

ABSTRACT
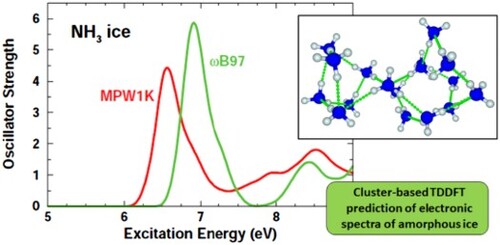
Time-dependent density functional theory (TDDFT) offers a tractable means to predict electronic excitation spectra of ices if sufficient care is taken in selecting the DFT functional, basis sets, and the number of excited states. In this work 48 functionals in conjunction with various correlation consistent basis sets were benchmarked for predicting electronic spectra. A training set of important astromolecules was chosen: carbon monoxide, water, hydrogen cyanide, ammonia, methanol, and the hydroxyl radical. Vertical excitation energies between the ground and first excited state and some higher excited states were calculated using multireference configuration interaction calculations to serve as benchmarks for gas phase TDDFT calculations. When used in conjunction with aug-cc-pVDZ quality correlation consistent basis sets, two functionals, MPW1K and ωB97, emerged as very good choices to model electronic excitations for most of the molecules in the training set. Cluster calculation predictions for the UV spectra of amorphous ices of water and ammonia were performed at the TDDFT-MPW1K/AVDZ and TDDFT-ωB97/AVDZ levels and then compared against experimental data. The calculations qualitatively reproduce the shapes of the spectra for the most part and quantitatively predict the position of the critical first absorption peak.
GRAPHICAL ABSTRACT
Acknowledgements
This work commemorates the contributions of Tim Lee to science and the inspiration he provided to strive toward accuracy and integrity.
Disclosure statement
No potential conflict of interest was reported by the author(s).