
Introduction
Familial hypercholesterolaemia (FH) is associated with severely elevated serum low-density lipoprotein cholesterol (LDL-C) levels that lead to premature onset coronary artery disease (CAD) and is responsible for 2-3% of the cases of myocardial infarction in individuals aged <60 years[Citation1]. In the majority (75-95%) of cases, FH is caused by a heterozygous pathogenic variant in the low-density lipoprotein receptor (LDLR), apolipoprotein B (APOB), or proprotein convertase subtilisin/kexin type 9 (PCSK9) genes with an autosomal dominant pattern of inheritance with an incidence of 1 in 250 individuals [Citation1,Citation2]. In rare cases, biallelic pathogenic variants (homozygous or compound heterozygous) in the LDLR, APOB, PCSK9, or low-density lipoprotein receptor adaptor protein 1 (LDLRAP1) genes have been reported, with an estimated incidence varying from 1 in 1 million in the Lebanese population to 1 in 275,000 in the French Canadian population, resulting in homozygous FH (HoFH) [Citation1,Citation3]. In the untreated state, Atherosclerotic Cardiovascular Disease (ASCVD) events have been reported in children younger than 6 years with a mean age of death at 18–20 years[Citation3]. Timely diagnosis and appropriate management are necessary to prevent complications.
We present a case of a young girl with HoFH caused by a homozygous deletion in the LDLR gene.
Case details
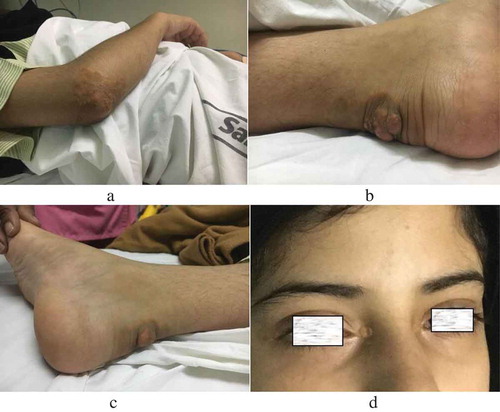
A 17-year-old girl, the resident of Yemen, born in a consanguineous family, presented to the outpatient department for an opinion on multiple swellings on her body since the age of 2 years. General examination revealed multiple xanthomas on the extensor aspect of both elbows, heels, and xanthelasmas on the inner canthus, and lower eyelids of both eyes (). Corneal arcii suggestive of accelerated atherosclerosis were also noted. Blood pressure and heart rate were normal.
Figure 1. A. Xanthomas on elbow. B, C. Achilles tendon xanthomas on the heel. D. Xanthelasma palpebrum.
Blood glucose, renal function, routine urine examination, and thyroid function were within normal limits. Her serum fasting lipid profile revealed extremely elevated total cholesterol, LDL-C, and apolipoprotein B with marginally raised triglycerides (). Her results, along with those of her family members, are shown in . LDL-C was measured by direct estimation. The Friedewald equation, which is commonly used in most settings for estimating LDL-C levels (in mg/dl), is based on a fixed factor of 5 for the ratio of triglycerides to very low-density lipoprotein cholesterol (TG:VLDL-C); however, this ratio varies significantly across the range of triglyceride and cholesterol levels[Citation4]. Lp(a) values of the patient and her father are shown in . The Lp(a) levels of the patient, as ascertained by isoform insensitive assay, were 60 mg/dL. The Lp(a) assay was performed by immunoturbidimetry using a Biokit SA kit (Barcelona, Spain) on an Architect C4000 clinical chemistry analyser system (Abbott Core Laboratory, Abbott Park, Illinois, U.S.A).
Table 1. Fasting Lipid profile of the patient (at presentation and at follow-up after 8 weeks on rosuvastatin 40 mg and ezetimibe 10 mg), her parents and her brother.
Her carotid Doppler ultrasound demonstrated the presence of diffuse plaques in both carotid arteries causing 41–59% stenosis of the right internal carotid artery (ICA), 60-69% stenosis of the left ICA, and mild bilateral external carotid artery stenosis. Two-D echocardiography demonstrated anterior mitral valve leaflet prolapse, moderate aortic stenosis, and moderate aortic regurgitation with a left ventricular ejection fraction of 60%. Computed tomography (CT) coronary angiography could not be completed as the patient developed thrombophlebitis due to extravasation of the contrast agent.
Four days after the CT angiography, she presented to the emergency department with sudden onset of chest heaviness and was admitted to the intensive care unit with a diagnosis of the acute coronary syndrome. Coronary angiography showed 80% focal mid-segment stenosis of the right coronary artery, for which angioplasty was performed.
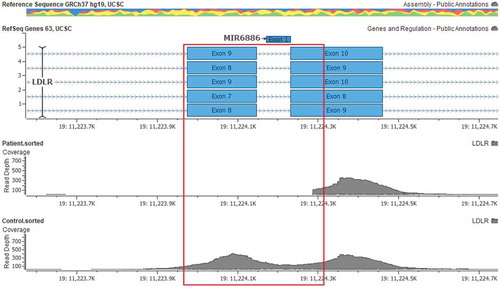
To evaluate the genetic aetiology, next generation sequencing (NGS) was performed using the TruSight One panel (cat no: 15,046,895, Illumina, CA, USA) using Illumina sequencing-by-synthesis chemistry on DNA extracted from the patient’s peripheral blood. In-house bioinformatic analysis was performed on the NGS data to identify clinically relevant variants in the coding regions and the exon-intron boundaries of the LDLR, APOB, PCSK9, and LDLRAP1 genes, which are associated with heterozygous FH (HeFH) or HoFH. No pathogenic or clinically relevant small sequence variants were detected within the coding regions or along the exon-intron boundaries of the LDLR, APOB, PCSK9, and LDLRAP1 genes. High-quality sequence data were obtained for the targeted regions (Q score >30 for 98% of the data). However, we did not obtain sequence data for exon 9 and the proximal region of exon 10 of the LDLR gene (). Raw sequence reads for exon 9 and the proximal region of exon 10 were identified in other control samples and there was a sharp border at the reappearance of reads at exon 10 of the LDLR gene in the present case. These findings were suggestive of a strong possibility of a homozygous gene deletion/rearrangement involving the LDLR gene.
Figure 2. Alignment snapshot of the LDLR gene. The deleted region (exon 9 and part of exon 10) are highlighted by a red box.
The presence of consanguinity, tendon xanthomas, accelerated atherosclerosis at 17 years of age, serum LDL-C levels of 646 mg/dL (16.7 mmol/L), and the results of the molecular genetic assay were consistent with the diagnosis of HoFH in accordance with the Simon Broome criteria[Citation5]. Further, both her parents had serum LDL-C levels >190 mg/dL (4.9 mmol/L) (), which was suggestive of them being heterozygous carriers for the above-mentioned deletion in the LDLR gene and hence being HeFH patients, requiring intensive lipid-lowering treatment. At the time of diagnosis, the age of the mother was 45 years and that of the father was 54 years. The patient had a 19-year-old brother with LDL-C levels of 166 mg/dL (4.2 mmol/L).
The results for the parents, however, could not be confirmed by a molecular assay. The patient was discharged on a daily dose of rosuvastatin 40 mg, ezetimibe 10 mg, aspirin 75 mg, metoprolol 25 mg and ticagrelor 90 mg. After 8 weeks on the suggested medications, only a 16% reduction was observed in her serum LDL-C levels. The family confirmed that the patient was compliant with the prescribed medications (). The family was lost to follow-up after this.
Discussion
We present a 17-year-old girl, born in a consanguineous family, diagnosed with HoFH probably caused by a biallelic deletion in the LDLR gene. Both her parents’ serum lipid profiles were consistent with them being HeFH patients. In cases with FH, high cholesterol may manifest as xanthomas on the eyelids, tendons of the elbows, knees, hands and feet [Citation2,Citation6]. Her 54-year-old father had no significant physical findings; her 45-year-old mother and 19-year-old brother were not available for physical examination. Tendon xanthomas are pathognomonic of FH, especially HoFH, and differentiate them from familial combined hyperlipidaemia but the absence of tendon xanthomas does not rule out the possibility of FH[Citation7].
Accelerated atherosclerosis of the coronary, carotid, and lower limb arteries may result in cardiovascular disease occurrence, recurrent transient ischaemic attacks, stroke, intermittent claudication, or gangrene at a younger age[Citation8]. Valvulopathy in FH may be a combination of aortic root thickening, aortic valve calcification, stenosis, or regurgitation and mitral regurgitation[Citation9]. These progress rapidly with age in spite of treatment with statins[Citation9]. Elevated Lp(a) levels are commonly observed in HoFH and are an independent risk factor for rapid progression of aortic valve calcification [Citation9,Citation10].
Total cholesterol levels in HeFH cases are generally between 350 and 550 mg/dL (9.0–14.2 mmol/L), whereas in HoFH, this value is between 650 and 1000 mg/dL (16.9–25.8 mmol/L)[Citation2]. A majority of patients with HoFH experience severe CAD by their mid-twenties and the chances of requiring coronary bypass surgery and mortality are high in the second decade of life[Citation11]. Untreated HeFH cases among males and females develop CAD before the age of 55 and 60, respectively, but can be treated with cholesterol-lowering medication in addition to lifestyle modifications to attenuate the development of atherosclerosis[Citation12]. Additional agents like bile acid sequestrants and a cholesterol absorption inhibitor (ezetimibe) may be required if adequate lipid control is not achieved with the maximum tolerated dose of statin[Citation2].
Three groups have developed clinical diagnostic tools for FH: Simon Broome criteria[Citation5], Dutch Lipid Network[Citation13], and US MEDPED[Citation14] criteria. Of these, the Simon Broome criteria are the simplest to use[Citation15]. The Dutch Lipid Network takes molecular testing into consideration and uses a scoring system, which makes it cumbersome to use. The US MEDPED criteria include age- and relative-specific criteria for total cholesterol only.
In FH, LDL receptor (LDLR) function is reduced or absent, leading to reduced LDL removal from circulation. Normally, LDL remains in the blood for 2.5 days; however, in patients with FH, LDL remains in the blood for an average duration of 4.5 days[Citation16]. In cases with HoFH, LDL receptor levels are either zero (receptor negative) or greatly reduced (receptor defective)[Citation2]. Biallelic pathogenic/likely pathogenic variants in the LDLR gene are the most common genetic defects associated with HoFH. Approximately 1600 different mutations have been reported in the LDLR gene[Citation17]. More than 90% of these pathogenic variants are small sequence variants in the LDLR gene, whereas 2.5-10% of these variants are large deletions/duplications, involving whole exons or the complete gene[Citation1]. The latter cannot be diagnosed by capillary sequencing and usually require an orthogonal approach such as multiplex ligation-dependent probe amplification (MLPA), quantitative polymerase chain reaction (qPCR), long-range PCR, droplet digital PCR, or microarray. Typically, current commercially available NGS-based multigene panels are not customised for the evaluation of large deletion/duplication in the targeted region[Citation18]. However, when no clinically relevant sequence variants have been detected in the LDLR, PCSK9, or APOB genes, absence of reads aligning to the targeted region as well as a sharp margin at the border of the presumed deletion in clinically suspected cases of HoFH is strongly suggestive of homozygous deletion in the LDLR gene, as shown in the present case report[Citation19]. Various large deletions in the LDLR gene have been reported, including a deletion of more than 15 kb that encompasses the gene promoter and exon 1[Citation20]. Additionally, deletions involving exons 2–3, exon 5, exon 7, and exon 9–14 of the LDLR gene have also been reported [Citation21,Citation22]. However, a homozygous deletion of only exon 9 of the LDLR gene has not been previously reported. Exons 7–14 of the LDLR gene share 30% sequence homology to the human epidermal growth factor (EGF) precursor gene and this domain is called EGF precursor homology domain[Citation23]. The amino acid sequence in this domain is highly conserved with 70-86% sequence homology across six species (human, cow, rabbit, hamster, rat, and the toad Xenopus laevis). The EGF precursor homology domain is required for the release of lipoproteins during receptor recycling. The phenotype prediction of this novel homozygous deletion is difficult. Missense variants in exon 9 have previously been reported to result in recycle defective allele[Citation23]. Therefore, biallelic loss of exon 9 of the LDLR gene in the reported case may result in impaired release of LDL from the LDLR in the lysosome, thereby potentially interfering with recycling of the LDLR from the lysosome back to the cell surface. The EGF precursor homology domain also helps in binding of LDL on the cell surface by proper positioning of the ligand binding domain, which is encoded by exons 2–6 of the LDLR gene[Citation23]. Homozygous deletion of exon 9 of the LDLR gene may result in impaired function of the EGF precursor homology domain and might disrupt normal orientation of the ligand binding domain of the LDLR, thereby potentially resulting in loss of LDL binding to the LDLR.
Cholesterol-lowering agents like statins are the main therapy in both HeFH and HoFH cases, reducing cardiovascular complications and overall mortality[Citation24]. Cases of HoFH, however, do not readily respond to statin therapy since these drugs act by upregulating the function of intrinsic liver LDLR[Citation25].
Severe HoFH cases may require LDL apheresis from early childhood, which lowers the plasma LDL levels by 55-70% in the acute phase; long-term treatment promotes regression of xanthomas and slows the progression of atherosclerosis[Citation25]. Tendon xanthomas and xanthelasmata respond to LDL-C lowering agents. Xanthelasmas can be removed surgically; however, they are likely to recur[Citation26].
Other drugs for HoFH include lomitapide and mipomersen[Citation27]. Lomitapide inhibits microsomal triglyceride transfer protein (MTTP), which is necessary for very low-density lipoprotein (VLDL) assembly and secretion in the liver[Citation28]. Mipomersen inhibits translation of Apo B100[Citation29]. Alirocumab and evolocumab are monoclonal antibodies (mAbs) against proprotein convertase subtilisin/kexin type-9 (PCSK 9)[Citation30]. These mAbs bind PCSK9 in the blood and therefore inhibit the PCSK9-mediated degradation of LDLRs, thereby increasing the clearance of LDL from circulation. However, LDL apheresis and other recent therapeutic approaches are expensive and not readily available in India[Citation31]. Also, there may not be a very good response if the LDLR is ineffective.
An interesting finding was the increase in HDL-C levels by nearly 3 times after initiation of rosuvastatin. This may be explained to some extent by previously confirmed observations that statins vary widely in their ability to raise HDL-C levels and elevated HDL-C levels are positively dose-dependent on rosuvastatin[Citation32]. The marginally high triglyceride levels dropped after initiation of rosuvastatin and there is a possibility that the increase in HDL-C levels was owing to the well-documented inverse relationship between the concentrations of TG and HDL-C[Citation33]. However, this explanation may not account for the total increase in HDL-C levels. Our laboratory participates in quality assurance with Biorad EQAS (external quality assurance scheme). The high LDL values do not interfere with HDL measurement in our laboratory.
This case emphasises the importance of early diagnosis of FH followed by intensive statin treatment[Citation34]. Once the index case is identified, extended family screening for hypercholesterolaemia is recommended. Genetic confirmation can be considered when a mutation has been identified in the index patient. The Lipid Association of India (LAI) expert consensus recommends universal screening of all individuals at the age of 20 years, or earlier if there is a family history of HeFH, HoFH, or premature CAD[Citation35]. If all efforts are directed to implement this concept of screening, it may be possible to detect and treat more cases of FH in India.
This case report highlights the need for clinical diagnosis and appropriate treatment of HeFH or HoFH at an early age to avoid serious complications. Molecular confirmation of the diagnosis is required in children when the serum LDL-C levels are not in the diagnostic range and for families who want to opt for prenatal diagnosis.
Abbreviations
| Apo-B | = | Apolipoprotein B |
| ASCVD | = | Atherosclerotic Cardiovascular Disease |
| CAD | = | Coronary artery disease |
| CT | = | Computed tomography |
| FH | = | Familial hypercholesterolaemia |
| HDL-C | = | High density lipoprotein - cholesterol |
| HeFH | = | Heterozygous familial hypercholesterolaemia |
| HoFH | = | Homozygous familial hypercholesterolaemia |
| ICA | = | Internal carotid artery |
| LDL-C | = | Low-density lipoprotein-cholesterol |
| LDLR | = | Low-density lipoprotein receptor |
| LDLRAP 1 | = | Low-density lipoprotein receptor adaptor protein 1 |
| MTTP | = | Microsomal triglyceride transfer protein |
| NGS | = | Next generation sequencing |
| PCSK9 | = | Proprotein convertase subtilisin/kexin type 9 |
| PCR | = | Polymerase chain reaction |
| RCA | = | Right coronary artery |
| VLDL-C | = | Very low-density lipoprotein - cholesterol |
Consent
Consent was obtained from a family member to report this case. Also, Ethics Committee permission to proceed with publication was obtained (although this is not mandatory for our institution).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Youngblom E, Pariani M, Knowles JW. Familial Hypercholesterolemia. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. [Internet]. Seattle: University of Washington, Seattle; 2014 [Updated 2016]. 1993–2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK174884.
- Kasper DL, Fauci AS, Hauser S, et al. Harrison’s principles of internal medicine. 19th ed. Vol. 2. Ch 421. 2441. New York: McGraw-Hill; 2015.
- Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262–268.
- Martin SS, Blaha M, Elshazly MB, et al. Comparison of a novel method vs the Friedewald equation for estimating low-density lipoprotein cholesterol levels from the standard lipid profile. JAMA. 2013;310:2061–2068.
- Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific steering committee on behalf of the simon broome register group. Br Med J. 1991;303:893–896.
- Tsouli SG, Kiortsis DN, Argyropoulou MI, et al. Pathogenesis, detection and treatment of Achilles tendon xanthomas. Eur J Clin Invest. 2005;35:236–244.
- Mabuchi H. Half a century tales of familial hypercholesterolemia (FH) in Japan. J Atheroscler Thromb. 2017;24:189–207.
- Durrington P. Dyslipidaemia. Lancet. 2003;362:717–731.
- Fahed AC, Shibbani K, Andary RR, et al. Premature valvular heart disease in homozygous familial hypercholesterolemia. Cholesterol. 2017;2017:3685265.
- Vongpromek R, Bos S, Ten Kate GJR, et al. Lipoprotein(a) levels are associated with aortic valve calcification in asymptomatic patients with familial hypercholesterolaemia. J Int Med. 2015;278:166–173.
- Marais AD, Firth JC, Blom DJ. Homozygous Familial Hypercholesterolemia and its management. Semin Vasc Med. 2004;4:43–50.
- Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–3490.
- Dutch Lipid Network. World Health Organization. Familial hypercholesterolemia—report of a second WHO Consultation. Geneva, Switzerland: World Health Organization, 1999. (WHO publication no. WHO/HGN/FH/CONS/99.2).
- Williams RR, Hunt SC, Schumacher MC, et al. US Med Ped. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993;72:171–176.
- Austin MA, Hutter CM, Zimmern RL, et al. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidem. 2004;160:407–420.
- Repas TB, Tanner JR. Preventing early cardiovascular death in Familial Hypercholesterolemia. J Am Osteopath Assoc. 2014;114:99–108.
- Fahed AC, Khalaf R, Salloum R, et al. Variable expressivity and co‐occurrence of LDLR and LDLRAP1 mutations in familial hypercholesterolemia: failure of the dominant and recessive dichotomy. Mol Genetic Genomic Med. 2016;4:283–291.
- Zhao M, Wang Q, Wang Q, et al. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics. 2013;14(Suppl 11):S1.
- Giugliano T, Fanin M, Savarese M, et al. Identification of an intragenic deletion in the SGCB gene through a re-evaluation of negative next generation sequencing results. Neuromuscul Disord. 2016;26:367–369.
- Simard LR, Viel J, Lambert M, et al. The Delta>15 Kb deletion French Canadian founder mutation in familial hypercholesterolemia: rapid polymerase chain reaction-based diagnostic assay and prevalence in Quebec. Clin Genet. 2004;65:202–208.
- Horsthemke B, Dunning A, Humphries S. Identification of deletions in the human low density lipoprotein receptor gene. J Med Genetic. 1987;24:144–147.
- Nissen PH, Damgaard D, Stenderup A, et al. Genomic characterization of five deletions in the LDL receptor gene in Danish Familial Hypercholesterolemic subjects. BMC Med Genet. 2006;7:55.
- Hobbs HH, Russell DW, Brown MS, et al. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Ann Rev Genet. 1990;24:133–170.
- Pijlman AH, Huijgen R, Verhagen SN, et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in The Netherlands. Atherosclerosis. 2010;209:189–194.
- Cuchel M, Bruckert E, Ginsberg HN, et al. European atherosclerosis society consensus panel on familial hypercholesterolaemia. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146–2157.
- Burns T, Breathnach S, Cox N, et al. Rook’s Textbook of Dermatology, 8th Edition. Vol. 3. 58.84–58.87. Chichester, West Sussex: Wiley-Blackwell; 2010.
- Smith RJ, Hiatt WR. Two new drugs for homozygous familial hypercholesterolemia: managing benefits and risks in a rare disorder. JAMA Intern Med. 2013;173:1491–1492.
- Raal FJ. Lomitapide for homozygous familial hypercholesterolaemia. Lancet. 2013;381:7–8.
- Bell DA, Hooper AJ, Watts GF, et al. Mipomersen and other therapies for the treatment of severe familial hypercholesterolemia. Vasc Health Risk Manag. 2012;8:651–659.
- Everett BM M.D, Smith RJ, Hiatt WR. Reducing LDL with PCSK9 Inhibitors—the clinical benefit of lipid drugs. N Engl J Med. 2015;373:1588–1591.
- Iyengar SS, Puri R, Narasingan SN, et al. Lipid Association of India expert consensus statement on management of dyslipidemia in Indians Part 2. Clin Lipidol. 2017;12:56–109.
- Jones PH, Davidson MH, Stein EA, et al. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol. 2003;92:152–160.
- Barter PJ, Brandrup-Wognsen G, Palmer MK, et al. Effect of statins on HDL-C: a complex process unrelated to changes in LDL-C: analysis of the VOYAGER Database. J Lipid Res. 2010;51:1546–1553.
- Neil A, Cooper J, Betteridge J, et al. Reduction in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolemia: a prospective registry study. Eur Heart J. 2008;29:2625–2633.
- Iyengar SS, Puri R, Narasingan SN, et al. Lipid Association of India expert consensus statement on management of dyslipidemia in Indians: part 1. J Assoc Physicians India. 2016;64:7–52.