogram was drawn using maximum-likelihood algorithm. The bootstrap percentage values of 1000 replications are specified at the nodes. The scale denotes 0.05 substitutions per nucleotide position. Numbers inside the parentheses denote the relative abundance (%) of the ASVs.
Discussion
Recent industrial interests in sustainable bioconversion of seaweed waste in biorefineries necessitate the characterization of their microbiome. In this study, Sargassum waste samples were collected from two different beaches in Barbados, and the taxonomic composition and functional potentials of the microbiome were assessed. The resulting alpha diversity indices indicated a high diversity of prokaryotic and fungal communities associated with Sargassum waste. The high proportion of shared ASVs and occurrence of similar predominant prokaryotic and fungal taxa suggested that the microbiome of Sargassum waste, sampled from two different beaches, were relatively similar. Furthermore, the beta diversity indices of both the sites indicated that there were no significant differences among the resulting phylogenetic lineages, as well as abundance of prokaryotic and fungal communities. The resulting ASVs of prokaryotic (734) and fungal (179) communities were classified into 12 (predominantly Proteobacteria, followed by Bacteroidetes, Planctomycetes, Synergistetes, Chloroflexi, Spirochaetae, Actinobacteria, Chlamydiae, Verrucomicrobia, Lentisphaerae, Firmicutes and Atribacteria) and five phyla (predominantly Basidiomycota, followed by Ascomycota, Zygomycota, Chytridiomycota and Glomeromycota), respectively.
Previous studies have also documented Proteobacteria as the dominant bacterial phylum in other Sargassum species, such as S. hemiphyllum (Cleary & Huang, Citation2020), S. muticum (Serebryakova et al., Citation2018) and two drifting species (S. fluitans and S. natans) (Hervé, Lambourdière, René-Trouillefou, Devault, & Lopez, Citation2021). However, there is a difference at the family and genus level. The dominant bacterial families were reported as Microtrichaceae, Xenococcaceae, Flavobacteriaceae for S. ilicifolium (Oh, Bollati, Maithani, Huang, & Wainwright, Citation2021); Rhodobacteraceae and Saprospiraceae for two drifting species (S. fluitans and S. natans) (Torralba et al., Citation2017). Mei, Wu, Zhao, Yan, & Jiang (Citation2019) reported that Flavobacterium, Paracoccus, Kocuria, Pseudomonas and Bacteroides were the predominant bacterial genera of S. horneri microbiome.
Methanosarcina, Methanogenium and Methanosaeta were identified as the three predominant archaeal genera of Sargassum waste (relative abundance 1–3%). These three methanogenic archaeal genera, especially Methanosarcina, were reported to be tolerant to extreme conditions (Akyol, Ince, & Ince, Citation2019) and have industrial potential for biogenic methane production using Sargassum waste as the feedstock in anaerobic digesters (Leong & Chang, Citation2022; Thakur et al., Citation2022). Hervé, Lambourdière, René-Trouillefou, Devault, & Lopez (Citation2021) have reported the prevalence of methanogenic archaeal families, Methanomicrobiaceae, Methanosarcinaceae and Methanococcaceae in the biomasses of the pelagic species of Sargassum (S. fluitans and S. natans).
With regard to the fungal communities, previously, Wainwright, Bauman, Zahn, Todd, & Huang (Citation2019) reported the occurrence of four dominant fungal orders, such as Eurotiales, Hypocreals, Lecanorales and Pleosporales in the mycobiome of S. ilicifolium. Additionally, Euromycetes and Aspergillaceae were recorded as the most common class and family of S. ilicifolium mycobiome, respectively. The dominant fungal genera recorded here from Sargassum waste [Sigmoidea (24.2%), Tylopilus (2.55%) and Mucor (2.54%)] were previously reported as the potent degraders of polysaccharides by producing carbohydrate-active enzymes that convert renewable biomass into various value-added products, including biofuels, platform and fine chemicals (Chen, Taylor, Burke, & Cairney, Citation2001; Patyshakuliyeva, Falkoski, Wiebenga, Timmermans, & de Vries, Citation2019; Wolfe, Kuo, & Pringle, Citation2012).
In the present study, the metabolic functions of the metagenome of Sargassum waste were predicted. The prevalence of a high abundance of metabolic pathways associated with the biosynthesis of terpenoids and polyketides (subfunctions: enediyne biosynthesis; macrolide biosynthesis; polyketide sugar unit biosynthesis; type II polyketide biosynthesis; and terpenoid backbone biosynthesis), the biosynthesis of other secondary metabolites (subfunctions: β-lactams biosynthesis; other antibiotic biosynthesis; and other bacterial compound biosynthesis) and the metabolism of cofactors and vitamins (subfunction: cofactor and vitamin metabolism) indicates the potential of Sargassum waste associated microbial communities for the isolation of novel biologically active substances, including antibiotic (Keatinge-Clay, Maltby, Medzihradszky, Khosla, & Stroud, Citation2004; Schneider, Citation2005), anticancer (Van Lanen & Shen, Citation2008), antitumor (Lombo, Menendez, Salas, & Mendez, Citation2006; Shen, Liu, & Nonaka, Citation2003), immunosuppressant (Park et al., Citation2010), antiparasite (Gaynor & Mankin, Citation2003) and antioxidant (Gliszczynska-Swiglo, Citation2007) compounds. Additionally, the occurrence of metabolic pathways associated with the xenobiotic degradation (subfunctions: aromatics degradation) suggest the biotechnological importance of these microorganisms for environmentally friendly remediation of toxic aromatics including benzene (KEGG modules M00547, M00548), benzoate (KEGG module M00551), cymene (KEGG module M00419), phthalate (KEGG module M00636), terephthalate (KEGG module M00624) and toluene (KEGG module M00547) (Kanehisa, Sato, Kawashima, Furumichi, & Tanabe, Citation2016). Past studies of the metabolic prediction of functional genes of S. hemiphylum microbiome reported on their participation in biochemical pathways related to the biosynthesis of amino acids, antibiotics and secondary metabolites; the degradation of aromatic compounds; and the metabolism of amino acids, carbon and fatty acids (Cleary & Huang, Citation2020). Similarly, Chen et al. (Citation2022) have also documented the xenobiotic-degrading abilities of microbiota associated with S. thunbergii and S. muticum.
The alginate lyase-producing efficacy of the prokaryotic microbiome of Sargassum waste was also evaluated. Alginate lyases have potential applications in agriculture, bioenergy, food, medical diagnostic and pharmaceutical industries (Cheng, Jiang, Xu, Liu, & Mao, Citation2020; Mohapatra, Citation2020). The results revealed that the potent alginate lyases producers belonged to the members of five phyla, Bacteroides, Proteobacteria, Planctomycetes, Verrucomicrobia and Chlorobi. Previous findings have also documented Proteobacteria and Bacteroidetes as the major phyla of alginate-depolymerizing bacteria associated with the brown algae Ascophyllum nodosum (Martin et al., Citation2015) and Laminaria sp (Dong et al., Citation2012).
In conclusion, a next-generation sequencing approach was used in this study to investigate the taxonomical compositions of prokaryotic and fungal communities along with the functions of the Sargassum waste metagenome. This cultivation-independent study demonstrated the prevalence of phylogenetically diverse phyla of bacteria, archaea and fungal communities in Sargassum waste. The results of metagenome functional analysis indicate that Sargassum waste is a promising niche material for the isolation of potent microbial strains for the production of novel antibiotic, anticancer, antitumor, immunosuppressant, antiparasite and antioxidant compounds and for the bioremediation of xenobiotics. Additionally, these microbial associates have the potential to be used as novel biocatalysts for efficient and ecofriendly depolymerization of alginate and may therefore be useful for sustainable bioconversion of Sargassum waste into third-generation biofuels and novel biologically active alginate oligosaccharides of industrial importance.
Author contribution
BRM performed the experiments, analysed the data, and drafted and reviewed the manuscript.
Supplemental Material
Download MS Excel (313.4 KB)Acknowledgements
This research was supported by the UWI Campus Research Awards Fund.
Disclosure statement
No potential conflict of interest was reported by the author.
Supplementary data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/26388081.2023.2220384.
References
- Ai, N., Jiang, Y., Omar, S., Wang, J., Xia, L., & Ren, J. (2022). Rapid measurement of cellulose, hemicellulose, and lignin content in Sargassum horneri by near-infrared spectroscopy and characteristic variables selection methods. Molecules, 27, 335. doi:10.3390/molecules27020335
- Aires, T., Muyzer, G., Serrão, E. A., & Engelen, A. H. (2019). Seaweed loads cause stronger bacterial community shifts in coastal lagoon sediments than nutrient loads. Frontiers in Microbiology, 9, 3283. doi:10.3389/fmicb.2018.03283
- Akyol, Ç., Ince, O., & Ince, B. (2019). Crop-based composting of lignocellulosic digestates: Focus on bacterial and fungal diversity. Bioresources Technology, 288, 121549. doi:10.1016/j.biortech.2019.121549
- Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., & Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods, 13, 581–583. doi:10.1038/nmeth.3869
- Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D. … Knight, R. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7, 335–336. doi:10.1038/nmeth.f.303
- Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A. and Knight, R. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the USA, 108, 4516–4522.
- Carrillo-Domínguez, S., Rodríguez-Martínez, R. E., Díaz-Martínez, M., Magaña-Gallegos, E., & Cuchillo-Hilario, M. (2023). Potential application of pelagic Sargassum spp. in animal feeding. Journal of Applied Phycology, 35, 433–444. doi:10.1007/s10811-022-02877-x
- Chávez, V., Uribe-Martínez, A., Cuevas, E., Rodríguez-Martínez, R. E., van Tussenbroek, B. I., Francisco, V., & Masia, L. (2020). Massive influx of pelagic Sargassum spp. on the coasts of the Mexican Caribbean 2014–2020: Challenges and opportunities. Water, 12, 2908. doi:10.3390/w12102908
- Cheng, D., Jiang, C., Xu, J., Liu, Z., & Mao, X. (2020). Characteristics and applications of alginate lyases: A review. International Journal of Biological Macromolecules, 164, 1304–1320. doi:10.1016/j.ijbiomac.2020.07.199
- Chen, D. M., Taylor, A. F. S., Burke, R. M., & Cairney, J. W. G. (2001). Identification of genes for lignin peroxidases and manganese peroxidases in ectomycorrhizal fungi. The New Phytologist, 152, 151–158. doi:10.1046/j.0028-646x.2001.00232.x
- Chen, J., Zang, Y., Yang, Z., Qu, T., Sun, T. … Tang, X. (2022). Composition and functional diversity of epiphytic bacterial and fungal communities on marine macrophytes in an intertidal zone. Frontiers in Microbiology, 13, 3389. doi:10.3389/fmicb.2022.839465
- Cleary, D. F. R., & Huang, Y. M. (2020). A comparison of the prokaryotic communities associated with seven seaweed species, sediment, and seawater from the Penghu archipelago, Taiwan. Marine Biology Research, 16, 744–761. doi:10.1080/17451000.2020.1859119
- Davis, D., Simister, R., Campbell, S., Marston, M., Bose, S., McQueen-Mason, S. J., & Gallimore, W. A. (2021). Biomass composition of the golden tide pelagic seaweeds Sargassum fluitans and S. natans (morphotypes I and VIII) to inform valorisation pathways. The Science of the Total Environment, 762, 143134. doi:10.1016/j.scitotenv.2020.143134
- Davis, T. A., Volesky, B., & Mucci, A. (2003). A review of the biochemistry of heavy metal biosorption by brown algae. Water Research, 37, 4311–4330. doi:10.1016/S0043-1354(03)00293-8
- Del Río, P. G., Domínguez, E., Domínguez, V. D., Romaní, A., Domingues, L., & Garrote, G. (2019). Third generation bioethanol from invasive macroalgae Sargassum muticum using autohydrolysis pretreatment as first step of a biorefinery. Renewable Energy, 141, 728–735. doi:10.1016/j.renene.2019.03.083
- Dong, S., Yang, J., Zhang, X.-Y., Shi, M., Song, X.-Y., Chen, X.-L., & Zhang, Y.-Z. (2012). Cultivable alginate lyase-excreting bacteria associated with the arctic brown alga Laminaria. Marine Drugs, 10, 2481. doi:10.3390/md10112481
- Drula, E., Garron, M. L., Dogan, S., Lombard, V., Henrissat, B., & Terrapon, N. (2022). The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Research, 50, D571–D577. doi:10.1093/nar/gkab1045
- Fernando, P. S. I., Kim, K.-N., Kim, D., & Jeon, Y.-J. (2019). Algal polysaccharides: Potential bioactive substances for cosmeceutical applications. Critical Reviews in Biotechnology, 39, 99–113. doi:10.1080/07388551.2018.1503995
- Gaynor, M., & Mankin, A. S. (2003). Macrolide antibiotics: Binding site, mechanism of action, resistance. Current Topics in Medicinal chemistry, 3, 949. doi:10.2174/1568026033452159
- Gliszczynska-Swiglo, A. (2007). Folates as antioxidants. Food Chemistry, 101, 1480–1483. doi:10.1016/j.foodchem.2006.04.022
- Gower, J., & King, S. (2019). Seaweed, seaweed everywhere. Science, 365, 27. doi:10.1126/science.aay0989
- Hervé, V., Lambourdière, J., René-Trouillefou, M., Devault, D. A., & Lopez, P. J. (2021). Sargassum differentially shapes the microbiota composition and diversity at coastal tide sites and inland storage sites on Caribbean islands. Frontiers in Microbiology, 12, 701155. doi:10.3389/fmicb.2021.701155
- Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., & Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research, 44, D457–D462. doi:10.1093/nar/gkv1070
- Keatinge-Clay, A. T., Maltby, D. A., Medzihradszky, K. F., Khosla, C., & Stroud, R. M. (2004). An antibiotic factory caught in action. Nature Structural & Molecular Biology, 11, 888–893. doi:10.1038/nsmb808
- Kostas, E. T., Adams, J. M. M., Ruiz, H. A., Duran-Jimenez, G., & Lye, G. J. (2021). Macroalgal biorefinery concepts for the circular bioeconomy: A review on biotechnological developments and future perspectives. Renewable Sustainable Energy Reviews, 151, 111553. doi:10.1016/j.rser.2021.111553
- Laeliocattleya, R. A., Suloi, Y. A. F., Gayatri, P. P., Putri, N. A., & Anggraeni, Y. C. (2020). Fucoidan content from brown seaweed (Sargassum filipendula) and its potential as radical scavenger. Journal of Physics: Conference Series, 1430, 012023.
- Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D. … Huttenhower, C. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 31, 814–821. doi:10.1038/nbt.2676
- Langin, K. (2018). Seaweed masses assault Caribbean islands. Science, 360, 1157–1158. doi:10.1126/science.360.6394.1157
- Leong, Y. K., & Chang, J. S. (2022). Integrated role of algae in the closed-loop circular economy of anaerobic digestion. Bioresources and Technology, 360, 127618. doi:10.1016/j.biortech.2022.127618
- Lombo, F., Menendez, N., Salas, J. A., & Mendez, C. (2006). The aureolic acid family of antitumor compounds: Structure, mode of action, biosynthesis, and novel derivatives. Applied Microbiology and Biotechnology, 73, 1–14. doi:10.1007/s00253-006-0511-6
- Martin, M., Barbeyron, T., Martin, R., Portetelle, D., Michel, G., & Vandenbol, M. (2015). The cultivable surface microbiota of the brown alga Ascophyllum nodosum is enriched in macroalgal-polysaccharide-degrading bacteria. Frontiers in Microbiology, 6, 1487. doi:10.3389/fmicb.2015.01487
- Mei, X., Wu, C., Zhao, J., Yan, T., & Jiang, P. (2019). Community structure of bacteria associated with drifting Sargassum horneri, the causative species of golden tide in the Yellow Sea. Frontiers in Microbiology, 10, 1192. doi:10.3389/fmicb.2019.01192
- Merle, H., Resière, D., Mesnard, C., Pierre, M., Jean-Charles, A., Béral, L., & Nevière, R. (2021). Case report: Two cases of keratoconjunctivitis tied to Sargassum algae emanations. American Journal of Tropical Medicine and Hygiene, 104, 403–405. doi:10.4269/ajtmh.20-0636
- Milledge, J. J., Nielsen, B. V., & Bailey, D. (2016). High-value products from macroalgae: The potential uses of the invasive brown seaweed, Sargassum muticum. Reviews in Environmental Science and Biotechnology, 15, 67–88. doi:10.1007/s11157-015-9381-7
- Miranda, J. L. L., Celis, L. B., Estévez, M., Chávez, V., van Tussenbroek, B. I., Uribe-Martínez, A., & Cauich-Kantun, C. (2021). Commercial potential of pelagic Sargassum spp. in Mexico. Frontiers in Marine Science, 8, 768470. doi:10.3389/fmars.2021.768470
- Mohapatra, B. R. (2017). Kinetic and thermodynamic properties of alginate lyase and cellulase co-produced by Exiguobacterium species Alg-S5. International Journal of Biological Macromolecules, 98, 103–110. doi:10.1016/j.ijbiomac.2017.01.091
- Mohapatra, B. R. (2018). Biocatalytic efficacy of immobilized cells of Chryseobacterium sp. Alg-SU10 for simultaneous hydrolysis of urethane and urea. Biocatalysis and Biotransformation, 36, 307–315. doi:10.1080/10242422.2018.1445228
- Mohapatra, B. R. (2020). Biocatalytic characteristics of chitosan nanoparticle-immobilized alginate lyase extracted from a novel Arthrobacter species AD-10. Biocatalysis and Agricultural Biotechnology, 23, 101458. doi:10.1016/j.bcab.2019.101458
- Mohapatra, B. R. (2021a). Characterization of β-mannanase extracted from a novel Streptomyces species Alg-S25 immobilized on chitosan nanoparticles. Biotechnology & Biotechnological Equipment, 35, 150–161. doi:10.1080/13102818.2020.1858158
- Mohapatra, B. R. (2021b). Comparative kinetic analysis of alginate lyase and mannanase co-produced via solid-state fermentation of cow dung supplemented with seaweed wastes by a novel Streptomyces sp. Alg-S23. Biomass Conversion and Biorefinery, 11, 2013–2025. doi:10.1007/s13399-020-00658-5
- Mohapatra, B. R. (2021c). Solid-state fermentation conditions optimization, homology modelling and molecular docking of β-mannanase of a novel Streptomyces species LB66 isolated from Sargassum seaweed waste. Biocatalysis and Biotransformation, 41, 187–197. doi:10.1080/10242422.2021.2010719
- Mohapatra, B. R. (2022). Fermentation medium optimization, molecular modelling and docking analysis of the alginate lyase of a novel Pseudomonas sp. LB56 isolated from seaweed waste. Biotechnology & Biotechnological Equipment, 36, 281–291. doi:10.1080/13102818.2022.2071635
- Mohapatra, B. R., Schambeau, L., Pannell, L. K., & Kiene, R. P. (2017). Comparative proteomics of temperate and polar Phaeocystis species. Nova Hedwigia, 105, 15–27. doi:10.1127/nova_hedwigia/2016/0393
- Morand, P., & Merceron, M. (2005). Macroalgal population and sustainability. Journal of Coastal Research, 2005, 1009–1020. doi:10.2112/04-700A.1
- Nilsson, R. H., Larsson, K.-H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S. … Abarenkov, K. (2018). The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Research, 47, D259–D264. doi:10.1093/nar/gky1022
- Oh, R. M., Bollati, E., Maithani, P., Huang, D., & Wainwright, B. J. (2021). The microbiome of the reef macroalga Sargassum ilicifolium in Singapore. Microorganisms, 9, 898. doi:10.3390/microorganisms9050898
- Oviatt, C. A., Huizenga, K., Rogers, C. S., & Miller, W. J. (2019). What nutrient sources support anomalous growth and the recent Sargassum mass stranding on Caribbean beaches? A review. Marine Pollution Bulletin, 145, 517–525. doi:10.1016/j.marpolbul.2019.06.049
- Park, S. R., Han, A. R., Ban, Y. H., Yoo, Y. J., Kim, E. J., & Yoon, Y. J. (2010). Genetic engineering of macrolide biosynthesis: Past advances, current state, and future prospects. Applied Microbiology and Biotechnology, 85, 1227–1239. doi:10.1007/s00253-009-2326-8
- Patyshakuliyeva, A., Falkoski, D. L., Wiebenga, A., Timmermans, K., & de Vries, R. P. (2019). Macroalgae derived fungi have high abilities to degrade algal polymers. Microorganisms, 8, 52. doi:10.3390/microorganisms8010052
- Resiere, D., Valentino, R., Nevière, R., Banydeen, R., Gueye, P., Florentin, J., Cabié, A., Lebrun, T., Mégarbane, B., Guerrier, G., & Mehdaoui, H. (2018). Sargassum seaweed on Caribbean islands: An international public health concern. The Lancet, 392, 2691. doi:10.1016/S0140-6736(18)32777-6
- Rodríguez-Martínez, R. E., Roy, P. D., Torrescano-Valle, N., Cabanillas-Terán, N., Carrillo-Domínguez, S. … van Tussenbroek, B. I. (2020). Element concentrations in pelagic Sargassum along the Mexican Caribbean coast in 2018-2019. Peer J, 8, e8667. 2020 Feb 26. doi:10.7717/peerj.8667
- Schneider, G. (2005). Enzymes in the biosynthesis of aromatic polyketide antibiotics. Current Opinions in Structural Biology, 15, 629–636. doi:10.1016/j.sbi.2005.10.002
- Serebryakova, A., Aires, T., Viard, F., Serrao, E. A., Engelen, A. H., & Melcher, U. (2018). Summer shifts of bacterial communities associated with the invasive brown seaweed Sargassum muticum are location and tissue dependent. PLoS One, 13, e0206734. doi:10.1371/journal.pone.0206734
- Shen, B., Liu, W., & Nonaka, K. (2003). Enediyne natural products: Biosynthesis and prospects towards engineering novel antitumor agents. Current Medicinal Chemistry, 10, 2317–2325. doi:10.2174/0929867033456701
- Smetacek, V., & Zingone, A. (2013). Green and golden seaweed tides on the rise. Nature, 504, 84–88. doi:10.1038/nature12860
- Thakur, N., Salama, E.-S., Sharma, M., Sharma, P., Sharma, D., & Li, X. (2022). Efficient utilization and management of seaweed biomass for biogas production. Materials Today Sustainability, 18, 100120. doi:10.1016/j.mtsust.2022.100120
- Torralba, M. G., Franks, J. S., Gomez, A., Yooseph, S., Nelson, K. E., & Grimes, D. J. (2017). Effect of Macondo prospect 252 oil on microbiota associated with pelagic Sargassum in the northern Gulf of Mexico. Microbial Ecology, 73, 91–100. doi:10.1007/s00248-016-0857-y
- Van Lanen, S. G., & Shen, B. (2008). Biosynthesis of enediyne antitumor antibiotics. Current Topics in Medicinal Chemistry, 8, 448–459. doi:10.2174/156802608783955656
- Wainwright, B. J., Bauman, A. G., Zahn, G. L., Todd, P. A., & Huang, D. (2019). Characterization of fungal biodiversity and communities associated with the reef macroalga Sargassum ilicifolium reveals fungal community differentiation according to geographic locality and algal structure. Marine Biodiversity, 49, 2601–2608. doi:10.1007/s12526-019-00992-6
- Wang, M., Hu, C., Barnes, B. B., Mitchum, G., Lapointe, B., & Montoya, J. P. (2019). The great Atlantic Sargassum belt. Science, 365, 83–87. doi:10.1126/science.aaw7912
- Wolfe, B. E., Kuo, M., & Pringle, A. (2012). Amanita thiersii is a saprotrophic fungus expanding its range in the United States. Mycologia, 104, 22–33. doi:10.3852/11-056