
ABSTRACT
The accumulation of glucosylceramide (GlcCer), which is synthesized by UDP-glucose ceramide glucosyltransferase (UGCG), is associated with several diseases, including Gaucher disease and Parkinson’s disease. Since the inhibition of UGCG can be used to treat diseases caused by GlcCer accumulation, several UGCG inhibitors have been developed. In this study, we report on the inhibition of UGCG activity by cambinol, a sirtuin inhibitor. Unlike conventional UGCG inhibitors, cambinol has no structural similarity to GlcCer. LC-ESI MS/MS analysis revealed that the cellular GlcCer levels were reduced by cambinol with an increase in ceramide, the GlcCer precursor. Histidine 193 plays an important role in the inhibition of UGCG via a known UGCG inhibitor, D-PDMP. However, cambinol was found to inhibit UGCG activity in a histidine 193-independent manner. This study provides insights into the mechanism of inhibition of UGCG activity by cambinol, and provides a basis for the development of a cambinol-based novel UGCG inhibitor.
Graphic abstract
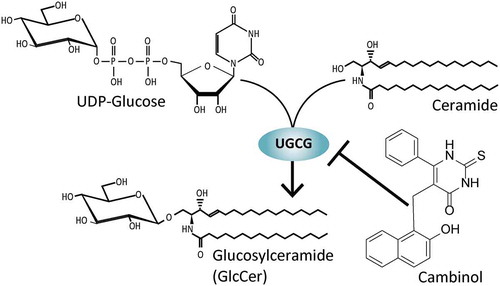
The sirtuin inhibitor cambinol inhibits the GlcCer synthase (UGCG) activity in an unconventional manner.
Glycosphingolipids (GSLs), which consist of oligosaccharides and ceramides, are ubiquitous in the plasma membrane and involved in a large number of cellular processes [Citation1–Citation3]. Most GSLs are generated from glucosylceramide (GlcCer) as a precursor. GlcCer is synthesized by GlcCer synthase (UDP-glucose:ceremide glucosyltransferase (UGCG), EC 2.4.1.80) from ceramide and UDP-glucose in the Golgi apparatus [Citation4]. The abnormal accumulation of GlcCer and GlcCer-based GSLs has been observed in several diseases, including Gaucher disease, Parkinson’s disease, and polycystic kidney disease [Citation5–Citation8]. As such, the characterization of the mechanism by which UGCG is regulated in vivo will improve our understanding of the pathology of these diseases and provide a basis for the development of UGCG inhibitors, paving the way for more effective treatments and medicines to treat these diseases.
We previously demonstrated that the cellular energy sensor, AMP-activated protein kinase (AMPK), and the Golgi-localized phosphoinositide, phosphatidylinositol 4 phosphate [PtdIns(4)P], regulate cellular GlcCer levels [Citation9,Citation10]. Although the precise mechanism is still unknown, we found that cellular UDP-glucose levels decreased under AMPK-activating conditions, leading to a reduction in GlcCer synthesis [Citation9]. PtdIns(4)P mildly attenuates UGCG activity by inhibiting the utilization of UDP-glucose by UGCG [Citation10]. In a previous study, we screened for several compounds and siRNAs that influenced the cellular GlcCer levels and found that resveratrol, a sirtuin-activating compound [Citation11], decreased the intracellular UGCG activity. This suggests that sirtuin, a NAD+-dependent deacetylase involved in the nutrient-responsive signaling pathways that is related to health and longevity [Citation12], may regulate the GlcCer metabolism. We investigated the relationship between UGCG and sirtuin by examining the effect of the sirtuin inhibitor, cambinol [5-(2-Hydroxynaphthalen-1-ylmethyl)-6-phenyl-2-thioxo-2,3-dihydro-1 H-pyrimidin-4-on] [Citation13] on the GlcCer metabolism and found that cambinol is a novel UGCG inhibitor with a mechanism that varies from conventional UGCG inhibitors.
Two oral UGCG inhibitors, miglustat and eliglustat, have been approved for use in decreasing the cellular levels of GlcCer in patients with Gaucher disease [Citation14]. Miglustat (Zavesca®, N-butyl-deoxynojirimycin, NB-DNJ) is an N-alkylated iminosugar, a synthetic analog of D-glucose that mimics the transition state of glucosyl synthase [Citation15]. N-(5ʹ-adamantane-1ʹ-yl-methoxy)-pentyl-1-deoxynojirimycin (AMP-DNM) is a more potent UGCG inhibitor than miglustat, although both possess similar structures [Citation16]. Eliglustat (Cerdelga®, Genz-112638) is a competitive UGCG inhibitor derived from a ceramide analog, D-PDMP (D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol) [Citation17], which is a well-recognized UGCG inhibitor that has been utilized to study the functions of GlcCer and GlcCer-derived GSLs [Citation18–Citation20]. As such, conventional UGCG inhibitors share a similar structure, comprising either glucose or ceramide, the components of GlcCer.
In this study, we report on the inhibition of UGCG by cambinol, which has no structural similarities with GlcCer, unlike conventional UGCG inhibitors. Our findings suggest that cambinol could become a leading compound in the development of a novel UGCG inhibitor.
Materials and methods
Materials
Cambinol, sirtinol, and D-PDMP were purchased from Abcam (Cambridge, UK). AMP-DMN and resveratrol were purchased from Cayman Chemical (Michigan, USA). Pre-coated Silica gel 60 TLC plates were purchased from Merck (Darmstadt, Germany). Fluorescent substrate 7-nitro-2,1,3-benzoxadiazole (NBD) C6-ceramide and NBD C6-ceramide complexed to BSA was purchased from Life Technologies (Carlsbad, USA). Antibodies against DYKDDDDK Tag (Rabbit IgG) and α-tubulin were purchased from Cell Signaling Technology (Massachusetts, USA) and Sigma Aldrich (Missouri, USA), respectively. Mouse embryonic fibroblast (MEF) of ugcg (+\+) and ugcg (‒/‒), which was established from ugcg (lox/lox) MEF by expressing Cre recombinase, were prepared as described in previous studies [Citation9,Citation21].
Cell culture and gene transfection
NIH3T3, HEK293, and MEF cells were cultured in DMEM supplemented 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37ºC in 5% CO2. HEK293 cells were transfected with TurboFect Transfection Reagent (Thermo Scientific) according to the manufacturer’s instructions.
Measurement of the UGCG activity
The intracellular UGCG assay was performed as previously described with some modifications [Citation22]. Cells were treated with 12.5, 25, 50, 100, or 200 μM cambinol for 1 h, or treated with 100 μM cambinol for 0.5, 1, 2, 4, or 8 h, then transferred to 250 μL DMEM 10% FBS containing 0.5 μM NBD C6-ceramide complexed to BSA; cambinol was dissolved in DMSO. After incubating for 1 h at 37ºC, the cells were rinsed with ice-cold PBS, scraped with 50 μL ice-cold water, and transferred to 1.5 mL tubes. Lipids were extracted using 190 μL chloroform/methanol (1/2, v/v). After incubation at room temperature for 10 min, 62.5 μL each of water and chloroform were added, then centrifuged at 12,000 × g for 5 min. The lower phase was collected and dried using a speed vac concentrator. The dried sample was dissolved in 20 μL of chloroform/methanol (1/2, v/v). Then, 3 μL of the sample was added to a TLC plate, which was developed with chloroform/methanol/water (65/25/4, v/v/v). The in vitro enzymatic activity was measured using a previously described method [Citation23]. The reaction products from the intracellular UGCG assay and in vitro assay were visualized using an LAS-3000 Luminescence Image Analyzer (Fujifilm) equipped with a blue-light-emitting diode (460 nm EPI) and an Y515-Di filter, and quantified in Image J 1.43 u software (NIH). The extent of the synthesis of NBD-labeled GlcCer was calculated using the following equation: GlcCer synthesis (%) = (peak area for NBD C6-GlcCer) × 100/(peak area for NBD C6-Cer + peak area for NBD C6-GlcCer + peak area for NBD C6-SM). The relative activity was recalculated by setting the control values as 100%.
Lipid extraction and quantification of sphingolipids by LC-ESI MS/MS
Cells were washed once with cold PBS, collected in 100 μL of cold PBS, and homogenized by sonication. Part of the sample (5 μL) was used in a bicinchoninic acid protein assay to determine the amount of protein. The total lipids were extracted by adding 375 μL of chloroform/methanol (1/2, v/v) containing 62.5 pmol of each component of Cer/Sph Mixture II (Avanti Polar Lipids, Inc.) as internal standard (IS) from the rest of the sample. The single-phase mixture was incubated at 48°C for 16 h. After cooling, 125 μL of water and chloroform were added. The lower phase was collected and dried using a speed vac concentrator. The dried sample was suspended in chloroform/methanol (2/1, v/v) containing 0.1 N KOH, incubated for 2 h at 37°C, and neutralized with an equal amount of acetic acid before adding water. The lower phase was withdrawn, dried, and resuspended in 200 μL of methanol before sonicating for 10 s, centrifuging at 14,000 × g for 5 min, and transferring the resulting supernatant to vials. The amount of GlcCer, LacCer, Cer, and SM were analyzed by LC-ESI MS/MS using a triple quadruple mass spectrometer 4000Q TRAP (SCIEX) coupled to an Agilent 1100 series HPLC system (Agilent). A binary solvent gradient with a flow rate of 0.2 mL/min was used to separate sphingolipids by normal-phase chromatography using an InertSustain® NH2 (2.1 × 150 mm, 5 μm bead size; GL Science). The gradient was started with 20% buffer B (methanol/water/formic acid, 89/9/1, v/v/v, with 20 mM ammonium formate) in buffer A (acetonitrile/methanol/formic acid, 97/2/1, v/v/v, with 5 mM ammonium formate) and reached 100% B in 4 min before maintaining at 100% B for 2 min. The gradient was then returned to the starting condition, and the column was equilibrated for 5 min before the next run. Sphingolipids containing C16:0, C18:0, C20:0, C22:0, C24:0, and C24:1 fatty acids were detected using a multiple reaction monitoring (MRM) method [Citation24]. Peak intensity of each sphingolipids were adjusted by using peak intensity of IS.
Evaluation of the inhibitory mechanism and IC50 of cambinol toward UGCG
The kinetic constants of UGCG were measured by using various concentrations of UDP-glucose (from 0.25 to 16 mM) and NBD-C6-Cer (from 1 to 64 μM). The fluorescent intensities of synthesized NBD C6-GlcCer by UGCG with or without cambinol were fitted to competitive or noncompetitive models using GraphPad Prism 6 (GraphPad Software Inc.). Various concentrations of cambinol, AMP-DNM, and D-PDMP were added to the reaction mixture of in vitro UGCG assay to evaluate their IC50, calculated using GraphPad Prism 6.
Construction of expression vectors
The H193A mutant of UGCG was generated by Phusion polymerase (Thermo Scientific) using the mutation primers GlcT H193A-S (ggaacttcagctccaagatac) and GlcT H193A-A (gtatcttggagctgaagttcc) and a C-terminal FLAG-tagged UGCG expression vector, prepared as described previously [Citation9], as a template. The amplified product was transformed into a plasmid vector using an In-Fusion HD Cloning kit (Clontech).
Immunoblot analysis
The transfected cells were lysed in ice-cold lysis buffer (20 mM Tris HCl buffer, pH 7.5) supplemented with 150 mM NaCl, 1 mM EDTA, 1% NP-40, and phosphatase and protease inhibitor cocktails (Roche Diagnostics), by sonication for 45 s. The cell debris was removed by centrifugation (18,000 × g for 10 min at 4°C). The amount of protein in the supernatant was determined using a bicinchoninic acid protein assay. Equal amounts of cellular protein were separated by SDS-PAGE using a Mini-PROTEAN TGX Gel (BioRad), followed by blotting on a PVDF membrane using Trans Blot Turbo (BioRad). The membrane was blocked in TBS-T supplemented with 5% BSA before incubating with antibodies against DYKDDDDK tag and α-tubulin (1:20000) for over 12 h at 4°C. The blot was washed with TBS-T and incubated with HRP-conjugated anti-mouse or rabbit IgG antibody (1:10000) (Cell Signaling Technology). The membrane was washed again with TBS-T. The protein levels were detected by chemiluminescence using Luminate Forte Western HRP substrate (Millipore) and LAS-3000.
Statistical analysis
All statistical analyses were performed using unpaired two-tailed Student’s t-tests. Data are expressed as the mean and standard deviation (SD) of at least three separate experiments. Statistical significance is indicated as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001, n.s., not significant.
Result and discussion
Effect of cambinol on intracellular UGCG activity
Several compounds with the potential to affect the intracellular GlcCer levels were screened to find novel regulatory factors of GlcCer synthesis in a cell-based GlcCer synthase assay [Citation22]. This was accomplished using cell-permeable fluorescent ceramide, which is transferred to the Golgi apparatus and can be incorporated into GlcCer and sphingomyelin. We found that resveratrol, a sirtuin-activating compound [Citation11], effectively reduced the levels of GlcCer synthase activity in NIH3T3 cells (). To determine whether the effect of resveratrol on GlcCer synthesis is caused by the activation of sirtuin, the sirtuin inhibitor, cambinol [Citation13], was added to the cell culture with resveratrol. We expected cambinol to attenuate the inhibitory effect of resveratrol on GlcCer synthesis. However, the intracellular GlcCer synthase activity was synergistically decreased by the addition of cambinol to cells treated with resveratrol (). These results suggested that the reduction in the synthesis of GlcCer caused by resveratrol occurs independently of the activation of sirtuin, indicating that cambinol itself inhibited GlcCer synthesis. In a previous study, we showed that GlcCer synthesis decreased under AMPK-activating conditions [Citation9]. Resveratrol is also capable of activating AMPK [Citation25]; thus, the reduction in GlcCer synthesis by resveratrol may be caused by AMPK activation. To verify the inhibitory effect of cambinol on cellular GlcCer synthesis, a cell-based UGCG assay using cell-permeable fluorescent ceramide was performed using HEK293 cells with or without cambinol treatment. TLC analysis showed that GlcCer synthesis in HEK293 cells was markedly suppressed by cambinol (). We found that intracellular UGCG activity decreased with cambinol in a concentration- and time-dependent manner (). On the other hand, sphingomyelin synthase (SMS) activity was not inhibited by cambinol (). These results suggest that cambinol may act as a UGCG inhibitor.
Figure 1. Effect of cambinol on the cellular UGCG activity.

Effect of cambinol on cellular sphingolipid levels
In agreement with the inhibition of UGCG activity, the levels of GlcCer in the HEK293 cells were dose-dependently decreased after treatment with cambinol (). The levels of ceramide, a precursor of GlcCer, increased in 100 μM cambinol treatment (), suggesting that ceramide accumulated by inhibiting GlcCer synthesis with cambinol. Unlike GlcCer levels, 25 or 50 μM cambinol treatment did not affect ceramide levels of HEK293 cells (). Because ceramide is the important cellular signal messenger, the levels of ceramide could be regulated by ceramide metabolizing enzymes such as ceramidase [Citation26]. It is assumed that the regulation mechanism of ceramide levels might suppress the accumulation of ceramide in the lower concentration of cambinol. The ceramide homeostasis might be disrupted by an excess of the cambinol (100 μM), leading the ceramide accumulation. On the other hand, the lactosylceramide (LacCer) and sphingomyelin levels were unchanged by cambinol treatment (), suggesting that cambinol is unable to inhibit the LacCer and sphingomyelin synthases.
Figure 2. Measurement of sphingolipid levels of cambinol-treated cells.

Inhibitory effect of cambinol on in vitro UGCG activity
To determine whether cambinol inhibits UGCG activity directly or indirectly, the effect of cambinol on the in vitro UGCG activity was investigated using the cell lysate of HEK293 as an enzyme source. D-PDMP and AMP-DNM, well-known inhibitors of UGCG, strongly inhibited the in vitro UGCG activity (). Although cambinol also inhibited in vitro UGCG activity, its inhibitory activity was much lower than that of conventional UGCG inhibitors (). The IC50 of cambinol, D-PDMP, and AMP-DNM were calculated as 86.7, 0.7, and 0.5 μM, respectively. In contrast, SMS activity was hardly inhibited by cambinol (). Next, the mechanism by which cambinol inhibits UGCG was examined using the nonlinear curve fitting of enzyme kinetics using UDP-glucose () and NBD-ceramide () as substrates. The values of NBD-GlcCer synthesis by UGCG in the presence of cambinol were well-fitted to the curve of the noncompetitive model for both substrates (). This indicates that cambinol is a noncompetitive inhibitor of UGCG, unlike conventional UGCG inhibitors, such as miglustat and eliglustat, which competitively inhibit UGCG activity [Citation15].
Figure 3. Inhibitory efficiency and mechanism of cambinol on UGCG activity.

Next, the structure-activity relationship was analyzed using a different sirtuin inhibitor, sirtinol [Citation27], which shares a 2-hydroxy-naphthaldehyde with cambinol (). In contrast to cambinol, sirtinol did not inhibit the UGCG activity (), indicating that the 2-hydroxy-naphthaldehyde moiety, which is necessary for the inhibition of sirtuin activity [Citation27], is not integral for the inhibition of UGCG activity. Although our results indicated that cambinol inhibits UGCG activity, the detailed mechanism or binding sites of cambinol remains unsolved at present. The tertiary structure of UGCG, which has yet been determined in spite of several trials [Citation28], will help to understand the inhibitory mechanism of cambinol on UGCG.
Significance of histidine 193 in UGCG for cambinol inhibitory activity
The histidine 193 residue (H193) of UGCG is integral for the activity of its inhibitor, D-PDMP [Citation29]. Thus, we investigated whether H193 also plays an important role in the inhibition of UGCG activity by cambinol. FLAG-tagged UGCG WT and H193A mutant, in which the histidine was replaced with an alanine, were expressed in HEK293 cells. Immunoblot analysis showed that the protein expression level of UGCG WT was almost the same as that of UGCG H193A (). In agreement with a previous report [Citation29], the UGCG H193A mutant became insensitive to D-PDMP when the cell lysate of HEK293 cells was used as an enzyme (). On the other hand, cambinol and AMP-DNM inhibited the UGCG activities of both the WT and H193A mutant (), indicating that the inhibitory mechanism of cambinol and AMP-DNM clearly varies from that of D-PDMP.
Figure 4. Dependency of histidine 193 in UGCG for the inhibition by cambinol.

Ceramide accumulation of cambinol-treated cells
Despite both cambinol and AMP-DNM decreasing the cellular GlcCer levels (), we found that their effects on the levels of ceramide were different. Ceramide was found to accumulate in cambinol-treated cells accompanied by a decrease in the GlcCer content, whereas AMP-DNM treatment did not change the amount of ceramide (), in agreement with the previous report [Citation30]. To confirm whether ceramide accumulation was caused by the inhibition of UGCG activity, the GlcCer and ceramide levels of ugcg (-/-) MEF were examined after treatment with cambinol. We confirmed that ceramide accumulated with as GlcCer decreased in ugcg (+/+) MEF after cambinol treatment (). On the other hand, cambinol did not increase the ceramide levels in ugcg (‒/‒), which is unable to synthesize GlcCer (). A previous study reported that cambinol also inhibits the activity of neutral sphingomyelinase (nSMase) 2 [Citation31], which hydrolyzes sphingomyelin to generate phosphocholine and ceramide under neutral pH conditions, suggesting the possibility that the cellular ceramide level would be decreased, not increased, after cambinol treatment due to the inhibition of nSMase 2 activity. However, transcriptome analyses of mammalian cells revealed that the mRNA expression level of nSMase 2 (Smpd 3) was extremely lower than that of UGCG (Ugcg) in MEF (Smpd3: 2 TPM, Ugcg: 187 TPM) [Citation32], and HEK293 cells (Smpd3: 0.7 TPM, Ugcg: 9.7 TPM) [Citation33], suggesting that the nSMSase 2 hardly affected the ceramide and sphingomyelin levels of the cells used in this study regardless of the presence or absence of cambinol. Thus, we consider that ceramide accumulation by cambinol depends on the inhibition of UGCG.
Figure 5. Involvement of UGCG in ceramide accumulation caused by cambinol.

In this study, we demonstrated that cambinol, a sirtuin inhibitor, inhibits UGCG activity, leading to a decrease in the cellular GlcCer levels and an increase in cellular ceramide. UGCG inhibitors are widely used for the treatment of Gaucher disease. We found that the cellular GlcCer levels were decreased after treatment with cambinol, suggesting that cambinol could be useful in the development of new drugs for the treatment of diseases caused by GlcCer accumulation. Two drugs that have been approved for the treatment of Gaucher disease, namely miglustat and eliglustat, show low brain penetration. Thus, these drugs are not effective for the alleviation of the symptoms of type 3 Gaucher disease related to the central nervous system [Citation34,Citation35] nor the symptoms of Parkinson’s disease, which is genetically correlated with Gaucher disease [Citation36]. Thus, a UGCG inhibitor that is able to pass through the blood-brain barrier (BBB) is required for treatment. As reported by Bilousova et al., orally administrated cambinol is able to reach the brain [Citation37], demonstrating the potential of cambinol or structurally-related compounds as BBB-permeable UGCG inhibitors. However, cambinol has some disadvantages as a UGCG inhibitor at present, such as the IC50 of cambinol for UGCG being much higher than that of D-PDMP or AMP-DNM, and the fact that cambinol inhibits not only UGCG but also sirtuin [Citation13] and nSMase2 [Citation31]. Thus, we propose for cambinol to be chemically modified to improve its inhibitory activity and remove any off-target effects.
Taken together, our results demonstrated that cambinol is a novel UGCG inhibitor despite having no structural similarities to GlcCer, unlike conventional UGCG inhibitors. As such, cambinol has the potential to become a leading compound in the development of new UGCG inhibitors with inhibitory mechanisms and pharmacokinetics that are different from those of conventional inhibitors.
Author contributions
Conceptualization: Y.I. and Y.H., Conducted the experiments: Y.I., Data analysis: Y.I., Supervision: M.I., Writing: Y.I. and M.I. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to the Support Unit for Bio-material Analysis, RIKEN Center for Brain Science for their assistance in the nucleotide sequencing analyses. This work was supported in part by the RIKEN Special Postdoctoral Researchers Program (to Y. I.), and a Grant-in-Aid for Young Scientists (B) (to Y. I.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Aerts JMFG, Artola M, van Eijk M, et al. Glycosphingolipids and infection. Potential new therapeutic avenues. Front Cell Dev Biol. 2019;7:324.
- Lahiri S, Futerman AH. The metabolism and function of sphingolipids and glycosphingolipids. Cell Mol Life Sci. 2007;64:2270–2284.
- Jennemann R, Gröne HJ. Cell-specific in vivo functions of glycosphingolipids: lessons from genetic deletions of enzymes involved in glycosphingolipid synthesis. Prog Lipid Res. 2013;52:231–248.
- Ishibashi Y, Kohyama-Koganeya A, Hirabayashi Y. New insights on glucosylated lipids: metabolism and functions. Biochim Biophys Acta. 2013;1831:1475–1485.
- Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet. 2008;372:1263–1271.
- Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11:986–998.
- Natoli TA, Modur V, Ibraghimov-Beskrovnaya O. Glycosphingolipid metabolism and polycystic kidney disease. Cell Signal. 2020;69:109526.
- Huebecker M, Moloney EB, Van Der Spoel AC, et al. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol Neurodegener. 2019;14:40.
- Ishibashi Y, Hirabayashi Y. AMP-activated protein kinase suppresses biosynthesis of glucosylceramide by reducing intracellular sugar nucleotides. J Biol Chem. 2015;290:18245–18260.
- Ishibashi Y, Ito M, Hirabayashi Y. Regulation of glucosylceramide synthesis by Golgi-localized phosphoinositide. Biochem Biophys Res Commun. 2018;499:1011–1018.
- Dai H, Sinclair DA, Ellis JL, et al. Sirtuin activators and inhibitors: promises, achievements, and challenges. Pharmacol Ther. 2018;188:140–154.
- Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471.
- Heltweg B, Gatbonton T, Schuler AD, et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006;66:4368–4377.
- Shayman JA, Larsen SD. The development and use of small molecule inhibitors of glycosphingolipid metabolism for lysosomal storage diseases. J Lipid Res. 2014;55:1215–1225.
- Loewith R, Riezman H, Winssinger N. Sphingolipids and membrane targets for therapeutics. Curr Opin Chem Biol. 2019;50:19–28.
- Wennekes T, Van Den Berg RJBHN, Donker W, et al. Development of adamantan-1-yl-methoxy-functionalized 1-deoxynojirimycin derivatives as selective inhibitors of glucosylceramide metabolism in man. J Org Chem. 2007;72:1088–1097.
- Shayman JA. Developing novel chemical entities for the treatment of lysosomal storage disorders: an academic perspective. Am J Physiol - Ren Physiol. 2015;309:F996–F999.
- Shayman JA, Lee L, Abe A, et al. Inhibitors of glucosylceramide synthase. Methods Enzymol. 2000;311:373–387.
- Inokuchi J, Radin NS. Preparation of the active isomer of 1-phenyl-2-decanoylamino-3-morpholino-1-propanol, inhibitor of murine glucocerebroside synthetase. J Lipid Res. 1987;28:565–571.
- Inokuchi J, Ode T, Hara-Yokoyama M. Pharmacological modulation of glycosphingolipid metabolism. Methods Mol Biol. 2018;1804:401–410.
- Watanabe S, Endo S, Oshima E, et al. Glycosphingolipid synthesis in cerebellar purkinje neurons: roles in myelin formation and axonal homeostasis. Glia. 2010;58:1197–1207.
- Gupta V, Patwardhan GA, Zhang QJ, et al. Direct quantitative determination of ceramide glycosylation in vivo: A new approach to evaluate cellular enzyme activity of glucosylceramide synthase. J Lipid Res. 2010;51:866–874.
- Hayashi Y, Horibata Y, Sakaguchi K, et al. A sensitive and reproducible assay to measure the activity of glucosylceramide synthase and lactosylceramide synthase using HPLC and fluorescent substrates. Anal Biochem. 2005;345:181–186.
- Shaner RL, Allegood JC, Park H, et al. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J Lipid Res. 2009;50:1692–1707.
- Price NL, Gomes AP, Ling AJY, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–690.
- Morad SAF, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer. 2013;13:51–65.
- Grozinger CM, Chao ED, Blackwell HE, et al. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem. 2001;276:38837–38843.
- Shinoda T, Shinya N, Ito K, et al. Cell-free methods to produce structurally intact mammalian membrane proteins. Sci Rep. 2016;6:1–15.
- Wu K, Marks DL, Watanabe R, et al. Histidine-193 of rat glucosylceramide synthase resides in a UDP-glucose- and inhibitor (D-threo-1-phenyl-2-decanoylamino-3-morpholinopropan-1-ol)-binding region: a biochemical and mutational study. Biochem J. 1999;341:395.
- Casson L, Howell L, Mathews LA, et al. Inhibition of ceramide metabolism sensitizes human leukemia cells to inhibition of BCL2-like proteins. Starczynowski DT, editor. PLoS One. 2013;8:e54525.
- Figuera-Losada M, Stathis M, Dorskind JM, et al. Cambinol, a novel inhibitor of neutral sphingomyelinase 2 shows neuroprotective properties. PLoS One. 2015;10:e0124481.
- Lienert F, Mohn F, Tiwari VK, et al. Genomic prevalence of heterochromatic H3K9me2 and transcription do not discriminate pluripotent from terminally differentiated cells. Reik W, editor. PLoS Genet. 2011;7:e1002090.
- Thul PJ, Akesson L, Wiking M, et al. A subcellular map of the human proteome. Science. 2017;356:eaal3321.
- Schiffmann R, Fitzgibbon EJ, Harris C, et al. Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol. 2008;64:514–522.
- Shayman JA. ELIGLUSTAT TARTRATE: glucosylceramide synthase inhibitor treatment of type 1 Gaucher disease. Drugs Future. 2010;35:613–620.
- Sardi SP, Clarke J, Kinnecom C, et al. CNS expression of glucocerebrosidase corrects α-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci U S A. 2011;108:12101–12106.
- Bilousova T, Elias C, Miyoshi E, et al. Suppression of tau propagation using an inhibitor that targets the DK-switch of nSMase2. Biochem Biophys Res Commun. 2018;499:751–757.