
ABSTRACT
Maltol derivatives are used in a variety of fields due to their metal-chelating abilities. In the previous study, it was found that cytochrome P450 monooxygenase, P450nov, which has the ability to effectively convert the 2-methyl group in a maltol derivative, transformed 3-benzyloxy-2-methyl-4-pyrone (BMAL) to 2-(hydroxymethyl)-3-(phenylmethoxy)-4H-pyran-4-one (BMAL-OH) and slightly to 3-benzyloxy-4-oxo-4 H-pyran-2-carboxaldehyde (BMAL-CHO). We isolated Pseudomonas nitroreducens SB32154 with the ability to convert BMAL-CHO to BMAL-COOH from soil. The enzyme responsible for aldehyde oxidation, a BMAL-CHO dehydrogenase, was purified from P. nitroreducens SB32154 and characterized. The purified BMAL-CHO dehydrogenase was found to be a xanthine oxidase family enzyme with unique structure of heterodimer composed of 75 and 15 kDa subunits containing a molybdenum cofactor and [Fe-S] clusters, respectively. The enzyme showed broad substrate specificity toward benzaldehyde derivatives. Furthermore, one-pot conversion of BMAL to BMAL-COOH via BMAL-CHO by the combination of the BMAL-CHO dehydrogenase with P450nov was achieved.
GRAPHICAL ABSTRACT
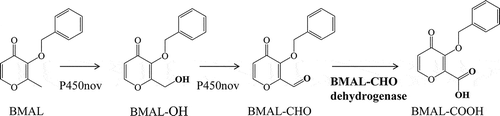
Enzymatic conversion of BMAL to BMAL-COOH by P450nov from Novosphingobium sp. SB32149 and BMAL-CHO dehydrogenase from Pseudomonas nitroreducens SB32154
Maltol (3-hydroxy-2-methyl-4-pyrone) derivatives are used in the food, beverage, cosmetic, and pharmaceutical industries due to their metal-chelating abilities; in particular, the hydroxypyrone moiety of maltol is known to have potent transition metal-chelating capability [Citation1]. Maltol has also been used as a ligand for vanadyl ions in insulin-enhancing agents for the treatment of diabetes mellitus, and a bis-maltol-vanadium complex showed improved pharmacokinetic parameters when compared to vanadium alone [Citation2]. Maltol derivatives have tyrosinase inhibitory activity due to their ability to chelate copper at the active site of the enzyme. Tyrosinase is involved in melanogenesis, enzymatic browning of fruits and vegetables, parasite encapsulation, and sclerotization in insects; hence, maltol derivatives are used as skin whiteners, food preservatives, and insecticides [Citation3,Citation4]. The inhibitor activity for metalloproteinase is also found in maltol derivatives and enhanced by extending their 2-methyl side chain, which non-covalently interacts with specific subsites neighboring the active site of the enzyme [Citation5,Citation6]. We previously reported the modification of this 2-methyl side chain using cytochrome P450 monooxygenase [Citation7]. Finally, in a jar fermenter reaction under controlled culture conditions, 5.2 g/L 2-(hydroxymethyl)-3-(phenylmethoxy)-4 H-pyran-4-one (BMAL-OH) was produced from 3-benzyloxy-2-methyl-4-pyrone (BMAL) as a substrate with P450nov mutant.
The present study focused on the oxidation of BMAL-OH into 3-benzyloxy-4-oxo-4 H-pyran-2-carboxylic acid (BMAL-COOH) via 3-benzyloxy-4-oxo-4 H-pyran-2-carboxaldehyde (BMAL-CHO). The 2-carboxylic acid group in the final reaction product could be a chemical modifying point for expanding the functional diversity of maltol derivatives. As P450nov showed BMAL-OH-oxidizing activity to BMAL-CHO, BMAL-CHO-oxidizing activity was targeted for the analysis (). We isolated Pseudomonas nitroreducens SB32154 with the ability to convert BMAL-CHO to BMAL-COOH from soil.
Figure 1. Scheme of BMAL-COOH production from BMAL. P450nov from Novosphingobium sp. SB32149 catalyzed the hydroxylation of BMAL and the oxidation of BMAL-OH. BMAL-CHO dehydrogenase from Pseudomonas nitroreducens SB32154 catalyzed the dehydrogenation of BMAL-CHO into BMAL-COOH.

Aldehyde oxidase is a candidate enzyme responsible for BMAL-CHO conversion to BMAL-COOH. Aldehyde oxidase is a molybdenum-containing enzyme and belongs to xanthine oxidase family. Aldehyde oxidases catalyze various aldehydes oxidation. For example, membrane-bound aldehyde dehydrogenase from Acetobacter pasteurianus SKU1108 oxidize acetaldehyde to acetic acid [Citation8]. Aldehyde oxidase from Methylobacillus sp. KY4400 oxidizes various aldehydes [Citation9,Citation10]. Aldehyde oxidoreductase from Escherichia coli oxidizes aromatic aldehydes [Citation11]. Aldehyde oxidase from Burkholderia sp. AIU 129 oxidizes glycolaldehyde to glycolate [Citation12]. Aldehyde oxidoreductase from Desulfovibrio desulfuricans ATCC 27774 oxidizes acetaldehyde, propioaldehyde, butanaldehyde, and benzaldehyde [Citation13]. Addition to aldehyde oxidases, xanthine oxidase family contains various oxidoreductase such as xanthine dehydrogenase from E. coli and Rhodobacter capsulatus [Citation14,Citation15], nicotinate dehydrogenase from Eubacterium barkeri [Citation16], CO dehydrogenase from Hydrogenophaga pseudoflava and Oligotropha carboxidovorans OM5 [Citation17,Citation18], caffeine dehydrogenase from Pseudomonas sp. CBB1 [Citation19], and quinoline 2-oxidoreductase from Pseudomonas putida [Citation20], 4-hydroxybenzoyl-CoA reductase from Thauera aromatica [Citation21].
In this study, molybdenum-containing aldehyde dehydrogenase was purified, characterized, and identified as the enzyme responsible for BMAL-CHO oxidation in P. nitroreducens SB32154. The enzyme was applied to the synthesis of BMAL-COOH from BMAL in the coupled reaction with P450nov catalyzing BMAL conversion to BMAL-CHO.
Materials and methods
Materials
BMAL-OH, BMAL-CHO, and BMAL-COOH were synthesized as described previously [Citation22,Citation23]. Soil samples as a source of microorganisms were collected from Kinki region of Japan.
Enrichment culture for BMAL-OH-assimilating microbial consortia
Approximately 1 mg of each soil sample was suspended in 1 mL M9 minimal medium in 96-well assay blocks (1 mL; Corning, New York, NY, USA). The suspensions were sonicated and centrifuged, and then the supernatants were added to 0.5 mL enrichment medium (pH 7.0) containing 11.28 g/L M9 minimal salts (5× powder), 0.49 g/L MgSO4·7H2O, 14.7 mg/L CaCl2·2H2O, 1 mL/L metal mixture (0.1 g/L H3BO3, 0.5 g/L FeSO4·7H2O, 0.05 g/L KI, 2 g/L CoCl2·2H2O, 0.2 g/L CuSO4·5H2O, 2 g/L MnCl2·4 H2O, 4 g/L ZnSO4·7H2O, 1 g/L H2SO4), and 500 mg/L BMAL-OH as a sole carbon source in 96-well assay blocks (1 mL). Enrichment cultivation was carried out in a 96-well plate shaker at 30°C, and 10% of the volume of the culture was transferred to fresh enrichment medium every 2 days. After passaging twice, the resultant culture was centrifuged, and the supernatant was mixed with an equal volume of ethanol. BMAL-OH metabolites in the solution were analyzed by high-performance liquid chromatography (HPLC). The BMAL-COOH-producing microorganisms were stored in nutrient agar broth (Becton Dickinson, Franklin Lakes, NJ, USA).
Isolation of BMAL-OH- and BMAL-CHO-oxidizing microorganism from BMAL-OH-assimilating microbial consortia
BMAL-OH-assimilating microbial consortia were streaked on BMAL-OH medium plates and isolated. The isolates were inoculated into BMAL-OH or BMAL-CHO medium containing 11.28 g/L M9 minimal salts (5× powder), 2 g/L succinate, 0.25 g/L yeast extract, 0.49 g/L MgSO4·7H2O, 14.7 mg/L CaCl2·2H2O, 1 mL/L metal mixture as described above, and 200 mg/L BMAL-OH or BMAL-CHO and were cultivated in a 96-well plate shaker at 30°C. After cultivation for 7 days, the culture was centrifuged, and the supernatant was mixed with an equal volume of ethanol. BMAL-COOH in the solution was analyzed by HPLC. Microbial identification was carried out by 16S rDNA sequence analysis using a MicroSeq Full Gene 16S rDNA Bacterial Identification kit (Applied Biosystems, Foster City, CA, USA).
Assay of BMAL-CHO dehydrogenase
BMAL-CHO dehydrogenase activity was assayed by the method of Mohapatra et al. [Citation24] with some modifications. The standard assay mixture contained 0.24 mg/mL BMAL-CHO, 0.29 mM 2,6-dichlorophenolindophenol (DCIP), and 0.48 mM phenazine methosulfate (PMS) in 20 mM potassium phosphate buffer (pH 6.0). The reaction was started by the addition of enzyme solution (1.9 µg/mL of purified enzyme af final concentration) and carried out at 30°C for 30 min. The rate of decrease in the absorbance at 600 nm was followed against a blank in which 20 mM Tris-HCl buffer (pH 8.0) was added in place of enzyme. One unit of BMAL-CHO dehydrogenase activity was defined as 1 μmol DCIP reduced per minute per liter of enzyme solution. Protein concentration was quantified using Pierce Coomassie Protein Assay kit (Thermo Fisher Scientific, Waltham, MA, USA) with bovine serum albumin standard.
Purification of a BMAL-CHO-oxidizing enzyme from Pseudomonas nitroreducens SB32154
For the purification of a BMAL-CHO-oxidizing enzyme, P. nitroreducens SB32154 was cultivated in 1 L LB medium containing 10 mL ethanol. Cultivation was carried out at 30°C for 24 h with agitation at 80 rpm on a rotary shaker. Bacterial cells were harvested by centrifugation at 5,500 × g for 15 min at 4°C. The resulting cell pellets were resuspended in 20 mM Tris-HCl buffer (pH 8.0) containing 2 mM DTT and disrupted with a probe sonicator (Branson Sonifier 450; Emerson, St. Louis, MO, USA). The parameter was set at output of 6 at 20% duty cycle in 10 s pulse mode for 30 min. The cell debris was removed by centrifugation at 8,000 × g for 30 min at 4°C. The 0.22 µm filtrated cell-free supernatant was designated as crude BMAL-CHO dehydrogenase. All purification procedures were performed at 4°C using an ÄKTA FPLC system (GE Healthcare, Little Chalfont, UK).
The crude BMAL-CHO dehydrogenase was applied to a HiPrep DEAE 16/10 FF column (1.6 × 10 cm) (GE Healthcare) previously equilibrated with 20 mM Tris-HCl buffer (pH 8.0), which was used as a standard buffer for the following purification steps. After the column had been washed with 40 mL of this buffer, the enzyme was eluted with a linear gradient of NaCl (0–0.4 M) in 100 mL of 20 mM Tirs-HCl buffer (pH 8.0). The active fractions were combined and dialyzed against 5 L of 20 mM Tris-HCl buffer (pH 8.0) for 24 h using Slide-A-Lyze 10 kDa molecular weight cutoff cassettes (Thermo Fisher Scientific). The enzyme solution was applied to Mono Q 5/50 GL column (0.5 × 5 cm) (GE Healthcare) previously equilibrated with 20 mM Tris-HCl buffer (pH 8.0). After the column had been washed with 2 mL of 20 mM Tris-HCl buffer (pH 8.0), the enzyme was eluted with a linear gradient of NaCl (0–0.3 M) in 20 mL of 20 mM Tris-HCl buffer (pH 8.0). The active fractions were combined, added with the same volume of 1.6 M (NH4)2SO4, and then applied to HiTrap Butyl FF 5 mL column (1.6 × 2.5 cm) (GE Healthcare) previously equilibrated with 20 mM Tris-HCl buffer (pH 8.0) containing 0.8 M (NH4)2SO4. The enzyme was eluted with a linear gradient of (NH4)2SO4 (0.8–0 M) in 100 mL of 20 mM Tris-HCl buffer (pH 8.0). The active fractions were combined and concentrated, and the buffer was exchanged to 20 mM Tris-HCl buffer (pH 8.0) with 50 mM NaCl by ultrafiltration (Amicon Ultra; Merck Millipore, Burlington, MA, USA). The enzyme solution was applied to a Superdex 200 10/300 GL (1.0 × 30 cm) (GE Healthcare) previously equilibrated with 20 mM Tris-HCl buffer (pH 8.0) containing 50 mM NaCl and eluted with the same buffer. The active fractions were used for characterization. The purity of the proteins in the active fractions was checked by SDS-PAGE using a 12.5% polyacrylamide gel stained with Coomassie Brilliant Blue R-250. The protein bands were cut from the gel and transferred to a PVDF membrane. The N-terminal amino acid sequences of the proteins were determined by automated Edman degradation with a PSQQ-30 Protein Sequencer (Shimadzu).
Characterization of BMAL-CHO dehydrogenase
The effect of different electron acceptors was evaluated with the purified enzyme from P. nitroreducens SB32154 in the standard reaction mixture with each electron acceptor instead of DCIP and PMS. The reaction was carried out at 30°C for 30 min. The enzyme activity was evaluated by the production of BMAL-COOH using liquid chromatography-mass spectrometry (LC-MS).
To determine the substrate specificity, the assay conditions were the same as mentioned above, except that different aldehydes were used as the substrate instead of BMAL-CHO. Separate blanks with individual substrates of aldehyde were also prepared. The kinetic parameters, such as Michaelis-Menten constant (Km) and maximum reaction velocity (Vmax) values against aldehyde substrates, were calculated using XLfit (IDBS, Guilford, UK).
Cloning and expression of recombinant BMAL-CHO dehydrogenase in Escherichia coli
Whole-genome sequencing (WGS) of P. nitroreducens SB32154 was conducted by Genaris, Inc. (Yokohama, Japan). Primers were designed to amplify the BMAL-CHO dehydrogenase sequence from P. nitroreducens SB32154 genomic DNA (i.e. Fw: 5ʹ-TCACATATGATTACCGTGAACCTGAACGGCAAGGAC-3ʹ and Rev: 5ʹ-ATACTCGAGTCAGGCCTGCAACTGATTGCCGATC-3ʹ). The polymerase chain reaction-amplified product was ligated into expression vector pET17b (Novagen, Madison, WI, USA) using NdeI and XhoI restriction enzyme sites. The resulting plasmid was purified and then used to transform E. coli BL21 STAR (DE3) (Thermo Fisher Scientific). The transformed cells were cultured in Luria-Bertani (LB) medium at 37°C for 2 h with shaking at 150 rpm, and then isopropyl-β-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM. After adding IPTG, the transformed cells were cultivated at 25°C for 24 h with shaking at 150 rpm. The profile of protein expression was checked on denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The activity of BMAL-CHO conversion was checked with disrupted recombinant E. coli cells by the method indicated above and BMAL-COOH production was measured by LC-MS.
LC-MS analysis
The analysis of BMAL derivatives was performed by ultra high performance liquid chromatography (UPLC) (Agilent 1290 Infinity; Agilent, CA, USA) on a ACQUITY UPLC HSS C18 column (2.1 mm × 50 mm) (Waters, MA, USA) with a linear gradient of acetonitrile (10–40%) containing 0.1% formic acid (FA) at a flow rate of 0.8 mL/min and detected at 280 nm. LC-MS data were recorded on an Agilent 1100 Series LC/MSD trap using ESI mode (Agilent). Drying gas temperature, 300°C; capillary voltage, 3.0 kV; Drying gas flow, 12 L/min, Drying gas pressure, 60 psig; Quad temperature, 100°C. Products were identified by comparison of its retention time and molecular mass with standards of BMAL-OH and BMAL-COOH.
One-pot reaction from BMAL to BMAL-COOH
The genes of P450nov and redox partner proteins were expressed in E. coli as described previously [Citation7]. To prepare the cells expressing P450nov, 1 mL of expressed recombinant E. coli was centrifuged and suspended in 500 µL phosphate-buffered saline. Reaction mixture contained 90% (v/v) the recombinant E. coli suspension, 1.5 µg/mL purified BMAL-CHO dehydrogenase from P. nitroreducens SB32154, and 0.25 mg/mL BMAL. The reaction was carried out at 28°C for 20 h with agitation at 1,400 rpm and BMAL-COOH production was measured by LC-MS.
Results
Screening of microorganisms producing BMAL-COOH from BMAL-OH
From a total of 665 soil samples, BMAL-COOH-producing microorganisms were screened using the enrichment medium with BMAL-OH as a sole carbon source. Out of these, three BMAL-OH-assimilating microbial consortia converted BMAL-OH to BMAL-COOH. From the three microbial consortia, three colonies producing BMAL-COOH were isolated. The three isolates showed not BMAL-OH-oxidizing but BMAL-CHO-oxidizing activity. According to 16S rDNA sequence analysis, these BMAL-CHO-oxidizing strains were identified as P. nitroreducens. The strain, SB32154, with the highest activity produced 85 mg/L BMAL-COOH from 100 mg/L BMAL-CHO after 24 h and was chosen for further investigation. We deposited the strain P. nitroreducens SB32154 to NITE Biological Resource Center (NBRC) as NBRC114008.
Identification of a BMAL-CHO-oxidizing enzyme of P. nitroreducens SB32154
The course of purification is summarized in . After a four-step purification procedure, the enzyme purity was increased approximately 139-fold (). The purified enzyme showed two major bands on SDS-PAGE ()). The molecular masses of these proteins were about 75,000 and 15,000. Amino acid sequence analysis of these proteins revealed the N-terminal amino acid sequence of the upper protein band of DFEPNAFVXRIAPDGTVT and that of lower protein band of MITVNLNGKDHELDAPGEM. These sequences matched that of a putative xanthine oxidase family protein molybdopterin-binding subunit from P. nitroreducens and that of a putative (2F-2S)-binding protein from Pseudomonas sp., respectively. To identify the nucleotide sequences of these proteins, the genome sequence of P. nitroreducens SB32154 was determined and the full-length amino acid sequences of the two subunits of BMAL-CHO-oxidizing enzyme were identified (GenBank accession numbers LC480925 and LC480926) ()). Those sequences were 99% identical to those of xanthine oxidase family proteins from P. nitroreducens (WP_088417191 and WP_026078846).
Table 1. Summary of the purification of BMAL-CHO dehydrogenase from P. nitroreducens SB32154.
Characterization of a BMAL-CHO-oxidizing enzyme of P. nitroreducens SB32154
The purified enzyme from P. nitroreducens SB32154 was used for its characterization. The purity is shown in lane5 of ). PMS served as an electron acceptor effectively (). Methylene blue, DCIP, and ferricyanide also served as an electron acceptor, but they were less effectively than PMS. Various aldehydes were evaluated as the substrates of purified BMAL-CHO dehydrogenase of P. nitroreducens SB32154 by measuring the reduction of DCIP (). The enzyme showed broad substrate specificity to benzaldehyde analogs but did not accept purines and pyrimidines as a substrate. Kinetic parameters, i.e., Km and Vmax were determined for purified BMAL-CHO dehydrogenase with some aldehyde and PMS. The kinetic analysis revealed that this aldehyde dehydrogenase has an apparent Km of 25 mM for BMAL-CHO (). The effects of temperature on the stability and activity of this enzyme were investigated (data not shown).
Figure 2. SDS-PAGE of enzyme fractions of each purification step and the operon of BMAL-CHO dehydrogenase genes. a) Lane M, broad-range protein molecular weight markers (Promega, Madison, WI, USA): 225,000, 150,000, 100,000, 75,000, 35,000, 25,000, 15,000, and 10,000; lane 1, cell-free extract; lane 2, fraction after weak ion exchange chromatography; lane 3, fraction after strong ion exchange chromatography; lane 4, fraction after hydrophobic interaction chromatography; lane 5, purified enzyme after gel filtration chromatography. b) The operon of BMAL-CHO dehydrogenase genes. GenBank accession numbers are LC480925 and LC480926.

Figure 3. Evaluation of BMAL-CHO dehydrogenase activity with various electron acceptors. The relative activities of BMAL-CHO dehydrogenase using various electron acceptors are presented, which were calculated based on BMAL-COOH production measured by peak area of 247 m/z mass spectrum, relative to using PMS as an electron acceptor. The reactions were carried out with of the reaction mixture containing 1.9 µg/mL of the purified enzyme, 0.25 mg/mL BMAL-CHO in DMSO, and 0.5 mM electron transporter at pH 6.0 at 30°C for 30 min.

Table 2. Substrate specificity of BMAL-CHO dehydrogenase.
Table 3. Km and Vmax values of BMAL-CHO dehydrogenase from P. nitroreducens SB32154.
Cloning and expression of recombinant BMAL-CHO dehydrogenase in E. coli
BMAL-CHO dehydrogenase was expected to consist of two subunits, i.e., molybdopterin subunit and (2Fe-2S)-binding subunit from the results of WGS. These genes were expressed in E. coli by transformation with a vector having these two subunit genes tandemly after T7 promoter in order of the genome sequence. Each gene was expressed (), and the E. coli expressing these two genes was applied to BMAL-CHO conversion. Host E. coli control did not produce BMAL-COOH from BMAL-CHO but produce BMA-OH. On the other hand, the E. coli transformant produced BMAL-COOH from BMAL-CHO (). From these results, the two subunit genes were confirmed to be responsible gene for BMAL-CHO dehydrogenase genes. BMAL-CHO dehydorogenase with his-tag did not show the activity.
Figure 4. SDS-PAGE of BMAL-CHO dehydrogenase expressed in E. coli. The molecular weight of expressed molybdenum-binding subunit was 75 kDa and that of (2 Fe-2S)-binding subunit was 15 kDa. Lane M, broad-range protein molecular weight markers (Promega, Madison, WI, USA): 225,000, 150,000, 100,000, 75,000, 35,000, 25,000, 15,000, and 10,000. Control was CFE of E. coli BL21 STAR (DE3) harboring pET17b. sup., supernatant; ppt., precipitation.

Figure 5. BMAL-CHO conversion by recombinant E. coli expressing BMAL-CHO dehydrogenase. The reactions were carried out with 20 µL of the reaction mixture containing 50% (v/v) the disrupted BMAL-CHO dehydrogenase-expressing recombinant E. coli solution, 0.25 mg/mL BMAL-CHO, 0.5 mM PMS, and 0.3 mM DCIP at pH 6.0 at 30°C for 30 min. BMAL derivative production was measured by 280 nm absorbance. The top chart indicates the reduction of BMAL-CHO by host E. coli cells, whereas the bottom chart indicates the production of BMAL-COOH by BMAL-CHO dehydrogenase-expressing recombinant E. coli cells. The peak at 2.2 min in the chart indicates the ethanol adducted hemiacetal form of BMAL-CHO.

One-pot production of BMAL-COOH from BMAL using P450nov and BMAL-CHO dehydrogenase
For one-pot enzymatic production of BMAL-COOH from BMAL, the combination with P450nov and BMAL-CHO dehydrogenase was evaluated. E. coli expressing P450nov (L188P/F218L), FDXnov, and FDRnov produced BMAL-OH from BMAL (P450nov only in ). When purified BMAL-CHO dehydrogenase of P. nitroreducens SB32154 was combined with the cell suspension of E. coli expressing P450nov (L188P/F218L), FDXnov, and FDRnov, a significant amount of BMAL-COOH was produced from BMAL as the substrate ().
Figure 6. One-pot enzymatic conversion of BMAL to BMAL-COOH. The reactions were carried out with 90% (v/v) P450nov-expressing recombinant E. coli suspension, 1.5 µg/mL purified BMAL-CHO dehydrogenase of P. nitroreducens SB32154, and 0.25 mg/mL BMAL at 28°C for 20 h. BMAL derivative production was measured by 280 nm absorbance. The top chart indicates the conversion of BMAL using only P450nov, whereas the bottom chart indicates the conversion of BMAL using P450nov and BMAL-CHO dehydrogenase.

Discussion
In this study, the aldehyde dehydrogenase from P. nitroreducens SB32154, which catalyzes the oxidation of BMAL-CHO to BMAL-COOH, was reported. This enzyme was a member of the xanthine oxidase family protein and consisted of two different subunits, i.e., iron-sulfur-binding subunit and molybdenum-binding protein subunit. Xanthine oxidase family protein exists in a great variety of organisms from bacteria to higher plants and humans and generally consisted of three different subunits, N-terminal iron-sulfur-binding subunit, intermediate FAD-binding subunit, and C-terminal molybdenum-binding subunit. Electrons that are passed to the molybdenum during oxidation are transferred to FAD via the iron sulfur centers. Finally, NAD+ or oxygen molecule, which is the final electron acceptor, is reduced [Citation25]. In contrast, the BMAL-CHO dehydrogenase, which did not contain FAD, could not use NAD+ or molecular oxygen as the final electron acceptor. The addition of flavin derivative such as riboflavin, FMN, and FAD to the purified enzyme did not increase BMAL-CHO oxidation activity, but methylene blue, DCIP, potassium ferricyanide, cytochrome c, and especially PMS succeeded in accepting electrons and increased BMAL-COOH production. The native final electron acceptor has not been revealed, but the bacterial respiratory chain may have been involved because of its ability of using cytochrome c as an electron acceptor. The xanthine oxidase family protein lacking FAD-binding subunit has also been reported from Pseudomonas diminuta 7 (Brevundimonas dimuta) [Citation26] and Desulfovibrio gigas [Citation27,Citation28]. These enzymes exhibited similar substrate specificity, such that benzaldehyde was a good substrate, but xanthine was not. Isoquinoline 1-oxidoreductase from B. diminuta 7 could hydroxylate N-heterocyclic compounds, but BMAL-CHO dehydrogenase could not. Aldehyde oxidoreductase from D. gigas could not use PMS as an electron acceptor, but BMAL-CHO dehydrogenase could. From such differences, BMAL-CHO dehydrogenase in this study could be a novel xanthine oxidase family protein that does not have an intermediate FAD-binding subunit. In addition, the phylogenetic neighbor–joining tree in shows that BMAL-CHO dehydrogenase was independent from other xanthine oxidase family proteins but relatively close to isoquinoline 1-oxidoreductase from B. dimuta and aldehyde dehydrogenase from A. pasteurianus.
Figure 7. Phylogenetic neighbor–joining tree of the large subunit of BMAL-CHO dehydrogenase and xanthine oxidase family proteins. The sequences were aligned using ClustalX BMAL-CHO dehydrogenase (LC480926); IorB, isoquinoline 1-oxidoreductase from Brevundimonas diminuta (Q51698) [Citation26]; AldH, membrane-bound aldehyde dehydrogenase from Acetobacter pasteurianus SKU1108 (APT_00973) [Citation8]; Aoml, aldehyde oxidase from Methylobacillus sp. KY4400 (Q84IX8) [Citation9]; PaoC, aldehyde oxidoreductase from Escherichia coli (P77489) [Citation11]; CutL, CO dehydrogenase from Hydrogenophaga pseudoflava (P19913) [Citation17]; CoxL, CO dehydrogenase from Oligotropha carboxidovorans OM5 (P19919) [Citation18]; XdhA, xanthine dehydrogenase from E. coli (Q46799) [Citation14]; CdhA, caffeine dehydrogenase from Pseudomonas sp. CBB1 (D7REY3) [Citation19]; QorL, quinoline 2-oxidoreductase from Pseudomonas putida (P72224) [Citation20]; HcrA, 4-hydroxybenzoyl-CoA reductase from Thauera aromatica (O33819) [Citation21]; NdhL, nicotinate dehydrogenase large from Eubacterium barkeri (Q0QLF2) [Citation16]; XdhB, xanthine dehydrogenase from Rhodobacter capsulatus (O54051) [Citation15]; MOD, aldehyde oxidoreductase from Desulfovibrio desulfuricans ATCC 27774 (Q9REC4) [Citation13]; Mop, aldehyde oxidoreductase from Desulfovibrio gigas (Q46509) [Citation27,Citation28].
![Figure 7. Phylogenetic neighbor–joining tree of the large subunit of BMAL-CHO dehydrogenase and xanthine oxidase family proteins. The sequences were aligned using ClustalX BMAL-CHO dehydrogenase (LC480926); IorB, isoquinoline 1-oxidoreductase from Brevundimonas diminuta (Q51698) [Citation26]; AldH, membrane-bound aldehyde dehydrogenase from Acetobacter pasteurianus SKU1108 (APT_00973) [Citation8]; Aoml, aldehyde oxidase from Methylobacillus sp. KY4400 (Q84IX8) [Citation9]; PaoC, aldehyde oxidoreductase from Escherichia coli (P77489) [Citation11]; CutL, CO dehydrogenase from Hydrogenophaga pseudoflava (P19913) [Citation17]; CoxL, CO dehydrogenase from Oligotropha carboxidovorans OM5 (P19919) [Citation18]; XdhA, xanthine dehydrogenase from E. coli (Q46799) [Citation14]; CdhA, caffeine dehydrogenase from Pseudomonas sp. CBB1 (D7REY3) [Citation19]; QorL, quinoline 2-oxidoreductase from Pseudomonas putida (P72224) [Citation20]; HcrA, 4-hydroxybenzoyl-CoA reductase from Thauera aromatica (O33819) [Citation21]; NdhL, nicotinate dehydrogenase large from Eubacterium barkeri (Q0QLF2) [Citation16]; XdhB, xanthine dehydrogenase from Rhodobacter capsulatus (O54051) [Citation15]; MOD, aldehyde oxidoreductase from Desulfovibrio desulfuricans ATCC 27774 (Q9REC4) [Citation13]; Mop, aldehyde oxidoreductase from Desulfovibrio gigas (Q46509) [Citation27,Citation28].](/cms/asset/94177c23-796b-421e-916e-eb8dd7676720/tbbb_a_1799749_f0007_b.gif)
In this paper, one-pot enzymatic conversion of BMAL to BMAL-COOH was also achieved by combining the BMAL-CHO dehydrogenase with P450nov, which could catalyze BMAL to BMAL-OH and had weak activity of BMAL-OH oxidation to BMAL-CHO. However, the conversion efficiency of this one-pot reaction was low, as BMAL-OH accumulated during the reaction and only a small amount of BMAL-COOH was produced from BMAL as substrate (). BMAL-OH accumulation was caused by the BMAL-CHO reduction activity of host E. coli cells () and weak BMAL-OH oxidation activity of P450nov. To reduce BMAL-CHO reduction activity, it is necessary to knock out the corresponding enzyme in host E. coli cells. To improve the BMAL-OH oxidation activity, P450nov is needed to be evolved, or a novel alcohol oxidoreductase, which can convert BMAL-OH to BMAL-CHO, is needed to be developed. As for BMAL-CHO dehydrogenase, the affinity for BMAL-CHO was very low (Km = 25 mM) compared to benzaldehyde derivatives (). It may be possible to improve substrate specificity if appropriate mutations were introduced into BMAL-CHO dehydrogenase. Alternatively, another potential microorganisms might be isolated if the screening is carried out with improved methods targeting more specific enzyme and with a variety of soils. As mentioned above, by improving the components of this one-pot enzymatic conversion of BMAL to BMAL-COOH via BMAL-CHO, BMAL-COOH is expected to be produced more effectively, which has potential applications in various fields.
Author contribution statement
Iori Kozono and Jun Ogawa conceived and designed research. Iori Kozono conducted experiments. Iori Kozno, Makoto Hibi, Michiki Takeuchi, and Jun Ogawa analyzed data. Iori Kozono, Michiki Takeuchi, and Jun Ogawa wrote the manuscript. All authors read and approved the manuscript.
Compliance with ethical standards
Conflict of interest: All authors declare that they have no conflict of interest.
Ethical approval: This article does not contain any studies with human participants performed by any of the authors.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Ki SK, Yamabe N, Hyun YK, et al. Role of maltol in advanced glycation end products and free radicals: in-vitro and in-vivo studies. J Pharm Pharmacol. 2008;60(4):445–452. .
- Thompson KH, Lichter J, LeBel C, et al. Vanadium treatment of type 2 diabetes: A view to the future. J Inorg Biochem. 2009;103(4):554–558. .
- Kim YJ, Uyama H. Tyrosinase inhibitors from natural and synthetic sources: structure, inhibition mechanism and perspective for the future. Cell Mol Life Sci. 2005;62(15):1707–1723.
- Bentley R. From miso, sake and shoyu to cosmetics: a century of science for kojic acid. Nat Prod Rep. 2006;23(6):1046–1062.
- Agrawal A, Romero-Perez D, Jacobsen JA, et al. Zinc-binding groups modulate selective inhibition of MMPs. Chem Med Chem. 2008;3(5):812–820. .
- Durrant JD, Oliveira CAF, Mccammon JA. Pyrone-based inhibitors of metalloproteinases types 2 and 3 may work as conformation-selective inhibitors. Chem Biol Drug Des. 2011;78(2):191–198.
- Kozono I, Mihara K, Minagawa K, et al. Engineering of the cytochrome P450 monooxygenase system for benzyl maltol hydroxylation. Appl Microbiol Biotechnol. 2017;101(17):6651–6658. .
- Yakushi T, Fukunari S, Kodama T, et al. Role of a membrane-bound aldehyde dehydrogenase complex AldFGH in acetic acid fermentation with Acetobacter pasteurianus SKU1108. Appl Microbiol Biotechnol. 2018;102(10):4549–4561. .
- Uchida H, Mikami B, Yamane-Tanabe A, et al. Crystal structure of an aldehyde oxidase from Methylobacillus sp. KY4400. J Biochem. 2018;163(6):321–328. .
- Yasuhara A, Akiba-Goto M, Fujishiro K, et al. Production of aldehyde oxidases by microorganisms and their enzymatic properties. J Biosci Bioeng. 2002;94(2):124–129. .
- Correia MA, Otrelo-Cardoso AR, Schwuchow V, et al. The Escherichia coli periplasmic aldehyde oxidoreductase is an exceptional member of the xanthine oxidase family of molybdoenzymes. ACS Chem Biol. 2016;11(10):2923–2925. .
- Yamada M, Adachi K, Ogawa N, et al. A new aldehyde oxidase catalyzing the conversion of glycoaldehyde to glycolate from Burkholderia sp. AIU 129. J Biosci Bioeng. 2015;119(4):410–415. .
- Rebelo J, Macieira S, Dias JM, et al. Gene sequence and crystal structure of the aldehyde oxidoreductase from Desulfovibrio desulfuricans ATCC 27774. J Mol Biol. 2000;297(1):135–146. .
- Xi H, Schneider BL, Reitzer L. Purine catabolism in Escherichia coliand function of xanthine dehydrogenase in purine salvage. J Bacteriol. 2000;182(19):5332–5341.
- Truglio JJ, Theis K, Leimkuhler S, et al. Crystal structures of the active and alloxanthine-inhibited forms of xanthine dehydrogenase from Rhodobacter capsulatus. Structure. 2002;10(1):115–125. .
- Wagener N, Pierik AJ, Ibdah A, et al. The Mo-Se active site of nicotinate dehydrogenase. Proc Natl Acad Sci USA. 2009;106(27):11055–11060. .
- Hanzelmann P, Dobbek H, Gremer L, et al. The effect of intracellular molybdenum in Hydrogenophaga pseudoflava on the crystallographic structure of the seleno-molybdo-iron-sulfur flavoenzyme carbon monoxide dehydrogenase. J Mol Biol. 2000;301(5):1221–1235.
- Dobbek H, Gremer L, Kiefersauer R, et al. Catalysis at a dinuclear [CuSMo(=O)OH] cluster in a CO dehydrogenase resolved at 1.1-Å resolution. Proc Natl Acad Sci USA. 2002;99(25):15971–15976. .
- Yu CL, Kale Y, Gopishetty S, et al. A novel caffeine dehydrogenase in Pseudomonas sp. strain CBB1 oxidizes caffeine to trimethyluric acid. J Bacteriol. 2008;190(2):772–776. .
- Bonin I, Martins BM, Purvanov V, et al. Active site geometry and substrate recognition of the molybdenum hydroxylase quinoline 2-oxidoreductase. Structure. 2004;12(8):1425–1435. .
- Unciuleac M, Warkentin E, Page CC, et al. Structure of a xanthine oxidase-related 4-hydroxybenzoyl-CoA reductase with an additional [4Fe-4S] cluster and an inverted electron flow. Structure. 2004;12(12):2249–2256. .
- Hider RC, Tilbrook GS, Liu Z Orally active iron (III) chelators. Patent US6448273 B1, 10 September 2002
- Seth MC, David TP Multidentate pyrone-derived chelators for medicinal imaging and chelation. Patent US20100098640 A1, 22 April 2010
- Mohapatra BR, Harris N, Nordin R, et al. Purification and characterization of a novel caffeine oxidase from Alcaligenes species. J Biotechnol. 2006;125(3):319–327. .
- Okamoto K, Kusano T, Nishino T. Chemical nature and reaction mechanisms of the molybdenum cofactor of xanthine oxidoreductase. Curr Pharm Des. 2013;19(14):2606–2614.
- Lehmann M, Tshisuaka B, Fetzner S, et al. Purification and characterization of isoquinoline 1-oxidoreductase from Pseudomonas diminuta 7, a novel molybdenum-containing hydroxylase. J Biol Chem. 1994;269(15):11254–11260.
- Thoenes U, Flores OL, Neves A, et al. Molecular cloning and sequence analysis of the gene of the molybdenum-containing aldehyde oxido-reductase of Desulfovibrio gigas. The deduced amino acid sequence shows similarity to xanthine dehydrogenase. Eur J Biochem. 1994;220(3):901–910. .
- Rebelo JM, Dias JM, Huber R, et al. Structure refinement of the aldehyde oxidoreductase from Desulfovibrio gigas (MOP) at 1.28 Å. J Biol Inorg Chem. 2001;6(8):791–800. .