Abstract
Background. Previous knowledge of cervical lymph node compromise may be crucial to choose the best treatment strategy in oral squamous cell carcinoma (OSCC). Here we propose a set four genes, whose mRNA expression in the primary tumor predicts nodal status in OSCC, excluding tongue. Material and methods. We identified differentially expressed genes in OSCC with and without compromised lymph nodes using Differential Display RT-PCR. Known genes were chosen to be validated by means of Northern blotting or real time RT-PCR (qRT-PCR). Thereafter we constructed a Nodal Index (NI) using discriminant analysis in a learning set of 35 patients, which was further validated in a second independent group of 20 patients. Results. Of the 63 differentially expressed known genes identified comparing three lymph node positive (pN +) and three negative (pN0) primary tumors, 23 were analyzed by Northern analysis or RT-PCR in 49 primary tumors. Six genes confirmed as differentially expressed were used to construct a NI, as the best set predictive of lymph nodal status, with the final result including four genes. The NI was able to correctly classify 32 of 35 patients comprising the learning group (88.6%; p = 0.009). Casein kinase 1alpha1 and scavenger receptor class B, member 2 were found to be up regulated in pN + group in contrast to small proline-rich protein 2B and Ras-GTPase activating protein SH3 domain-binding protein 2 which were upregulated in the pN0 group. We validated further our NI in an independent set of 20 primary tumors, 11 of them pN0 and nine pN + with an accuracy of 80.0% (p = 0.012). Conclusions. The NI was an independent predictor of compromised lymph nodes, taking into the consideration tumor size and histological grade. The genes identified here that integrate our “Nodal Index” model are predictive of lymph node metastasis in OSCC.
The presence of lymph node metastasis (pN +) is an important predictor of recurrence and shorter survival in head and neck squamous cell carcinoma (HNSCC). Unfortunately, pathological characteristics, imaging studies and specific biological markers studied so far do not predict the presence of lymph node metastasis optimally [Citation1,Citation2]. As a consequence, biological markers of lymph node metastasis are still needed to help design better treatment strategies in HNSCC.
More recently, attempts to identify such a marker have been made using methods capable of assessing global gene expression. By assuming that the determinants of metastatic behavior are already present in the primary tumors, three studies using different platforms had indicated sets of genes with 500, 102 and 20 genes, respectively [Citation3–5], capable of predicting lymph node metastases in squamous cell carcinoma of head and neck with an overall accuracy of approximately 80–87%. In addition, two studies using the same Affymetrix platform, showed non-overlapping different eight-gene sets, capable of correctly classifying primary oral tongue cancers patients according to lymph node status in 85% and 92.3% of patients, respectively [Citation6,Citation7].
Since these studies hold practical and cost constraints due to different platforms and the high number of genes, we sought to use another method of global screening with a great theoretical potential for the identification of abundant, as well as low-copy-number, mRNA transcripts, which is the Differential Display Reverse Transcription Polimerase Chain Reaction (DD RT-PCR) [Citation8]. Although not as high throughput as microarrays, it has a lower cost and the advantage of unbiased identification of both known genes and novel sequences.
Our goal in this study was to identify, by DD RT-PCR assay, in primary oral (excluding tongue) squamous cell carcinoma, a set of genes that could predict the presence of lymph node metastasis. We showed that just four genes, G3BP2, SCARB2, CSNK1A1 and SPRR2B, can classify patients as pN0 or pN +, with 80.0% accuracy.
Patients and methods
Patients
From September 1999 through October 2004, 69 patients with histologically proven diagnosis of OSCC except tongue OSCC, with no previous treatment and surgery with curative intent scheduled at Head and Neck Surgery Service of Hospital Heliópolis, São Paulo, Brazil, entered the study.
The first 49 patients were in the learning set, 35 of them being included in the discriminant analysis. Tumor samples of additional consecutive 20 patients were used for “Nodal Index” (NI) validation.
All patients were advised of the procedures and provided written informed consent in compliance with the Declaration of Helsinki and amendments and was conducted in according with good clinical practices. The study was approved by independent ethics committee at Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo and at Hospital Heliópolis.
Tumor fragments were collected by the pathologist during surgery and immediately frozen in liquid nitrogen. Tumor staging was conducted according to 5th edition of the UICC TNM and treatment conducted according to routine guidelines. All patients underwent primary tumor resection with therapeutic intent followed by adjuvant radiotherapy, when appropriate. The histological grade, lymph node status and tumor width were obtained from the surgical pathological report. The general patient characteristics are presented in
Table I. Clinicopathological parameters of OSCC patients.
In the patient learning group, there were 42 males and seven females. The age of patients ranged from 30 to 86 years (median 52 years). A total of 47 (95.9%) were tobacco smokers and 46 (93.8%) alcohol addicted. There were 31 patients with cancer arising from floor of mouth, eight patients with gingival cancer, six patients with retromolar trigone cancer, three patients with lip cancer and one patient with palate cancer.
In the NI validation group, there were 14 males and six females. The age of patients ranged from 42 to 73 years (median 55 years). Nineteen (95.0%) patients smoked tobacco and 17 (85.0%) consumed alcohol. Twelve patients had cancer arising from the floor of the mouth, four had gingival cancer and four retromolar trigone cancer.
RNA isolation
Total RNA was extracted and pooled from three primary tumors without compromised lymph nodes (pN0) and three primary tumors with compromised lymph nodes (pN +), using Trizol reagent (Invitrogen, Life Technologies, CA, USA) according to the manufacturer’s instructions. Residual DNA was removed from RNA preparations by digestion with DNAse I-RNAse free (Promega, WI, USA). The concentration and purity of RNA samples were determined by measuring absorbance at 260 and 280 nm using a spectrophotometer and RNA quality determined by denaturing agarose gel electrophoresis. All tumors contained at least 70% cancer cells.
Differential display analysis (DD RT-PCR)
Pooled RNA (200 ng) from three pN0 and three pN + tumors was used for reverse transcription, in the presence of 2.5 μM of three different anchored dT11 downstream primers and 200U Super script II (Promega) for 60 min at 37°C. After heat inactivation of the reverse transcriptase at 95°C, 1:10 cDNA product was used as a template for polymerase chain reactions (PCR). This reaction was carried out in the presence of 2.5 μM anchored downstream primers, 0.5 μM random upstream primers (13: 5’-CTGATCCATG-3’; 14: 5’-CTGCTCTCAA-3’ or 15: 5’-CTTGATTGCC-3’), 1 μCi [32P]-dCTP and 1 U Taq DNA polymerase sequence grade (Promega) in a total volume of 20 μl. The PCR profile was 30 s at 94°C, 2 min at 40°C and 1 min at 72°C for 40 cycles. An aliquot of PCR product was separated on a 6% denaturing polyacrilamide gel in TBE buffer at 50 W, which was then exposed to an x-ray film (Hyperfilm, GE Healthcare Biociences – formerly Amersham-Biociences, St. Giles, UK). Candidate bands with varying expression among samples were excised and eluted in 100 μl H2O by heating at 95°C for 10 min. Eluted cDNA was reamplified by PCR, subcloned into PCR® 4-TOPO.TA Cloning® kit for sequencing (Invitrogen) and sequenced. The sequences obtained were compared with GenBank sequences using a BLAST search program (http://www.ncbi.nlm.nih.gov/Blast). To validate the results, an initial screen was performed by Reverse Northern blots. Results were then confirmed either by Northern blot or real time RT-PCR analysis.
Reverse Northern
Two nylon membranes were prepared by spotting double dots of the 31 first sequenced cDNAs and one control probe (b-actin). Membranes were hybridized at 42°C with [32P]-dCTP labeled cDNAs probes for each RNA mix (pN0 and pN +) prepared in duplicate by reverse transcriptation. Band intensities were quantified by densitometric scanning (ImageMaster VDS software, version 2.0, GE Healthcare Biociences) and data expressed as a ratio to β-actin mRNA. All clones presenting a pN + /pN0 signal ratio ≥ 2.0 were considered as over expressed and the clones showing signal ratio ≤ 0.6 were considered as under expressed.
Northern blot
An amount of 15 μg of total RNA were eletrophoresed through 1% agarose gels and RNA transferred to nylon filters, which were hybridized for 20 h at 42°C with [32P]-dCTP labeled specific probes generated in DD RT-PCR assays. Hybridization with 18S ribosomal RNA probe (donated by Dr. N. Arnheim, Department of Biochemistry, State University of New York, USA) was subsequently done to check for the equivalence of RNA loading. Band intensities in autoradiograms were quantified by densitometric scanning and data expressed as the ratio of the specific mRNA to 18S rRNA.
Real-time Reverse Transcription Polimerase chain reaction (qRT-PCR)
For cDNA synthesis, 1 µg of total RNA was reverse transcribed in a solution of 20 μl containing 100 ng random hexamer primers, 20 μM dNTPs mixture, 4 μl 5X RT buffer, 1.5 mM MgCl2, 5 mM DTT and 100 U SuperScript III reverse transcriptase (Invitrogen) at 50°C for 1 h and the reaction was terminated at 85° for 10 min. Primers sequences were designed according to the protocols of http://www.ncbi.nlm.nih.gov/nucleotide using Primer 3 software (Whitehead Institute for Biomedical Research, (http://frodo.wi.mit.edu/) and were synthesized by IDT (Integrated DNA Technologies, Inc, IA, USA) ()
Table II. Primers, PCR conditions and expected PCR product lengths.
PCR reactions were done in a Rotor-Gene System (Corbett Research, DE). Thermocycling was done in a total volume of 20 µl containing 5 µl cDNA sample (diluted 1:10), 1.5 mM MgCl2, 0.2 µmol/l of each primers, 0.1 µl SYBR Green I (Molecular Probes, OR, USA) working dilution (1:100), 1.25U Platinum Taq DNA polymerase (Invitrogen), 1 × reaction buffer, 0.2 mM dNTPs mixture; 5% DMSO (Sigma, CA, USA) and 0.5 µl of 10 mg/ml bovine serum albumin (Promega). After 2 min at 95°C, the cycling conditions were as follows: 95°C for 15 s, 58–60°C for 30 s 72°C for 30 s for 40 cycles. For β-actin, the cycles occurred at 95°C for 60 s, 64°C for 60 s and 72°C for 60 s. All samples were tested in duplicate, and average values were used for quantification.
Relative expression of genes of interest was normalized to that of β-actin (primers : Left-5’AGAA AATCTGGCACCACACC3’,Right-5’AGAG GCGTACAGGGATAGCA3’), and gene expression in each sample was then compared with expression in a control adjacent mucosa. The comparative CT method (ΔΔCT) was used for quantification of gene expression and relative expression was calculated as 2–ΔΔCT.
Statistical analysis
The Mann-Whitney U-test was used to compare gene expression between different groups and a discriminant analysis was performed to determine which gene set best classifies patients as pN + or pN0. Statistical significance was defined as p < 0.1 and all p-values were two-sided. This 0.1 threshold was only considered to select the genes that would be used in a second step to compute the Nodal Index, which was further validated, as explained in the results section. Data analysis was carried out using SPSS software, version 10.0 released for Windows (SPSS Inc., Chicago, IL, USA).
Results
Differential display analysis and identification of the selected cDNAs
Our objective was to identify differentially expressed genes in pN0 versus pN + primary tumors. Adjacent mucosa was included in the assay as an internal control and will not be further discussed. Around 95% of the bands showed no difference in signal intensity across the different samples, as expected. An example of the differentially displayed RT-PCR products is shown in
Figure 1. Analysis of cDNA population in pN0, pN + and adjacent mucosa groups by DD RT-PCR. Total RNA isolated and pooled from two adjacent mucosa (A), three primary tumors without compromised lymph nodes (B) and three primary tumors with compromised lymph nodes (C) were reverse transcribed and PCR carried out as per the protocol described in the text. The heat denatured PCR products were electrophoresed on urea-polyacrylamide gel. The gels were dried and exposed to autoradiograph for 12–16 h at −70°C and developed until the DNA bands appeared on the film. The arrows indicate the transcripts which are differentially expressed in pN0 or pN + samples: 1: SCARB2; 2: G3BP2; 3: CSNK1A1; 4: SPRR2B.
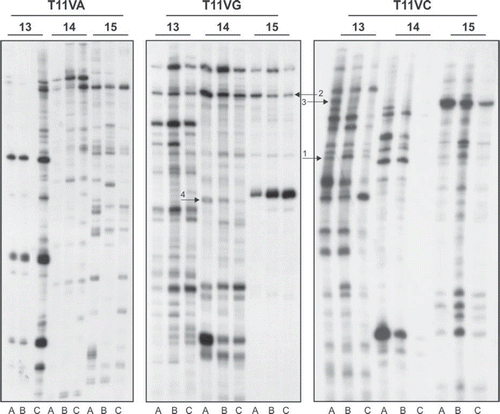
Considering the pN0 and pN + groups, a total of 99 differentially expressed bands were identified and 73 were further successfully reamplified, cloned and found to be suitable for sequencing analysis. The size of cDNA ranged from 140 to 650 bp. When compared with the GenBank database, we found that 10 clones presented less than 95% homology with human cDNA database sequences and 63 matched genes in Gene database. Since all DD assay used samples previously treated with DNAse enzyme, the 10 unmatched clones may represent novel genes that have not yet been identified. Of the 63 matched genes, 43 were known genes and 20 clones matched with ESTs or hypothetical proteins.
Validation of gene expression profiles
Reverse Northern analysis was performed on the first 31 cloned sequences and the results suggested that 21 genes would be differentially expressed. Since only three of these genes were confirmed to be differentially expressed by either Northern blot or qRT-PCR, we considered that reverse northern was not a good screening tool. We thus performed Northern blot or qRT-PCR directly to validate differentially expressed sequences. Of the 43 known genes, 22 were selected for further validation in 49 primary tumor tissue samples, by Northern analysis (10 genes) or qRT-PCR (12 genes) ()
Table III. Methods used to validate 23 genes found to be differentially expressed between pN0/pN + tumor samples, on initial analysis.
A total of six genes were confirmed to be differentially expressed between pN + /pN0 groups. Four were found to be up regulated in pN +, as compared to pN0 tumors: CSNK1A1 (casein kinase 1α1) (pN + median: 0.59 vs. 0.28 pN0 median; p = 0.013, Mann-Whitney U-test), SCARB2 (scavenger receptor class B, member 2) (0.21 vs. 0.07; p = 0.015), F3 [coagulation factor III (thromboplastin, tissue factor)] (0.62 vs. 0.39; p = 0.10) and POSTN (Periostin, osteoblast specific factor) (0.15 vs. 0.00; p = 0.10). Two genes were down regulated: SPRR2B (small proline-rich protein 2B (0.22 vs. 0.77; p = 0.042) and Ras-GTPase activating protein SH3 domain-binding protein 2 (G3BP2) (0.34 vs. 0.51; p = 0.055). The gene information, Gene ID access numbers and fold changes are summarized in
Table IV. Identity of the differentially expressed transcripts, based on the homology search in NCBI–BLAST, between pN + /pN0 OSCC patients.
Prediction of lymph metastasis by a four gene set
To develop a model for predicting nodal involvement, a discriminant analysis was used to determine which linear combinations of these genes best distinguished pN + from pN0 patients in the group of 35 patients, 19 with and 16 patients without compromised lymph nodes, in which all six gene expression were available. Starting with the six genes, an iterative algorithm was used to select for the gene set which yielded the best predictive result. The discriminant analysis yielded one canonical variable or function. The canonical variable accounted for 100% of the variance of the analysis and 0.552 of the eigen value when the expression of CSNK1A1, SCARB2, SPRR2B and G3BP2 mRNAs were associated. The canonical discriminant function coefficients were CSNK1A1 (2.248), SCARB2 (3.611), SPRR2B (−0.49), G3BP2 (0.046) and a constant (−1.145). The canonical discriminant function was z = 2.248(CSNK1A1) + 3.611(SCARB2)–0.49(SPRR2B)+ 0.046(G3BP2)–1.145 (Wilks’Λ = 0.644, χ2 = 13.619 and p = 0.009). We defined the z-value as the NI, with a value < 0, indicating a negative score and a value > 0 indicating a positive score (Supplementary Table to be found online, at http//www.informahealthcare.com/doi/abs/10.3109/0284186X.2011.620619). Each patient's NI was obtained and the results are plotted in . Among the 19 patients with positive scores, 17 (89.5%) presented lymph node metastasis and 14 (87.5%) of the 16 cases with negative scores were pN0 (p = 0.009, Fisher's exact test). Overall, 88.6% (32/35) of the original grouped patients were correctly classified, and our NI was well correlated with individual nodal status (). With the use of leave-one-out cross-validation procedure, 77.1% of patients were correctly classified, with sensibility 73.7% and specificity 83.3%.
Figure 2. The samples were rank-ordered by their Nodal Index, determined by the discriminant analysis. The samples with negative score indicated that the tumors were predicted to be free of lymph node metastasis (pN0). The samples with positive scores indicated that the tumors were predicted to metastasize to the cervical lymph node (pN +). A,B. The prediction results in learning cases: Fourteen of the 16 samples in pN0 group were negative and 17 of 19 samples in the pN + group were positive. C,D. The prediction results in test case group: seven of 11 samples in pN0 group were negative and all of samples in the pN + group were positive.
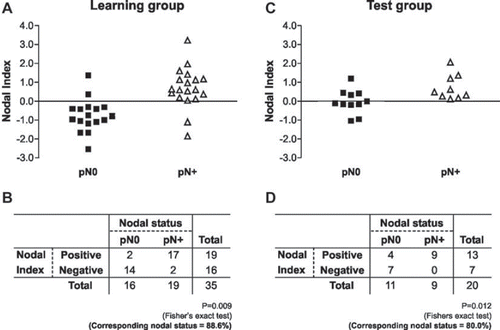
We next evaluated the prediction model, by applying our NI formula, in an independent test group composed of 20 primary tumors, 11 pN0 and nine pN + (). Seven lymph node negative primary tumors (7/11) and nine lymph node positive tumors (9/9) had their lymph node status predicted correctly by our model (overall: 80.0%; p = 0.012, ).
Finally, our NI was shown, in a logistic regression analysis model, to be an independent predictor of lymph node status taking into consideration known variables as tumor size and histological grade ().
Table V. Logistic regression analysis for lymph node status in 35 OSCC patients.
Discussion
In this study, we identified a small set of four genes, SPRR2B, CSNK1A1, SCARB2 and G3BP2, whose mRNA expression correctly classified 80.0% of patients according to their lymph node status. Previous studies had described sets with large number of genes (500 and 102) capable of predicting lymph nodal status in head and neck squamous cell carcinoma with 83% and 86% accuracy [Citation3,Citation4], which is similar to that described here. More recent publications had described small sets of genes with the same predictive strength [Citation5–7]. However, issues involving small number of patients [Citation5], lack of a validation set [Citation7] or type of tumor sample (whole tumor vs. laser-capture microdissection) [Citation6] complicates the conclusions.
Our study has important strengths. First of all, we have used whole tumor sections since the metastatic process seems to depend on both epithelial and stromal tumor components [Citation9]. Second, all patients in the learning and validation group were operable and enrolled prospectively, not to mention that four gene expression can be performed very easily, as long as these results were further validated. In lymph node positive cases, the correct prediction was in the range of 90–100%, including three patients with a clinically negative neck, data that has potential to change patient management.
We recognize however that our study has important limitations. One such limitation was that our NI validation cohort was still small and unfortunately NI was not available for all 49 patients in the learning set due to sample limitation. Second, our index resulted in a 22% false positive rate in node negative cases, which may lead to over treatment. Finally, part of the tests was done using Northern blotting which is still a time consuming method.
To our knowledge, no defined relationship between our four markers and metastasis in OSCC had been described until present. Concerning SPRR2B, a differentiation-associated gene, we have found it down regulated in pN + tumors as compared to pN0 tumors, suggesting that this marker decrease expression with progression in OSCC. In accordance with our data, an inverse correlation between the rate of metastasis and the degree of differentiation has been reported in HNSCC [Citation10] and previously published studies also showed SPRR2B low expression in HNSCC cell lines [Citation11] and in hypopharynx carcinoma [Citation12], even though this last study did not clarify the relationship between SPRR2B expression and clinicopathological findings. A down regulation of SPRR2 expression was also reported in scirrhous gastric cells with high metastatic potential [Citation13].
The remaining three genes in our NI are described as capable of promoting metastasis by either preventing cancer cell apoptosis or else by augmenting endothelial cell survival and favoring angiogenesis. We found a higher expression of isoform CSNK1A1 or CK1α in pN + primary tumors, which is in according with published data showing that CK1 isoforms, especially CK1α, seem to mediate resistance of tumor cells to apoptosis induced by tumor necrosis-factor-related apoptosis-inducing ligand (TRAIL) [Citation14]. CSNK1A1 seems also to influence Smad3 negatively affecting TGFβ signaling [Citation15], a key factor in HNSSC carcinogenesis, as shown by several investigators, including our own group [Citation16–18].
Our data showed an up regulation in pN + tumors of SCARB2, also known as CD36L2, HLGP85 or LIMPII. Even though we do not know if it affects HNSCC carcinogenesis, SCARB2 was previously described as functioning as a thrombospondin-1 receptor [Citation19], which may have an effect sequestering TSP1 and inhibiting TGF beta activation, as shown in a rat model [Citation20].
G3BP2, whose function was so far unknown, was isolated by sequence homology with G3BP1, a protein able to interact with the SH3 domains of the Ras-GTPase activating protein [Citation21]. G3BP2 overexpression seems to directly promote retention of IkBα in the cytoplasm interfering NF-kB/IkBα complexes [Citation22], whose aberrant activity has been described as critical in HNSSC carcinogenesis [Citation23,Citation24]. However, G3BP2 may exert a tumor suppressor role, which seems to be associated with MAP kinase-mediated VEGF down regulation [Citation25].
In conclusion, the coordinated expression of CSNK1A1, SCARB2, SPRR2B and G3BP2 mRNA may be predictive of the presence of lymph node metastasis in oral cancer. These genes could play relevant biological roles integrating TGFβ and NFKB gene networks, which are critical in HNSCC carcinogenesis. Our data can interpreted as a proof of principle concept, with further studies necessary for biological and clinical validation.
Supplementary Table I
Download PDF (1.5 MB)Acknowledgements
This study was supported by FAPESP 02/01738-9 grant.
Declaration of interest: The authors declare that they have no conflict of interest.
References
- De Bree R, Casteljins JA, Hoekstra OS, Leemans R. Advances in imaging in the work-up of head and neck cancer patients. Oral Oncol 2009;45:930–5.
- Kocaturk S, Yilmazer D, Onal B, Erkam U, Urunal B. Do micrometastases detected with cytokeratin immunoperoxidase reactivity affect the treatment approach to neck in supraglottic cancers? Otolaryngol Head Neck Surg 2003;128:407–11.
- Chung CH, Parker JS, Karaca G, Wu J, Funkhouser WK, Moore D, . Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell 2004;5:489–500.
- Roepman P, Wessels LF, Kettelarij N, Kemmeren P, Miles AJ, Lijnzaad P, . An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet 2005;37:182–6.
- Kato Y, Uzawa K, Saito K, Nakashima D, Kato M, Nimura Y, . Gene expression pattern in oral cancer cervical lymph node metastasis. Oncol Rep 2006;16:1009–14.
- Zhou X, Temam S, Oh M, Pungpravat N, Huang BL, Mao L, . Global expression-based classification of lymph node metastasis and extracapsular spread of oral tongue squamous cell carcinoma. Neoplasia 2006;8:925–32.
- Nguyen ST, Hasegawa S, Tsuda H, Tomioka H, Ushijima M, Noda M, . Identification of a predictive gene expression signature of cervical lymph node metastasis in oral squamous cell carcinoma. Cancer Sci 2007;98:740–6.
- Liang P, Pardee AB. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science 1992;257:967–71.
- Mullis TC, Tang X, Chong KT. Expression of connective tissue growth factor (CTGF/CCN2) in head and neck squamous cell carcinoma. J Clin Pathol 2008;61:606–10.
- Remmert S, Rottmann M, Reichenbach M, Sommer K, Friedrich HJ. Lymph node metastasis in head-neck tumors. Laryngorhinootologie 2001;80:27–35.
- Jeon GA, Lee JS, Patel V, Gutkind JS, Thorgeirsson SS, Kim EC, . Global gene expression profiles of human head and neck squamous carcinoma cell lines. Int J Cancer 2004; 112:249–58.
- Lemaire F, Millon R, Young J, Cromer A, Wasylyk C, Schultz I, . Differential expression profiling of head and neck squamous cell carcinoma (HNSCC). Br J Cancer 2003;89:1940–9.
- Hippo Y, Yashiro M, Ishii M, Taniguchi H, Tsutsumi S, Hirakawa K, . Differential gene expression profiles of scirrhous gastric cancer cells with high metastatic potential to peritoneum or lymph nodes. Cancer Res 2001;61:889–95.
- Izeradjene K, Douglas L, Delaney AB, Houghton JA. Casein kinase I attenuates tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by regulating the recruitment of fas-associated death domain and procaspase-8 to the death-inducing signaling complex. Cancer Res 2004;64: 8036–44.
- Guo X, Waddell DS, Wang W, Wang Z, Liberati NT, Yong S, . Ligand-dependent ubiquitination of Smad3 is regulated by casein kinase 1 gamma 2, an inhibitor of TGF-beta signaling. Oncogene 2008;27:7235–47.
- Lu SL, Herrington H, Reh D, Weber S, Bornstein S, Wang D, . Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev 2006;20:1331–42.
- Mangone FR, Walder F, Maistro S, Pasini FS, Lehn CN, Carvalho MB, . Smad2 and Smad6 as predictors of overall survival in oral squamous cell carcinoma patients. Mol Cancer 2010;9:106–15.
- Logullo AF, Nonogaki S, Miguel RE, Kowalski LP, Nishimoto IN, Pasini FS, . Transforming growth factor beta (TGFbeta1) expression in head and neck squamous cell carcinoma patients as related to prognosis. J Oral Pathol Med 2003;32:139–45.
- Crombie R, Silverstein R. Lysosomal integral membrane protein II binds thrombospondin-1. Structure-function homology with the cell adhesion molecule CD36 defines a conserved recognition motif. J Biol Chem 1998;273: 4855–63.
- Chen Y, Wang X, Weng D, Tao S, Lv L, Chen J. A TSP-1 functional fragment inhibits activation of latent transforming growth factor-beta1 derived from rat alveolar macrophage after bleomycin treatment. Exp Toxicol Pathol 2009;61: 67–73.
- Parker F, Maurier F, Delumeau I, Duchesne M, Faucher D, Debussche L, . A Ras-GTPase-activating protein SH3-domain-binding protein. Mol Cell Biol 1996;16:2561–9.
- Prigent M, Barlat I, Langen H, Dargemont C. IkappaBalpha and IkappaBalpha/NF-kappa B complexes are retained in the cytoplasm through interaction with a novel partner, Ras GAP SH3-binding protein 2. J Biol Chem 2000;275: 36441–9.
- Bindhu OS, Ramadas K, Sebastian P, Pillai MR. High expression levels of nuclear factor kappa B and gelatinases in the tumorigenesis of oral squamous cell carcinoma. Head Neck 2006;28:916–25.
- Allen CT, Ricker JL, Chen Z, Van Waes C. Role of activated nuclear factor-kappaB in the pathogenesis and therapy of squamous cell carcinoma of the head and neck. Head Neck 2007;29:959–71.
- Wu WZ, Sun HC, Shen YF, Chen J, Wang L, Tang ZY, . Interferon alpha 2a down-regulates VEGF expression through PI3 kinase and MAP kinase signaling pathways. J Cancer Res Clin Oncol 2005;131:169–78.