
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
We investigate the problem of detecting toxicogenomic associations that generalize across organisms, that is, statistical dependencies between transcriptional responses of multiple organisms and toxicological outcomes. We apply an interpretable probabilistic model to detect cross-organism toxicogenomic associations and propose an approach for drug toxicity analysis based on the interactive retrieval of drugs with similar toxicogenomic properties. We show that our approach can give relevant information about the properties of a drug even when direct prediction of toxicity is not feasible. Moreover, we show that a search from a cross-organism database can improve accuracy in the analysis.
Abbreviations
| ATC | = | anatomical therapeutic chemical |
| DILI | = | Drug-induced liver injury |
| FDA | = | Food and Drug Administration |
| GFA | = | group factor analysis |
| GSEA | = | gene set enrichment analysis |
| QSAR | = | quantitative structure-activity relationship |
| TGP | = | Japanese Toxicogenomics Project |
Introduction
Evaluation of potential toxicity of new drugs and other chemical compounds is highly important for safety reasons. The toxic effects of new drugs cannot be tested directly on humans due to the obvious ethical issues, and new drugs thus go through a series of in silico and in vitro analyses, and then an animal experimentation phase. Organisms from yeastCitation1 to the worm C. elegans,Citation2 zebrafishCitation3 and murine animalsCitation4 are used in the drug development process, starting with simple organisms and moving toward organisms more similar to humans. All toxic effects are not visible in all the model organisms and experimental setups, and many of the effects are discovered only when the compound is experimented on humans. Even after the drug has entered the market, weak or rare effects can be discovered among the large population of consumers.
The earlier the toxic responses can be detected, the more potential harm can be avoided and resources saved. Computational tools for predictive toxicity have been developed and applied at each stage of the drug development cycle.Citation5,6 Quantitative structure-activity relationship (QSAR) assessment has traditionally been the most prominent in silico toxicity prediction procedure, where toxicological profiles, such as lethal concentrations, are predicted based on structural descriptors of the compounds.Citation7 Recently, the focus has shifted to identification of critical perturbations in biological pathways that lead to adverse outcomes, based on high-throughput screening methods.Citation8
Toxicogenomics
Toxicogenomics has emerged in the cross-section of toxicology and bioinformatics, with the aim of finding predictive associations between transcriptomic and toxicological responses.Citation9,10 The rationale is that drug-treatment transcriptional data consist of various response patterns, some of which are related to drug toxicity. The identification of these toxicity-associated transcriptional response patterns is essential for understanding the molecular mechanisms behind toxicity and for enabling the prediction of toxicity.Citation11 However, distinguishing toxic adverse effects from intended therapeutic effects and from various types of noise factors, such as batch effects, is highly non-trivial. Moreover, transcriptomic response patterns vary over tissues and cell types, making this more complicated. As toxicogenomic studies are typically performed in vitro, it would be important to identify those toxicogenomic associations that generalize to humans as well.
The ToxCast projectCitation12 is an example of large-scale high-throughput in vitro screening for predicting in vivo toxicity. The TG-GATEs database from the Japanese toxicogenomics projectCitation13 is another interesting toxicogenomic resource with transcriptional drug-treatment data available from organisms both in vitro and in vivo. Additionally, the database includes toxic outcome observations such as blood level measurements and observed liver injuries from rats in vivo.
Liver toxicity is among the most common types of drug toxicity in humans.Citation5 The drug-induced liver injury (DILI) labelingsCitation14 have been designed to describe the risk of hepatotoxicity in humans: The labels are continuously updated as the Food and Drug Administration (FDA) acquires more information about the potential side effects of the drugs on the market. The DILI labels are available for most of the drug compounds with experimental data at the TG-GATEs database.
Data translation with machine learning
The next step that follows detection of responses to drug compounds in a model organism is translation of these responses to humans. In this work, we build on the hypothesis that responses shared across organisms are more likely to generalize to humans as well. This is analogous to searching for conserved genomic regions or responses, but on the more abstract level of statistical relationships in the response profiles.
To detect “conserved responses,” we need to examine databases of drug-response experiments from multiple model organisms, or “domains.” The conserved response patterns can then be utilized to make predictions about the human response based on experimental data from model organisms, that is, to carry out “data translation” from one domain to another.
We define “data translation” as an analog of language translation: of finding how a phenomenon in one domain or organism is expressed in another, assuming it generalizes across domains, and then predicting it. Data translation is a key part of “translational medicine,” which involves many additional aspects.
In summary, our goal is to develop machine learning methods for discovering responses conserved across organisms and for generalizing the responses to humans. The generalization of the responses has so far been an unsolved problem. For discovering conserved responses, Le and Bar-JosephCitation15 have presented an approach for clustering genes across organisms based on their response patterns. Suvitaival et al.Citation16 focused on quantifying the responses to external covariates, such as the drug treatment, that are conserved across organisms. Both of these approaches assume that a group of genes responds to the covariate in a coherent fashion.
In this article, we assume that drug responses can be modeled as factors, each of which describes a biological process that is disturbed by the treatment. Individual genes may be members of many of these processes and the genes may be different across organisms. Also the level and direction of responses may vary across genes and organisms while still following the abstract conserved pattern.
Generative model for cross-organism toxicogenomics
Inspired by the CAMDA challenge,Citation17 we address the following research questions: 1) Can we associate drug-induced toxicological responses observed in humans or rats to changes observed at the molecular level, and are these associations predictive? 2) Can we find toxicogenomic associations that are conserved across organisms? Could these associations be utilized to replace animal studies with in vitro assays?
In other words, we seek simultaneous associations between transcriptional data and toxicological outcome data, and between transcriptional data from multiple organisms. Associations that generalize both across organisms and across levels of biological complexity have the potential of enabling the data translation between the molecular level and the organ level or the population level.
The biological properties and their resemblance to the human vary across the cells extracted from animals grown in vivo and cell lines grown in vitro. Even though this resemblance to the human is still largely unkown, they all are grown with the purpose of experimenting chemical compounds intended for human use. By taking a data-driven approach to identifying conserved responses, we do not make prior assumptions about the organisms’ similarity to the human. To stress these points, we refer to each of the types of biological sample as a model organism, even though a cell line is not an entire representation of the animal from which it is originally extracted from. Moreover, we view a cell line grown in vitro as a different model organism than what a cell extract from an animal of the same species grown in vivo is.
We propose a generative model-based approach to answer the two research questions. To do this, we make the following modeling assumptions: 1) The data consist of drug-induced transcriptional responses patterns, that is, consistent gene expression changes for a subset of the drugs and genes, and noise from various sources. 2) Drugs may activate multiple response patterns, and the patterns may be partially overlapping in terms of affected genes. 3) We are especially interested in response patterns that are associated with observed toxic outcomes and are conserved across organisms.
It turns out that a recently introduced model family, group factor analysisCitation18 (GFA), when applied to toxicogenomic data, matches these assumptions. It is a multi-view model that in an unsupervised fashion detects statistical dependencies between multiple data sets having co-occurring samples. In this context, samples correspond to drug treatments, which are the same in all the data sets. We call the data sets “views,” because they are matched by their samples.
The associations found by the model are represented by factors that are interpretable in terms of factor loadings of the data variables, in this case genes. This interpretability allows the user to formulate testable hypotheses, for instance about the mechanisms of action of a drug and about their association to toxicological outcomes. The associations can also be used for predicting one data view based on another, for example, predicting toxic outcomes based on transcriptomic responses.
For cross-organism toxicogenomic analysis, group sparsity is an especially useful feature of GFA. The model can distinguish patterns that are shared across all the data sources from patterns that are specific to a single source or shared by a subset of the sources. In this paper, we will apply GFA to studying biological responses that are conserved across organisms.
Results
We demonstrate the potential of the model to detect responses that generalize across organisms in two practical use cases with the TG-GATEs data,Citation13 consisting of three sets of transcriptional drug-treatment measurements: human in vitro, rat in vitro and rat in vivo. In Case 1, the task is to find associations between transcriptional changes and pathological findings from in vivo rat livers. In Case 2, the task is to search for drugs having a similar risk of DILI in humans at the population level, based on data about transcriptional changes in model organisms.
Case 1: Finding associations between transcriptomic responses and pathological findings
In the first case, we are interested in two types of associations to start with: First, associations between the molecular level and the organ-level, and second, molecular-level associations between the different organisms. In order to detect responses that are most likely to generalize to humans, we require both of these constraints to hold for the associations that we focus on. Focusing on these maximally conserved associations will also be beneficial for filtering out structured noise that arises from the laboratory effects and from the properties of the model organisms.
Applying GFA to the combination of three transcriptomic data sets and pathological findings for rat in vivo, we obtain a set of factors that capture the required kind of associations. Each factor is interpretable as a biological process associated with specific pathological findings at the organ-level and is generalized across a subset of the organisms at the molecular level (). This result indicates that the model learns biologically meaningful response structure in the transcriptomic data. For example, Factor B associates changes in metabolic processes to degeneration in the liver tissue, while Factor C associates changes in the cell-cycle to increased mitosis in the liver.
Figure 1. The model detects drug response patterns that generalize across organisms and are associated to organ-level changes driven by toxicity. Also the biological interpretation of the associations represented by a factor generalizes across organisms: changes at the molecular level are interpretable as a biological process. The “eye diagram” shows identified associations between pathological findings (left) and enriched gene ontology (GO) terms (right), represented by factors of the model (middle). Line widths between pathological findings and factors indicate the magnitude of factor loadings learned by the model. Line widths between factors and GO terms indicate the strength of the enrichment. Associations are shown individually for each organism and factor: organisms are indicated as small nodes attached to the nodes of the factors. Factors are named alphabetically from A to H; organisms are human in vitro (1), rat in vitro (2) and rat in vivo (3).
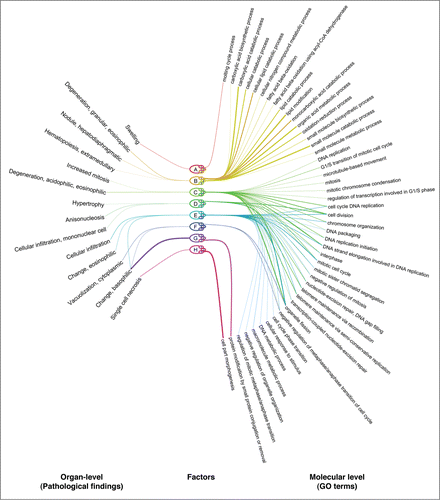
Although the associations are biologically meaningful, given the small amount of available data, their predictive power is not significant (results not shown; the low power was not due to the method, which was tested additionally using a standard L1-regularized regression model). As more toxicogenomic data accumulates, the predictive power of the associations needs to be revisited.
Case 2: Modeling-based data retrieval for human drug toxicity analysis
Direct prediction of toxicity for a new drug is not a trivial task, but we have demonstrated that the detected conserved associations are biologically meaningful. Predicting the toxicity of a drug on humans is even more difficult due to the lack of direct experimental data. Analyzing drug toxicity in humans is possible indirectly, using available drug toxicity classifications of approved drugs. These data are not perfect, however, as the toxic potential of many drugs has been over-estimated for increased safety.Citation14 Some drugs have been categorized as risky based on only indirect evidence of other drugs, with similar therapeutic potential or chemical properties, having shown toxic outcomes.
Interactive toxicity analysis framework
We propose an alternative approach for the risk-analysis of a novel drug by formulating the prediction task as an information retrieval problem. We assume that transcriptomic response data in existing databases of model organism experiments carries relevant information on drug toxicity in humans. The level of relevance may, however, vary across different experimental practices and model organisms. For instance, in vivo experiments are likely to be more informative than in vitro experiments.
The interactive toxicity analysis takes place through a table-lookup procedure: Given a query compound and a measure of similarity, the expert receives a ranked list of database compounds in the order of the similarity of transcriptomic response. To the extent there are associations between the molecular level and the organ-level, the properties of the top-ranked database compounds are likely to be similar to the query compound. Based on the list, an expert user can then construct a hypothesis about the expected properties of the drug and about the uncertainty around these properties. In an illustrative example of the retrieval result for a query (), many of the top-ranked drug compounds retrieved from the database are shown to share toxic and therapeutic properties with the query.
Table 1. An example retrieval result shows notable similarity to the query both by toxic and therapeutic properties
The idea of searching for similar drugs has earlier been introduced as “connectivity mapping”Citation19 and applied to drug discovery and drug repositioning.Citation20,21 It has also been applied to drug toxicity analysis.Citation22,23 Recently, Xing et al.Citation24 introduced an online resource for making queries to the TG-GATEs database. We use the retrieval method behind that tool as one of the two baseline approaches in the experiments that follow. In the connectivity mapping approaches the similarity measure for the retrieval relevance is based on the gene set enrichmentCitation25 computed on the list of the most differentially expressed genes for the query drug. These approaches have either focused on a single cell type or simply averaged over multiple cell types, neglecting the likely differences between organisms.
We propose to carry out toxicity analysis by modeling-based retrieval that takes into account the translatability of data between different organisms. In particular, we use the GFA to detect shared transcriptomic responses between the three model organisms in the database: human in vitro, rat in vitro and rat in vivo. Now, we can examine the similarity in the responses in the lower-dimensional latent space of the model. More importantly, we can focus our examination into the part of the latent space that is shared between the model organisms (details in the Materials and Methods). The shared latent factors describe the drug-responses that are conserved across the model organisms, and thus are likely to have potential for the generalization to humans as well.
We evaluate the retrieval with ground truth from the DILI label and concern classes,Citation14 as well as with more detailed information about the drugs’ mechanism of action, described by the anatomical therapeutic chemicalCitation26 (ATC) classes. We compare with rank-based connectivity mappingCitation19 and simple correlation between the differential expression profiles. As a measure of performance, we use mean average precision.
Retrieval from single-organism database
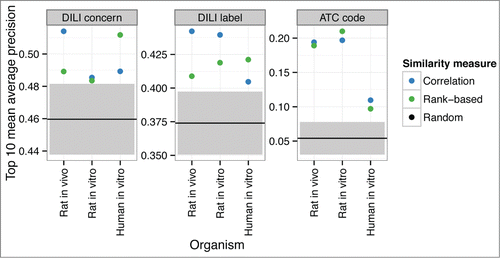
Transcriptomic drug response data are informative about both the toxicity and mechanisms of action (), resulting from off-target and on-target effects of the drug, respectively. For all organisms, types of validation classes and used similarity measures, retrieval based on the transcriptomic database lead to a higher performance than expected by chance. This indicates that the transcriptomic response data on model organisms is informative of the toxicity of the drugs on humans at the population level. However, the results are not conclusive of the relative performance of the individual organisms. Retrieval performance is observed to be almost as sensitive to the choice of the similarity measure as it is to the choice of the organism.
Figure 2. All model organisms are informative of the human population-level risk of toxicity. The figure shows how much information the retrieved similar drugs give about the DILI concern, DILI label and ATC level four class, of the query drug. The figure shows the top-10 mean average precision (y-axis) for each organism (x-axis) when used for the retrieval. Retrieval based on differential expression data gives above-random results for each organism using both the correlation and rank-based similarity measure. For the randomized results, shaded areas indicate the 95% confidence intervals.
Retrieval from cross-organism database
We study the potential of cumulating biological information from existing model organism experiments to increase the amount of knowledge that can be extracted from human in vitro experiments. We focus on human in vitro experiments, because they are more ethical and less expensive than in vivo experiments and could potentially replace in vivo animal studies in the future.
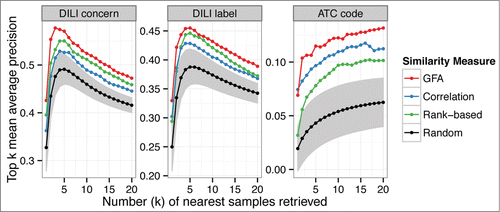
We examine model-based retrieval performance from a cross-organism database of transcriptional measurements, given a human in vitro sample as a query. The results show that retrieval performance is improved by using the cross-organism database of experiments compared with single-organism retrieval, when the retrieval is based on responses conserved across the model organisms (). The outcome is consistent on all the three validation classes. This is indirect evidence for the hypothesis that compared with organism-specific responses, conserved responses of model organisms are more likely to generalize to humans at the population level.
Figure 3. GFA-based cross-organism approach leads to a higher performance in the retrieval of similar compounds to a human in vitro query. The figure shows the top-k mean average precision as a function of the number k of retrieved highest-ranking samples. GFA utilizes the cross-organism associations learned from the database while the other methods rely on the human in vitro data only. For the randomized results, shaded areas indicate the 95% confidence intervals.
Discussion
We have analyzed drug toxicity using a new machine learning approach that identifies cross-organism toxicogenomic associations. This is a key step toward developing methods for predictive toxicology. The identification of associations that generalize reliably across multiple organisms, especially from in vitro to in vivo, is essential for toxicity analysis. This approach has potential for predicting drug toxicity in humans based on in vitro experiments, thus reducing the need for animal studies in vivo.
The TG-GATEs data set with experiments on three model organisms has given us the opportunity to take a data-driven approach for cross-organism toxicogenomics. The group factor analysis model for toxicogenomic responses is flexible about the type of responses: neither genes nor biological pathways are restricted to be the same between the organisms. Minimum two model organisms are needed for identifying conserved responses. A new experiment in one organism can then be generalized via retrieval. The model can operate in the “small n, large p” regime thanks to the probabilistic approach and the sparsity assumptions.
We have shown how our probabilistic model finds biologically relevant associations between transcriptomic drug responses and pathological findings from rats, and that many of these associations generalize across in vivo and in vitro organisms. However, the predictive performance of these linear associations is very limited, probably due to limited amount of data, as the pathological findings have been observed only for a few rat samples.
Since quantitative linear prediction of toxicological outcomes is limited in performance, we propose an alternative toxicity analysis scheme. It is based on information retrieval, where the task is to search for the most relevant drugs from the database of existing experiments, given a new query drug. Based on the most relevant drugs retrieved, the user can then construct a hypothesis of the toxicity and other properties of the query drug. This can support expert decision making.
We first studied the retrieval performance using the differential gene expression data only, and confirmed earlier findingsCitation22,23 about the suitability of the retrieval approach to the task of identification of toxic drug compounds. We then showed that when we do retrieval based on cross-organism associations, we were able to improve the retrieval performance, as compared with single-organism retrieval. This indicates that the cross-organism associations detected by the model are relevant for human toxicity and give hope that the in vivo animal studies could be replaced with in vitro studies in the future.
Materials and Methods
We report the pre-processing done for the data before modeling, the model description, and the technical details of the two experiments (Cases 1 and 2). The details of Cases 1 and 2 are described in the subsections Model-based exploratory analysis and Retrieval of relevant experiments, respectively.
Data pre-processing
The data set of the Japanese Toxicogenomics Project (TGP) includes transcriptional data from three model organisms: primary hepatocyte cells from humans and rats grown in vitro, and similar cells extracted from rats in vivo. The conditions of the experiment can be summarized as three experimental covariates: administered drug compound, its dosage and time from the administration of the compound. For the analysis in this work, we selected the subset of experimental covariate levels that are observed in all three organisms. This set includes 119 drug compounds administered at two dosage levels (middle and high) and measurements made at two time points after the treatment (8/9 h and 24 h). Histopathology of the liver had been examined from the extracted livers in the rat in vivo experiments at the same time points and dosage levels, providing a pathological finding class and severity grading for each sample. The data were downloaded from the website of the CAMDA challenge,Citation27 where the transcriptional observations were provided in a FARMS-summarizedCitation28 format.
For the modeling task, we considered each treatment—a combination of compound, dose and time—as a single sample in the model. We selected transcriptomic probes, which have non-zero variance across the samples and which appear in all the three transcriptomic microarray data sets. This was done to make the data sets from different organisms balanced in their size in order to allow a fair comparison between the relevant information content in them. However, the model itself does not require the variables of the data sets to be matched and the analysis could alternatively be done on all probes as well.
We computed the average differential expression of the treated samples against the corresponding control samples. We represented the pathological finding classes for each sample as a grade-weighted count. As the four data matrices (differential gene expression X(human in vitro), X(rat in vitro), and X(rat in vivo), as well as pathological findings Y) are now matched by their samples, we call the matrices different views of the data.
Model
We have N observation vectors , corresponding to measured transcriptional and toxicological responses to drug treatments indexed as n = 1, …, N. Observations from one measurement type m are concatenated as columns of a data set X(m). All data sets are matched by co-occurring observations, that is, they can be regarded as views. We assume the transcriptomic data contain complex drug-induced response patterns embedded in measurement noise. We are interested in finding these patterns and, more importantly, in associating them to toxic outcomes. Response patterns that are present in multiple views provide valuable information for interpretation and data translation. The task suits well to the problem formulation of GFA,Citation18 which learns associations between matched data sets.
GFA is formulated as a Bayesian latent factor model, where the data are explained by factors. Each observation from the mth view is generated from a multivariate normal distribution
(1)
(1) where zn are the latent factors for the nth observation, W(m) are the factor loadings for the mth view, and the noise covariance matrix is assumed to be diagonal,
, with a view-specific precision τm. The main task is to learn how factors are associated with the views: each factor describes associations between any combination of the views. Thus, some factors are shared across all the views, some are shared by a subset of the views, and the rest are specific to a single view. For a view m that is not associated with factor k, the kth column of W(m) is automatically set to zero by the model. With variables from each view seen as groups, this is equivalent to group-sparse factor loadings.
GFA learns the associations by employing a group-sparse prior distribution for the factor loadings. That is, each column of W(m) is generated from a normal distribution(2)
(2) where precision
is drawn from a gamma prior distribution,
(3)
(3) with small values for the shape parameters α0 and β0. Gamma distribution is conjugate to normal distribution with a known mean. When the prior and the likelihood are conjugate, posterior inference through Gibbs sampling is possible, as the posterior is of the same form as the likelihood and the parameters of the posterior distribution can be directly calculated based on the parameters of the prior and the likelihood. The model learns the sought-for associations for factor k by setting the
of non-associated views m close to zero, thus pushing all the elements in the factor loadings for those views jointly to zero. To complete the model description, a conjugate gamma prior,
(4)
(4) is set for the noise precisions, and the latent variables are generated from a normal distribution
(5)
(5)
Factors capture response patterns in the observed data, for instance, sets of genes in the transcriptomic views that respond to sets of drug-treatments in a coherent fashion. Some of these patterns are shared across views. Each factor and the corresponding loadings are assumed to represent a biological process and we are interested in interpreting them. Thus, each factor is assumed to be related to a sparse set of drugs and each loading to a sparse set of variables, for example genes. Further, we assume that each drug induces a sparse set of response patterns corresponding to sparsity of zn. Motivated by these assumptions, we modify the priors for GFA in a way that leads to a more easily interpretable model.
We extend the plain GFA by assuming that, in addition to the group sparsity, both the factors and the factor loadings are element-wise sparse. With this extension, the GFA model becomes a multi-view biclustering model, generalizing the factor analysis-based multiplicative biclustering model (FABIA)Citation29 to multiple views of the data. Further, FABIA and GFA with the element-wise sparsity structure extend the Bayesian plaid modelCitation30 from additive responses to multiplicative responses.
We modify the priors of the GFA model to achieve the element-wise sparsity for the factors and the factor loadings by drawing them both from a two-component mixture distribution. In the mixture, the first component corresponds to a delta distribution δ0 with a peak at zero, and the second to a normal distribution with a zero mean and an unknown precision. This construction corresponds to a spike-and-slab prior,Citation31,32 where the spike is a delta distribution and the slab is a normal distribution.
Mathematically, the spike-and-slab prior for the factors is written as(6)
(6) and for the factor loadings as
(7)
(7)
Binary variables and
indicate whether zn,k and
, respectively, are set to zero or drawn from a normal distribution. The
are drawn from a Bernoulli distribution,
(8)
(8) where the expectation
is specific to each factor k and view m. The
is drawn from a β distribution
(9)
(9) with shape parameters a0 and b0. The β prior distribution is conjugate to the Bernoulli distribution, leading to a posterior, which is Bernoulli-distributed. A similar construction is used for the
but now the expectation is shared across observations. When
is close to zero, the kth column of W(m) is suppressed to zero jointly, implementing group sparsity. We also find shared noise for each view too limiting and instead allow variable-wise independent noise by assuming a non-isotropic diagonal Σ(m) whose elements are drawn independently from a gamma distribution.
Since all the priors are conjugate, we implement inference using Gibbs sampling. The sampler learns the model for the TG-GATEs data set overnight on a standard desktop computer. A variational Bayesian approximation, presented for the vanilla GFA model earlier,Citation18 may be useful for larger data sets. An implementation of the model and a demo are available at http://research.ics.aalto.fi/mi/software/GFAtoxgen/.
Model-based exploratory analysis
We study the biological interpretability of the learned associations which are represented by factors of the model. More specifically, we focus on factors that are shared across all the views. In order to do that, we need to define a threshold for a factor to be considered shared by the views. We consider the kth factor as shared, if in each of the m views there exists at least one non-zero value in the loadings vector of the kth factor. In Case 1, we study associations that generalize across the transcriptomic views X(human in vitro), X(rat in vitro) and X(rat in vivo), and the pathology view Y.
For the interpretation of the model, we want to study the importance of individual variables of the observed data to the detected association. For the kth factor representing an association between the views, we do this by examining its loadings across the m views.
For biological interpretation, we rank variables of the observed data for each factor-view pair (k,m). The ranking is done by sorting the loadings by their magnitude. For the transcriptomic data views, this procedure leads to a ranked list of transcriptomic microarray probes. The drug-response behavior of the top-ranked probes can be seen as being explained by the factor based on which the ranking was done.
To detect biological processes, whose changes in the mth transcriptomic view are explained by the kth factor, we computed the hyper-geometric enrichment testCitation25 for gene ontology (GO) terms of the transcriptomic probes for the factor-transcriptomic view pair. The P values of the test were controlled for false discovery with the Benjamini-Hochberg correctionCitation33 at the level 0.05. Associations between the enriched pathways and pathological findings were reported in based on factor loadings of the pathology view.
Retrieval of relevant items
Retrieval means the search of relevant items given a query item. Given the query, the relevance of the items in the database is computed based on a similarity measure, and the items are retrieved in the ranked order of similarity.
In Case 2, the items are drug-treatments. We retrieved drug-treatments relevant to the query treatment from the database based on their similarity in transcriptomic responses, either using a single-view database X(human in vitro), X(rat in vitro) or X(rat in vivo), or using a multi-view database consisting of all the three transcriptomic views.
For single-view retrieval, we considered two similarity measures. In the first measure (“correlation”), similarity is defined simply as the correlation between the transcriptomic profiles of the query and the database from the organism in question. As the second measure (“rank-based”), we used a ranked-based approach, also known as connectivity mapping.Citation19 To compute the similarity of the items, we followed the procedure by Iorio et al.Citation20 In brief, we used a signature of the 250 most differentially expressed genes, and computed the average enrichment score similarity between the query signature and the entire ranked list of genes of each of the database items.
Multi-view database
The simple approach used to compare the query against a single-view database is not directly applicable, when the database and query come from different views or from a different set of views. In either of the cases, we can utilize GFA to detect cross-view associations that then enable the data translation between the query and the database domains and allow us to retrieve relevant items across views.
The database contains data matrices representing views m = 1, …, M. In each view, items are organized as rows and variables as columns. Items are co-occurring between the views. The query item X(query) may be observed in a subset of the database views. In the experiment of this article, the query item is an observation vector from the human in vitro transcriptomic view, while the database consist of all the three transcriptomic views.
Since the data domains of the query and the database now are different, similarity search cannot be done in the original data domain as it was done with a single-view database. Latent representation of GFA allows us to carry out the similarity search between items that are observed in different domains. First, we learn a GFA model for the database items. Then, using the learned factors, we learn a latent representation for the query item. Having a latent representation for both the query item and the database items, we can carry out the similarity search in the latent space of the model. Again, we use correlation as a similarity measure, but now in the latent space instead of the original data domain.
Validation
We validate the retrieval outcome using external information for the items. First, we use the DILI label and concern classes,Citation14 which describe the toxic risks of the drugs observed for the large population of consumers. Second, we use the ATC codesCitation26 at level 4 to give more detailed information about the drugs’ mechanisms of action.
We measure the retrieval performance in terms of mean average precision at retrieving items with the same class with the query. We compare the retrieval performance to the performance that follows the randomization of the class information. For the randomization, we report the mean and confidence intervals with the width of two standard deviations.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
The Academy of Finland (Finnish Centre of Excellence in Computational Inference Research COIN, 251170; Computational Modeling of the Biological Effects of Chemicals, 140057), Finnish Doctoral Programme in Computational Sciences FICS, and Helsinki Doctoral Programme in Computer Science.
References
- Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997; 278:1064-8; PMID:9353181; http://dx.doi.org/10.1126/science.278.5340.1064
- Kaletta T, Hengartner MO. Finding function in novel targets: C. elegans as a model organism. Nat Rev Drug Discov 2006; 5:387-98; PMID:16672925; http://dx.doi.org/10.1038/nrd2031
- Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nat Rev Drug Discov 2005; 4:35-44; PMID:15688071; http://dx.doi.org/10.1038/nrd1606
- Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov 2006; 5:741-54; PMID:16915232; http://dx.doi.org/10.1038/nrd2110
- Collins FS, Gray GM, Bucher JR. Toxicology. Transforming environmental health protection. Science 2008; 319:906-7; PMID:18276874; http://dx.doi.org/10.1126/science.1154619
- Hardy B, Apic G, Carthew P, Clark D, Cook D, Dix I, Escher S, Hastings J, Heard DJ, Jeliazkova N, et al. Toxicology ontology perspectives. ALTEX 2012; 29:139-56; PMID:22562487; http://dx.doi.org/10.14573/altex.2012.2.139
- Willighagen EL, Wehrens R, Buydens LMC. Molecular chemometrics. Crit Rev Anal Chem 2006; 36:189-98; http://dx.doi.org/10.1080/10408340600969601
- Krewski D, Westphal M, Al-Zoughool M, Croteau MC, Andersen ME. New directions in toxicity testing. Annu Rev Public Health 2011; 32:161-78; PMID:21219154; http://dx.doi.org/10.1146/annurev-publhealth-031210-101153
- Chen M, Zhang M, Borlak J, Tong W. A decade of toxicogenomic research and its contribution to toxicological science. Toxicol Sci 2012; 130:217-28; PMID:22790972; http://dx.doi.org/10.1093/toxsci/kfs223
- Zhou T, Chou J, Watkins PB, Kaufmann WK. Toxicogenomics: transcription profiling for toxicology assessment. In: Luch A, editor. Vol. 1, Molecular, Clinical and Environmental Toxicology; Basel (Switzerland): Birkhäuser; 2009. p. 325-366. (Experientia Supplementum; vol. 99)
- Hartung T, van Vliet E, Jaworska J, Bonilla L, Skinner N, Thomas R. Systems toxicology. ALTEX 2012; 29:119-28; PMID:22562485; http://dx.doi.org/10.14573/altex.2012.2.119
- Thomas RS, Black MB, Li L, Healy E, Chu T-M, Bao W, Andersen ME, Wolfinger RD. A comprehensive statistical analysis of predicting in vivo hazard using high-throughput in vitro screening. Toxicol Sci 2012; 128:398-417; PMID:22543276; http://dx.doi.org/10.1093/toxsci/kfs159
- Uehara T, Ono A, Maruyama T, Kato I, Yamada H, Ohno Y, Urushidani T. The Japanese toxicogenomics project: application of toxicogenomics. Mol Nutr Food Res 2010; 54:218-27; PMID:20041446; http://dx.doi.org/10.1002/mnfr.200900169
- Chen M, Vijay V, Shi Q, Liu Z, Fang H, Tong W. FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov Today 2011; 16:697-703; PMID:21624500; http://dx.doi.org/10.1016/j.drudis.2011.05.007
- Le H-S, Bar-Joseph Z. Cross species expression analysis using a Dirichlet process mixture model with latent matchings. In: Lafferty JD, Williams CKI, Shawe-Taylor J, Zemel RS, Culotta A, editors. Advances in Neural Information Processing Systems 23. 24th Annual Conference on Neural Information Processing Systems; 2010 Dec 6–9; Vancouver, BC, Canada. Red Hook, NY: Curran Associates; 2011. p. 1270-1278
- Suvitaival T, Huopaniemi I, Oresic M, Kaski S. Cross-species translation of multi-way biomarkers. Honkela T, Duch W, Girolami M, Kaski S, editors. Artificial Neural Networks and Machine Learning – ICANN 2011. 21st International Conference on Artificial Neural Networks; 2011 June 14–17; Espoo, Finland. Berlin/Heidelberg (Germany): Springer; 2011. Part I: p. 209-216. (Lecture Notes in Computer Science; vol 6791)
- The CAMDA Organizing Committee. The CAMDA challenges [Internet]. [cited 2013 Sep 23]. Available from: http://dokuwiki.bioinf.jku.at/doku.php/contest_dataset
- Virtanen S, Klami A, Khan SA, Kaski S. Bayesian group factor analysis. In: Lawrence N, Girolami M, editors. JMLR W&CP 22. 15th International Conference on Artificial Intelligence and Statistics; 2012 Apr 21–23; La Palma, Canary Islands. JMLR; 2012. p. 1269-1277
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet J-P, Subramanian A, Ross KN, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006; 313:1929-35; PMID:17008526; http://dx.doi.org/10.1126/science.1132939
- Iorio F, Rittman T, Ge H, Menden M, Saez-Rodriguez J. Transcriptional data: a new gateway to drug repositioning? Drug Discov Today 2013; 18:350-7; PMID:22897878; http://dx.doi.org/10.1016/j.drudis.2012.07.014
- Qu XA, Rajpal DK. Applications of Connectivity Map in drug discovery and development. Drug Discov Today 2012; 17:1289-98; PMID:22889966; http://dx.doi.org/10.1016/j.drudis.2012.07.017
- Caiment F, Tsamou M, Jennen D, Kleinjans J. Assessing compound carcinogenicity in vitro using connectivity mapping. Carcinogenesis 2014; 35:201-7; PMID:23940306; http://dx.doi.org/10.1093/carcin/bgt278
- Smalley JL, Gant TW, Zhang S-DD. Application of connectivity mapping in predictive toxicology based on gene-expression similarity. Toxicology 2010; 268:143-6; PMID:19788908; http://dx.doi.org/10.1016/j.tox.2009.09.014
- Xing L, Wu L, Liu Y, Ai N, Lu X, Fan X. LTMap: a web server for assessing the potential liver toxicity by genome-wide transcriptional expression data. J Appl Toxicol 2013; (Forthcoming); PMID:24022982; http://dx.doi.org/10.1002/jat.2923
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102:15545-50; PMID:16199517; http://dx.doi.org/10.1073/pnas.0506580102
- ATC classification index with DDDs 2013. Oslo: WHO Collaborating Centre for Drug Statistics Methodology. 2012 [cited 2013 Sep 23]. Available from: http://www.whocc.no/atc_ddd_index/
- The CAMDA Organizing Committee. Preprocessed TGP data [Internet]. 2013 [cited 2013 Mar 1]. Available from: http://dokuwiki.bioinf.jku.at/doku.php/tgp_prepro/
- Hochreiter S, Clevert D-A, Obermayer K. A new summarization method for Affymetrix probe level data. Bioinformatics 2006; 22:943-9; PMID:16473874; http://dx.doi.org/10.1093/bioinformatics/btl033
- Hochreiter S, Bodenhofer U, Heusel M, Mayr A, Mitterecker A, Kasim A, Khamiakova T, Van Sanden S, Lin D, Talloen W, et al. FABIA: factor analysis for bicluster acquisition. Bioinformatics 2010; 26:1520-7; PMID:20418340; http://dx.doi.org/10.1093/bioinfor-matics/btq227
- Caldas J, Kaski S. Bayesian biclustering with the plaid model. In: Proceedings of the IEEE Workshop on Machine Learning for Signal Processing (MLSP); 2008 Oct 16–19; Cancun, Mexico. IEEE 2008; p. 291-6
- Ishwaran H, Rao JS. Spike and slab variable selection: Frequentist and Bayesian strategies. Ann Stat 2005; 33:730-73; http://dx.doi.org/10.1214/009053604000001147
- Mitchell T, Beauchamp JJ. Bayesian variable selection in linear regression. J Am Stat Assoc 1988; 83:1023-32; http://dx.doi.org/10.1080/01621459.1988.10478694
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc, B 1995; 57:289-300