
ABSTRACT
An enzyme belonging to glycoside hydrolase family 68 (GH68) from Beijerinckia indica subsp. indica NBRC 3744 was expressed in Escherichia coli. Biochemical characterization showed that the enzyme was identified to be a β-fructosyltransferase (BiBftA). Crystallization of a full-length BiBftA was initially attempted, but no crystals were obtained. We constructed a variant in which 5 residues (Pro199-Gly203) and 13 residues (Leu522-Gln534) in potentially flexible regions were deleted, and we successfully crystallized this variant BiBftA. BiBftA is composed of a five-bladed β-propeller fold as in other GH68 enzymes. The structure of BiBftA in complex with fructose unexpectedly indicated that one β-fructofuranose (β-Fruf) molecule and one β-fructopyranose molecule bind to the catalytic pocket. The orientation of β-Fruf at subsite −1 is tilted from the orientation observed in most GH68 enzymes, presenting a second structure of a GH68 enzyme in complex with the tilted binding mode of β-Fruf.
Graphical Abstract
Structure of the GH68 enzyme unexpectedly indicated that one β-fructofuranose and one β-fructopyranose bind to the catalytic pocket.
Enzymes in the glycoside hydrolase family 68 (GH68) catalyze the hydrolysis and transfructosylation of sucrose [Citation1]. Some GH68 enzymes exhibit high transfructosylation activity to produce fructooligosaccharides (FOS), which is predominantly a mixture of the trisaccharide 1-kestose, the tetrasaccharide nystose, and the pentasaccharide fructosylnystose [Citation2]. FOS, especially 1-kestose, have been shown to have prebiotic effects on human health by stimulating the growth of beneficial gut bacteria [Citation3–5]. Enzymes that act on the fructosyl moiety of sucrose are classified into three glycoside hydrolase families, GH32, GH68, and GH100, in the CAZy database (http://www.cazy.org/) [Citation6]. Enzymes belong to GH100, composed of an (α/α)6 barrel fold, are structurally unrelated to those belong to GH32 and GH68; GH100 is categorized into clan GH-G in the CAZy database, and is proposed to adopt an inverting catalytic mechanism [Citation7]. On the other hand, GH32 and GH68 enzymes contain a five-bladed β-propeller domain, and are categorized as members of clan GH-J. GH32 and GH68 share similar catalytic mechanisms, which operate via a double displacement reaction involving three carboxylic acid residues [Citation1,Citation8].
Beijerinckia indica is a Gram-negative nitrogen-fixing bacterium that can grow on a variety of organic acids, sugars, and alcohols [Citation9,Citation10]. The whole genome of B. indica subsp. indica ATCC 9039 has been sequenced [Citation10], and B. indica was shown to possess genes for three GH32 enzymes and one GH68 enzyme. Here, we show that a GH68 enzyme from B. indica subsp. indica NBRC 3744 catalyzes transfructosylation to produce FOS from sucrose. The enzyme, designated here as BiBftA, displays 65% sequence identity to Gluconacetobacter diazotrophicus levansucrase (GdLsdA) [Citation11] and 57% identity to Microbacterium saccharophilum β-fructofuranosidase (MsFFase) [Citation12,Citation13]. We determined the crystal structure of BiBftA and compared it with other GH68 enzymes.
Materials and methods
Construction of BiBftA expression plasmids
The strain B. indica subsp. indica NBRC 3744 was obtained from NITE Biological Resource Center (Chiba, Japan). DNA primers for the PCR amplification of BiBftA was designed using the sequence of Bind_2021 (DDBJ/EMBL/GenBank ID, CP001016.1) of B. indica subsp. indica ATCC 9039 as the reference source. The DNA fragment encoding BiBftA was amplified by colony-directed PCR using the primers 5ʹ-GGT TAC CCG ATA CCG ACT CCG CAT TCG GGA CAA GCC TAT GAT CC-3ʹ and 5ʹ-TTA CTG GCC GTT CGT GAC ACC ATG GCC ATT AAC TTG GCC AAG CGC GGG AAG AT-3ʹ. The obtained DNA fragment was sequenced, and the nucleotide sequence data was deposited in the DDBJ/EMBL/GenBank database under accession number LC522529. Five bases were different from those of the sequence of B. indica subsp. indica ATCC 9039. Codons for the five amino acid residues from the strain ATCC 9039 were GCG (Ala47), GGC (Gly429), AAT (Asn451), ACT (Thr479), and AGA (Arg495), and the codons for the corresponding residues from the strain NBRC 3744 were GCA (Ala47), GGT (Gly429), AAC (Asn451), CCT (Pro479), and GGA (Gly495); the underlined letters indicate the bases that differed.
To subclone the PCR product, the PCR primers were designed based on the signal peptide prediction using the Signal-P4 server (https://www.cbs.dtu.dk/services/SignalP/)[Citation14]. The N-terminal signal sequence of BiBftA was predicted to comprise the 29 amino acid residues MASRSFNVCIRSLIAGSLLTATALSAQAQ and was omitted in the primer design. The primers 5′-TCT AAA GGA TCC TCG GGT TAC CCG ATA CC-3′ and 5′- T ACC AGA CTC GAG TTA CTG G − 3′ (BamHI and XhoI restriction sites are underlined) were used for PCR amplification. The amplified product was ligated into the pET-32a(+) vector (Merck Millipore, Darmstadt, Germany). Oligonucleotide-directed mutagenesis was carried out using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA) and primers. To remove the sequence encoding residues 199–203, the 5′- C GCC CGC TTC GTG GGC GGG CCC CAA CAC GC −3′ and 5′- G GTA GGC GTG TTG GGG CCC GCC CAC GAA GC −3′ primers were used. To remove the sequence encoding residues 522–534, the 5′-CC AAT CTT CCC GCG TAA CTC GAG CTC CAC C-3′ and 5′-G GTG GAG CTC GAG TTA CGC GGG AAG ATT GG-3′ primers were used. Constructs were verified by DNA sequencing.
Protein expression and purification
Escherichia coli BL21 (DE3) was used as the host strain for protein expression. The transformant was grown at 37°C in 1 L of medium containing 10 mg/mL tryptone, 5 mg/mL yeast extract, 10 mg/mL sodium chloride, and 50 μg/mL ampicillin. When the culture reached an absorbance of 0.6 at 600 nm, protein expression was induced with isopropyl-β-D-thiogalactopyranoside at a final concentration of 0.1 mM, and the culture was incubated for an additional 18 h at 18°C. The cells were harvested by centrifugation at 4,000 g for 5 min, resuspended in 30 mL of 20 mM Tris–HCl (pH 8.0), and then disrupted by sonication. After centrifugation at 12,000 g for 20 min to remove insoluble material, the supernatant was applied onto a nickel nitrilotriacetic acid agarose column (10 mL; QIAGEN, Hilden, Germany) that was equilibrated with the 20 mM Tris-HCl (pH 8.0). The column was washed with 20 mM Tris-HCl (pH 8.0), and thioredoxin-His-tagged BiBftA was eluted with 50 mM imidazole in 20 mM Tris-HCl (pH 8.0). To cleave the thioredoxin-His tag from the protein, thrombin protease (1 U; Fujifilm Wako, Osaka, Japan) was added to 10 mg of thioredoxin-His-tagged BiBftA in 20 mM Tris-HCl buffer (pH 8.0) and incubated for 18 h at 22°C. Then, this BiBftA and protease mixture was applied to an HiTrap Q HP column (5 mL; GE Healthcare, Chalfont St Giles, UK) that was equilibrated with 20 mM Tris-HCl (pH 8.0). BiBftA was eluted with a linear 100–300 mM NaCl gradient in 20 mM Tris–HCl (pH 8.0) at a flow rate of 2 mL min−1 at 4°C. Protein elution was monitored by absorbance at 280 nm. Protein purity was confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Figure S1).
Crystallization, data collection, and structure determination
The purified BiBftA was concentrated to 20 mg mL−1 in 10 mM Tris-HCl (pH 7.0) using an Amicon Ultra-15 Centrifugal Filter Unit 30 K (Merck Millipore) and was crystallized at 20°C using the hanging drop vapor diffusion method in which 1.0 μL of protein solution was mixed with an equal volume of crystallization reservoir solution containing 30% (w/v) polyethylene glycol 10,000, 0.2 M magnesium chloride, 0.1 M potassium sodium tartrate, and 0.1 M Tris-HCl (pH 8.0). The crystal was cryoprotected in the reservoir solution supplemented with 30% (w/v) glycerol. To obtain the structure in complex with fructose (Fru), the crystal was soaked in the reservoir solution supplemented with 30% (w/v) Fru, which also acted as a cryoprotectant. The crystals were then flash-cooled to 100 K in a nitrogen gas stream. Diffraction data were collected using a BL17A beamline at Photon Factory (Tsukuba, Japan). Data images were processed with XDS [Citation15] and scaled with SCALA [Citation16] in the CCP4 program suite [Citation17]. The structures were solved by the molecular replacement method with MOLREP [Citation18] in CCP4. To solve the unliganded structure, a model of GdLsdA (PDB ID, 1W18) [Citation11] was employed as a probe model. Automated model building was carried out with the ARP/wARP program [Citation19]. Refinement was performed using REFMAC5 [Citation20] in CCP4, and manual adjustment and rebuilding of the model were carried out using COOT [Citation21]. Validation of the structures was carried out using RAMPAGE [Citation22] in CCP4. The figures were produced using LigPlot+ [Citation23] and PyMOL (https://www.pymol.org/). The data collection and refinement statistics are summarized in . The coordinates and structure factors were deposited in the Protein Data Bank under the accession codes 6M0D and 6M0E.
Table 1. Data collection and refinement statistics
Measurement of BiBftA enzyme activity
BiBftA activity was measured using 2% sucrose as the substrate and measuring the amount of glucose liberated. This enzymatic reaction was carried out in 50 mM sodium phosphate buffer (pH 6.0) at 30°C in a total volume of 250 μL, and 100 μL of the reaction mixture was taken after 10-min incubation and the reaction was stopped by adding 50 μL of 0.2 M NaOH, 5 μL of the solution was mixed with 200 μL of Glucose C-II Test Kit solution (Fujifilm Wako) and incubated for 5 min at 37°C, and absorbance at 505 nm was measured. A glucose standard was used to determine enzyme activity. One unit (U) of enzymatic activity was defined as the amount of enzyme needed to liberate 1 μmol min−1 of glucose from sucrose.
The optimal pH was determined in 50 mM sodium phosphate-citrate buffer (pH 3.0–5.5), sodium phosphate buffer (pH 6.0–7.0), and Tris-HCl buffer (pH 8.0–9.0). pH stability was determined by incubating 50 μL of BiBftA in 62.5 mM sodium phosphate-citrate buffer (pH 3.0–5.5), sodium phosphate buffer (pH 6.0–7.0), and Tris-HCl buffer (pH 8.0–9.0) at 30°C for 1 h, cooling the solution in an ice bath, diluting the solution with water, and measuring residual BiBftA activity. The optimal temperature was assayed at various temperatures (30–80°C) in 50 mM sodium phosphate buffer (pH 6.0) for 10 min. To measure the thermostability, BiBftA in 10 mM Tris-HCl buffer (pH 7.0) was incubated at 30–80°C for 30 min, cooled in an ice bath, and then residual activity was measured.
The kinetic parameters for glucose liberated from sucrose were determined as follows. Initial rates of the enzymatic reaction against substrate concentration were measured using sucrose (0.4, 1, 2, 4, 10, 20, 40 and 100 mM) in 50 mM sodium phosphate buffer (pH 6.0) at 30°C in a total volume of 1 mL, and 100 μL of the reaction mixture was taken after 5-, 10-, 15-, 20-, and 25-min incubations, and mixed with 50 μL of 0.2 M NaOH. Then 5 μL of the solution was mixed with 200 μL of Glucose C-II Test Kit solution and analyzed as the aforementioned procedure. The parameters were calculated by nonlinear regression analysis using the program KaleidaGraph (Synergy Software, Reading, PA, USA).
The protein concentration of the purified BiBftA was determined by measuring the absorbance at 280 nm using the molar extinction coefficient [tag-removed full-length BiBftA, 1 mg/mL = 1.499; BiBftA deletion variant (BiBftA ΔAΔC), 1 mg/mL = 1.497], which was calculated using the ExPASy ProtParam server (https://web.expasy.org/protparam/).
Thin layer chromatography (TLC)
Purified, tag-removed full-length BiBftA (0.72 mg/mL final concentration) was incubated with 30% (w/v) sucrose in sodium phosphate buffer (pH 6.0) at 37°C for 18 h. Samples were removed at several timepoints, were immediately heated at 100°C for 5 min, and then were analyzed by TLC on a Silica Gel 60 plate (Merck Millipore) using a running solvent with 1-butanol/ethanol/water (5:5:3, v/v/v). Spots were detected by charring with 5% sulfuric acid in methanol.
Results and discussion
Mutagenesis for crystallization
The thioredoxin-His-tagged BiBftA protein was affinity-purified and digested with thrombin, and then the untagged BiBftA was further purified. We first attempted to crystallize full-length BiBftA, but crystals were not obtained. The amino acid sequence of BiBftA was then aligned with the amino acid sequences of the GH68 enzymes MsFFase, GdLsdA [Citation11], Erwinia amylovora levansucrase (EaLsc) [Citation24], and Bacillus subtilis levansucrase (BsSacB) [Citation25] (), and the alignment suggested that a sequence of 5 amino acid residues, PYADG (residues 199–203; yellow in ; designated loop-A), and a sequence of 13 C-terminal residues, LGQVNGHGVTNGQ (residues 522–534; yellow in ), in BiBftA may be flexible regions. These two regions are not conserved among the GH68 enzymes and were predicted to be located far from the active site. We hypothesized that these two regions prevented BiBftA crystal growth due to their flexibility [Citation26,Citation27], thus we created a variant containing deletions of both of the 5 residue and 13 residue regions. The BiBftA deletion variant (BiBftA ΔAΔC) crystallized successfully ()).
Figure 1. Comparison of amino acid sequences of BiBftA and related GH68 enzymes. Sequences were aligned using the Clustal Omega server (https://www.ebi.ac.uk/Tools/msa/clustalo/), and manual adjustments were made based on crystal structures. The three catalytic residues are highlighted in red. Residues shown in ) are highlighted in green. Four conserved motifs, A-D, in GH32 and GH68 enzymes [Citation1] are boxed. Nine loops, 1–9, at the rim of the active site funnel [Citation24] are underlined in blue. The sequences (5 residues in loop-A and 13 C-terminal residues) that were deleted to improve crystallization are highlighted in yellow
![Figure 1. Comparison of amino acid sequences of BiBftA and related GH68 enzymes. Sequences were aligned using the Clustal Omega server (https://www.ebi.ac.uk/Tools/msa/clustalo/), and manual adjustments were made based on crystal structures. The three catalytic residues are highlighted in red. Residues shown in Figure 4(c) are highlighted in green. Four conserved motifs, A-D, in GH32 and GH68 enzymes [Citation1] are boxed. Nine loops, 1–9, at the rim of the active site funnel [Citation24] are underlined in blue. The sequences (5 residues in loop-A and 13 C-terminal residues) that were deleted to improve crystallization are highlighted in yellow](/cms/asset/4fcc5d85-701f-4246-844c-6e541245aa80/tbbb_a_1804317_f0001_oc.jpg)
Figure 2. Crystals and overall structure of BiBftA ΔAΔC. (a) Crystals of BiBftA ΔAΔC. (b) Overall structure of BiBftA ΔAΔC shown as a ribbon model. The five blades (i–v) comprising the β-propeller fold are shown in blue (I, III, and V) or cyan (II and IV). The N-terminal region that wraps around the β-propeller fold is shown in green. Three loops, loops 5, 7, and 8, are shown in orange. Magnesium ions on the molecular surface are shown as magenta balls. Disordered loop-A is shown as a dotted line. (c) Stereo view of the superimposed Cα backbones in BiBftA ΔAΔC (red), MsFFase (green), and BsSacB (blue). The small domain-like structure in the C-terminus of MsFFase is indicated with a green arrow

Enzymatic properties of full-length BiBftA
The enzymatic activity of full-length BiBftA was measured by monitoring glucose liberation from sucrose, and the effects of pH and temperature on BiBftA activity were determined. The optimum pH was found to between pH 5–6 ()), and BiBftA was stable (residual activity > 75%) from pH 3–9 ()). The optimal temperature was 60°C ()), and BiBftA was stable (residual activity > 80%) at 60°C for 30 min ()). The specific activity of full-length BiBftA on the hydrolysis of 2% sucrose at 30°C was 307.7 ± 4.9 U mg−1. The specific activity of BiBftA ΔAΔC was 328.7 ± 4.9 U mg−1 suggesting that the deletions did not affect enzymatic activity.
Figure 3. Enzymatic properties of full-length BiBftA. (a) Optimal pH was measured in 50 mM of sodium phosphate-citrate buffer (black circles), sodium phosphate buffer (white circles), and Tris-HCl buffer (black squares). (b) pH stability was determined by incubating BiBftA in 62.5 mM of sodium phosphate-citrate buffer (black circles), sodium phosphate buffer (white circles), and Tris-HCl buffer (black squares). (c) Optimal temperature. (d) Thermostability. In (a)–(d), error bars represent standard deviations. (e) BiBftA enzymatic activity with various sucrose concentrations. (f) TLC analysis of the BiBftA and sucrose reaction. Symbols: M, malto-oligosaccharide markers; G1, glucose; G2, maltose; G3, maltotriose; G4, maltotetraose; S, sucrose; F, fructose; K, 1-kestose

The kinetic parameters of full-length BiBftA based on glucose liberation were determined ()), and the Km, kcat, and kcat/Km values were 16.0 ± 1.1 mM, 305.0 ± 7.7 s−1, and 19.1 s−1 mM−1, respectively. Km values for GH68 enzymes have been shown to range between 6.6–160 mM, and the Km and kcat values of EaLsc were determined to be 33.6 mM and 502.7 s−1 at pH 6.5 and 37°C [Citation28]. These results suggest that the kinetic parameters for BiBftA hydrolysis of sucrose are similar to other GH68 enzymes.
The products of full-length BiBftA activity on 30% sucrose were analyzed by TLC ()), and a spot corresponding to the trisaccharide 1-kestose was observed, suggesting that the sugar is either 1-kestose, 6-kestose, neokestose or a mixture of these three. During the late stage of the reaction, another spot was detected at the origin of the TLC. This spot, corresponding to a polysaccharide, may be identified as either levan, inulin, or a mixture of both. In the reaction of Bacillus levansucrases with sucrose, major products are identified as glucose, fructose, and levan [Citation29,Citation30]. In contrast, the spot corresponding to the trisaccharide still remained in the reaction of BiBftA after sucrose was consumed. Spots corresponding to tetrasaccharide and pentasaccharide were faint.
Overall structure of BiBftA ΔAΔC
The crystal structures of unliganded BiBftA ΔAΔC and the BiBftA ΔAΔC-Fru complex were determined at 2.2-Å and 1.35-Å resolutions, respectively. The crystals belong to the space group P212121, which contain a single BiBftA molecule in an asymmetric unit. The Ramachandran plot was calculated with RAMPAGE of CCP4 and indicated that no residues in unliganded BiBftA ΔAΔC were outliers, whereas one residue, Asp285, in BiBftA ΔAΔC-Fru was an outlier. Asp285 is one of the catalytic residues in BiBftA, and the electron density for this residue was well defined. The 2|Fo|-|Fc| electron density contoured at 1 σ showed continuous density for most main chain atoms, but residues 197–211 (unliganded BiBftA ΔAΔC) and residues 197–209 (BiBftA ΔAΔC-Fru) in loop-A were not visible and are represented by the dotted line in ). As described in the previous section, we predicted that loop-A is flexible, thus we deleted 5 residues (residues 199–203) to improve crystal growth, and 10 residues (residues 197–198 and 204–211) and 8 residues (residues 197–198 and 204–209) were not visible in the unliganded BiBftA ΔAΔC and BiBftA ΔAΔC-Fru structures, respectively.
BiBftA is composed of a five-bladed (I–V) β-propeller fold and several subcomponents ()), which is similar to other GH68 enzymes. Each of the blades is composed of four β-strands, A, B, C, and D. The N-terminal region, including ~60 residues, wraps around the β-propeller fold (green in )). In addition to the N-terminal region, the loops between β-strands IIIB-IIIC (residues 307–342; loop 5 in ), IVB-IVC (396–406; loop 7 in ), and IVD-VA (434–458; loop 8 in ) are relatively long; the N-terminal region and the three loops between β-strands form a subdomain-like structure. Two and three magnesium ions were found on the molecular surfaces of the unliganded BiBftA ΔAΔC and BiBftA ΔAΔC-Fru structures, respectively, most likely due to the high concentration (200 mM) of magnesium chloride used for crystallization and thus are likely artifacts. GH68 enzymes are retaining glycoside hydrolases with two aspartic acid residues and one glutamic acid residue forming the catalytic triad [Citation1,Citation25]. On the basis of homology with other GH68 enzymes, Asp100, Asp285, and Glu377 were identified as the catalytic nucleophile, the transition state stabilizer, and the general acid/base, respectively.
A structural homology search was carried out using the DALI server (http://ekhidna2.biocenter.helsinki.fi/dali/) [Citation31], and the Z-scores and the sequence similarities are listed in . High similarities were found between GH68 enzymes from Gram-negative bacteria, GdLsdA, MsFFase, and EaLsc. BiBftA showed a relatively high similarity with an enzyme from a Gram-positive bacterium, BsSacB [Citation25]. The Cα backbone of BiBftA was compared with the Cα backbones of MsFFase [Citation13] and BsSacB [Citation25] ()). The β-propeller fold structures superimposed well even though the subcomponent structures were distinct from each other. MsFFase has a small domain-like structure in the C-terminus, while the C-terminal 13 amino acid residues of BiBftA were removed to improve the crystallinity. In addition to GH68, BiBftA shares homology with other enzymes possessing the five-bladed β-propeller fold structure, such as GH43, GH32, GH117, GH62, and GH130, as reported previously [Citation32].
Table 2. Summary of the structural similarity search using the DALI server
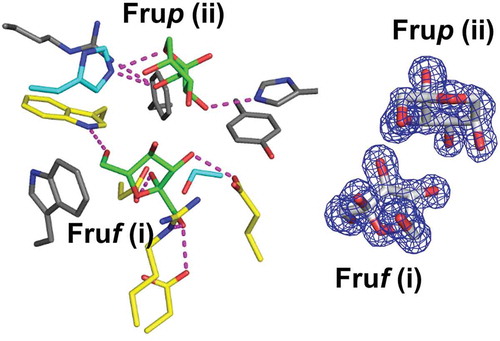
In the structure of BiBftA ΔAΔC-Fru, 5 Fru molecules indicated by (i)–(v) in ) were observed. Fru (i) was identified as β-fructofuranose (β-Fruf), whereas Fru (ii)–(v) were modeled as β-fructopyranose (β-Frup) ()). Electron density |Fo|-|Fc| maps of Fruf (i) and Frup (ii)–(iv) were clearly seen at the contour level of 3 σ, whereas Frup (v) was present at the lower contour level. Fruf (i) and Frup (ii) bound to the active site, whereas Frup (iii)–(v) were located on the molecular surface of BiBftA.
Figure 4. Fru-bound structure of BiBftA ΔAΔC. (a) The positions of five Fru molecules, Fruf (i), Frup (ii), Frup (iii), Frup (iv), and Frup (v) in BiBftA ΔAΔC are shown in magenta. (b) |Fo|-|Fc| omit maps contoured at 3 σ. (c) Schematic of the amino acid residues interacting with Fruf (i) and Frup (ii). Colors: black circle, carbon atom; blue circle, nitrogen atom; red circle, oxygen atom; cyan circle, water molecule; green dashed line, hydrogen bond; red crown, hydrophobic interaction

Comparison of the active sites of BiBftA and related enzymes
The complexed BiBftA ΔAΔC-Fru structure ()) was compared with other complexed GH68 enzyme structures. Two types of GH68 enzymes complexed with Fru have been reported. The structure of MsFFase in complex with Fru showed that a β-Fruf molecule was located at subsite −1 in the active site ()) [Citation12]. The orientation of β-Fruf at subsite −1 in MsFFase is almost identical to that of a β-Fruf residue of sucrose found at subsite −1 in BsSacB [Citation12]. The sucrose-bound structure of Lactobacillus johnsonii GH68 inulosucrase has also indicated that the orientation of sucrose is very similar to that observed in BsSacB [Citation33]. In comparison, the structural study of EaLsc soaked with sucrose indicated that, although a β-Fruf molecule was observed at subsite −1, the orientation of β-Fruf bound to EaLsc was tilted ~125°. In addition, an α-glucopyranose molecule was present at a position separate from the β-Fruf molecule in the EaLsc structure ()). Wuerges et al. have proposed that the structure of EaLsc soaked with sucrose is a snapshot of the products of sucrose hydrolysis trapped into the active site [Citation24], whereas the binding of β-Fruf to MsFFase is the substrate mode.
Figure 5. Stereo views of residues in the active sites of BiBftA ΔAΔC-Fru (a) and the corresponding residues in MsFFase (PDB code 3VSS) (b) and in EaLsc (PDB code 4D47) (c). Residues that form direct hydrogen bonds with Fruf (i) and Frup (ii), three aromatic residues, Trp189, Tyr460, and Phe473, and Ser461 in BiBftA ΔAΔC are indicated. Five residues that form direct hydrogen bonds with Fruf (i) in BiBftA ΔAΔC and the corresponding residues in (b) and (c) are shown in yellow. His137 and Ser461 in BiBftA ΔAΔC and the corresponding residues in (b) and (c) are shown in cyan. Fru and glucose (Glc) molecules are shown in green. Direct hydrogen bonds between amino acid residues and sugar molecules are indicated by magenta dotted lines. β-Fruf at subsite −1 in (b) is indicated as −1

The position and orientation of Fruf (i) bound to BiBftA ΔAΔC ()) matches well with β-Fruf bound to EaLsc ()). In BiBftA ΔAΔC, atom O1 of Fruf (i) interacts with two residues, Asp285 and Arg284, and atoms O2 and O3 of Fruf (i) interact with Asp100 and Glu377, respectively, via direct hydrogen bonds ()). The corresponding residues in EaLsc, Asp203, Arg202, Asp46, and Glu287, interact with β-Fruf at subsite −1 in the same manner ()). Also, the position and orientation of the pyranose ring of Frup (ii) in BiBftA ΔAΔC were similar to the position and orientation of α-glucopyranose in the EaLsc structure.
The interactions between BiBftA ΔAΔC and the two Fru molecules, Fruf (i) and Frup (ii), were analyzed with the program Ligplot+ ()). Five residues of BiBftA, Trp99, Asp100, Arg284, Asp285, and Glu373 (yellow in ), formed direct hydrogen bonds with Fruf (i). Asp100, Asp285, and Glu377 are the catalytic residues of GH68, and Trp99 and Arg284 are fully conserved among GH68 enzymes [Citation1,Citation34,Citation35]. Three residues, Arg136, His137, and His395, formed direct hydrogen bonds with Frup (ii) in BiBftA, but these residues are not conserved in BsSacB (). In the structure of EaLsc, atom O3 of α-glucopyranose interacts with atom NE2 of His305 (corresponding to His395 in BiBftA) via a direct hydrogen bond, and the same interaction was observed in BiBftA ΔAΔC-Fru (between atom O3 of Frup (ii) and atom NE2 of His395). Aside from this, the hydrogen bonding pattern between BiBftA ΔAΔC and Frup (ii) ()) is different from that between EaLsc and α-glucopyranose ()), most likely due to the structural difference between β-fructopyranose and α-glucopyranose.
The binding of β-Fruf at subsite −1 is completely different in BiBftA ΔAΔC than in MsFFase. Atom O1 of β-Fruf in MsFFase forms direct hydrogen bonds with two residues, Asp112 (corresponding to Asp100 in BiBftA) and Ser458 (Ser461 in BiBftA). Atom O6 of β-Fruf in MsFFase forms a direct hydrogen bond with His147 (His137 in BiBftA) ()). However, His137 and Ser461 in BiBftA ΔAΔC (cyan in ) did not form any hydrogen bonds with Fruf (i). The His137 side chain in BiBftA ΔAΔC adopted a different orientation than His147 in MsFFase.
Comparison of the structures of unliganded form and complex form with Fru
The structures of unliganded BiBftA ΔAΔC and BiBftA ΔAΔC-Fru were compared. The BiBftA ΔAΔC structure, like the EaLsc structure, has nine loops, 1–9, at the rim of the funnel of the catalytic β-propeller structure (, ), and it was proposed that the length of loops, especially loop 8 in EaLsc, determined the chain lengths of the transfructosylation products [Citation24]. A conformational change in loop 1 in BiBftA ΔAΔC-Fru (residues 127–138) was observed ()), and Arg136 and His137 located in this loop 1 were shifted away from Frup (ii) ()). The conformational change in loop 1 in BiBftA ΔAΔC-Fru resulted in the opening of the catalytic pocket; the distances between His137-Nε2 and His395-Nε2 in unliganded BiBftA ΔAΔC and in BiBftA ΔAΔC-Fru were 7.0 Å and 7.8 Å, respectively. These results may suggest that the BiBftA ΔAΔC-Fru structure reveals a snapshot of the discharging process of the reaction product, but it is still unclear how the structural difference of β-Fruf at subsite −1 is related to the catalytic mechanism of GH68 enzymes. In addition to loop 1, conformational changes in the N-terminal region, loop 2, loop 3, and loop 5 were observed, but these regions did not interact directly with Fruf (i) and Frup (ii). Unlike loop 1, the conformation of loop 8 in BiBftA ΔAΔC-Fru and in unliganded BiBftA ΔAΔC was almost identical ()).
Table 3. Numbers of amino acid residues in the loops at the rim of the active site funnel in some GH68 enzymes
Figure 6. Comparison of unliganded BiBftA ΔAΔC and BiBftA ΔAΔC-Fru structures. (a) The conformational change in BiBftA ΔAΔC upon binding of Fru. Colors: blue, loop 1 and loop 8 of unliganded BiBftA ΔAΔC; green, unliganded BiBftA ΔAΔC; red, loop 1 and loop 8 of BiBftA ΔAΔC-Fru; orange, BiBftA ΔAΔC-Fru; magenta, Fruf (i) and Frup (ii). (b) Stereo view of the active site of unliganded BiBftA ΔAΔC (blue) and BiBftA ΔAΔC-Fru (red). Fruf (i), Frup (ii), three catalytic residues (D100, D285, E377), and residues within 4 Å of Frup (ii) are shown

Implications for the structure–function relationship of GH68
The findings of this study suggest that BiBftA catalyzes transfructosylation to primarily produce 1-kestose from sucrose. The transfructosylation reactions of GH68 enzymes are divergent. BsSacB [Citation30,Citation36] and Bacillus megaterium levansucrase [Citation29] primarily produce the polysaccharide levan from sucrose, whereas MsFFase mainly catalyzes hydrolysis when only sucrose was present as the substrate [Citation37]. Despite the divergence of GH68 enzymes, the residues at subsites −1 and +1 are highly conserved [Citation8,Citation38]. It has been suggested that the lengths of the loops, especially loop 8, at the rim of the funnel of the catalytic β-propeller structure determine the chain lengths of the transfructosylation products [Citation24]. The lengths of the nine loops in BiBftA and GdLsdA are identical (, ), thus they both probably primarily produce the short length oligosaccharide 1-kestose. In this study, the conformational change in loop 1 in BiBftA upon binding Fru indicates that loop 1 may also be important in determining the manner of transfructosylation. The length of loop 1 is divergent in GH68 enzymes, and BiBftA Arg136 and His137, which are located in loop 1, interact with Frup (ii) via hydrogen bonds ()).
Although residues at subsites −1 and +1 are fully conserved in GH68 enzymes from Gram-negative bacteria, β-Fruf at subsite −1 binds in two distinct binding modes, the substrate mode and so-called the tilted binding mode [Citation12,Citation24]. The orientation of β-Fruf in the tilted binding mode is completely rolled over from the orientation in the substrate mode. To our knowledge, such a drastic change in sugar binding at subsite −1 has never been reported in other carbohydrate active enzymes other than GH68. This study shows the first structure of a GH68 enzyme in complex with a combination of β-Fruf and β-Frup in the catalytic pocket. There is, however, a possibility that the binding of Fruf (i) and Frup (ii) observed in this study is an artifact due to the crystallization condition which is highly different from the physiological one. It is unlikely that Frup (ii) functions as an acceptor molecule for the transfructosylation reaction because of the tilted binding of Fruf (i) at subsite −1.
Major members of the GH68 enzyme family produce levan after production of FOS. Analysis of levan production by BsSacB has shown that BsSacB produces a high-molecular weight levan and a low-molecular weight levan, probably through different mechanisms [Citation39–41]. However, the mechanisms of levan production remain unclear. Recently, a structure of Erwinia tasmaniensis levansucrase in complex with levanbiose has indicated that levanbiose is bound to an exposed pocket on the surface of the enzyme, and no levanbiose molecule is observed in the inner part of the β-propeller [Citation42]. Arg377 in Erwinia tasmaniensis levansucrase interacts with levanbiose via hydrogen bonds, and this Arg377 residue is conserved in BiBftA as Arg480, but no fructose molecule was observed near Arg480 in BiBftA. It also remains to be determined whether the two binding modes of β-Fruf at subsite −1 and the presence of β-Frup in the catalytic pocket are related to two distinct mechanisms of levan production.
Conclusions
In this study, the GH68 enzyme from B. indica subsp. indica NBRC 3744 was expressed in Escherichia coli, and the properties of the purified enzyme were investigated. The enzyme was identified as a β-fructosyltransferase, suggesting that the enzyme, BiBftA, may be useful for the production of FOS from sucrose. To improve the crystallinity of BiBftA, two putative flexible regions, residues 199–203 and residues 522–534, were removed by site-directed mutagenesis, and the variant enzyme, BiBftA ΔAΔC, was successfully crystallized. In the BiBftA ΔAΔC-Fru structure, one β-Fruf and one β-Frup were found in the catalytic pocket. The orientation of β-Fruf at subsite −1 was tilted when compared with the orientation observed in most GH68 enzymes. In addition, the binding of the two Fru molecules in BiBftA differed from the Fru binding observed in previous studies. Further studies are needed to elucidate why multiple binding modes of Fru are present in GH68 enzymes.
Author contributions
T. Tochio, K. Hirano, T. Fujii, K. Tamura, and T. Tonozuka planned the experiments. T. Fujii, K. Hirano, and K. Tamura constructed the expression plasmids. M. Nagaya crystallized the enzyme. M. Nagaya and T. Tonozuka determined the structure. J. Kitamura and R. Kawai determined the enzymatic properties. T. Tonozuka wrote the manuscript with support from T. Fujii, J. Kitamura, R. Kawai, and A. Nishikawa. All authors read and approved the final manuscript.
BiBftA_FigureS1.pdf
Download PDF (1.8 MB)Disclosure statement
The authors disclose the following conflicts of interest: Katsuaki Hirano, Tadashi Fujii, and Takumi Tochio work for B Food Science Co. Ltd., a manufacturer of sugars, and Keisuke Tamura works for MicroBiopharm Japan Co. Ltd., a manufacturer of biochemical reagents.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Lammens W, Le Roy K, Schroeven L, et al. Van den Ende W. Structural insights into glycoside hydrolase family 32 and 68 enzymes: functional implications. J Exp Bot. 2009;60:727–740.
- Velázquez-Hernández ML, Baizabal-Aguirre VM, Bravo-Patiño A, et al. Microbial fructosyltransferases and the role of fructans. J Appl Microbiol. 2009;106:1763–1778.
- Tochio T, Kadota Y, Tanaka T, et al. 1-Kestose, the smallest fructooligosaccharide component, which efficiently stimulates Faecalibacterium prausnitzii as well as Bifidobacteria in humans. Foods. 2018;7:140.
- Koga Y, Tokunaga S, Nagano J, et al. Age-associated effect of kestose on Faecalibacterium prausnitzii and symptoms in the atopic dermatitis infants. Pediatr Res. 2016;80:844–851.
- Tochio T, Kitaura Y, Nakamura S, et al. An alteration in the cecal microbiota composition by feeding of 1-kestose results in a marked increase in the cecal butyrate content in rats. PLoS One. 2016;11:e0166850.
- Garron ML, Henrissat B. The continuing expansion of CAZymes and their families. Curr Opin Chem Biol. 2019;53:82–87.
- Xie J, Cai K, Hu HX, et al. Structural analysis of the catalytic mechanism and substrate specificity of anabaena alkaline invertase InvA reveals a novel glucosidase. J Biol Chem. 2016;291:25667–25677.
- Nagaya M, Kimura M, Gozu Y, et al. Crystal structure of a β-fructofuranosidase with high transfructosylation activity from Aspergillus kawachii. Biosci Biotechnol Biochem. 2017;81:1786–1795.
- Becking JH. Studies on nitrogen-fixing bacteria of the genus Beijerinckia: I. Geographical and ecological distribution in soils. Plant Soil. 1961;14:49–81.
- Tamas I, Dedysh SN, Liesack W, et al. Complete genome sequence of Beijerinckia indica subsp. indica. J Bacteriol. 2010;192:4532–4533.
- Martínez-Fleites C, Ortíz-Lombardía M, Pons T, et al. Crystal structure of levansucrase from the Gram-negative bacterium Gluconacetobacter diazotrophicus. Biochem J. 2005;390:19–27.
- Tonozuka T, Tamaki A, Yokoi G, et al. Crystal structure of a lactosucrose-producing enzyme, Arthrobacter sp. K-1 β-fructofuranosidase. Enzyme Microb Technol. 2012;51:359–365.
- Ohta Y, Hatada Y, Hidaka Y, et al. Enhancing thermostability and the structural characterization of Microbacterium saccharophilum K-1 β-fructofuranosidase. Appl Microbiol Biotechnol. 2014;98:6667–6677.
- Petersen TN, Brunak S, von Heijne G, et al. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786.
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132.
- Evans P. Scaling and Assessment of Data Quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82.
- Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242.
- Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66:22–25.
- Langer GG, Hazledine S, Wiegels T, et al. Visual automated macromolecular model building. Acta Crystallogr D Biol Crystallogr. 2013;69:635–641.
- Murshudov GN, Skubák P, Lebedev AA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367.
- Emsley P, Lohkamp B, Scott WG, et al. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501.
- Lovell SC, Davis IW, Arendall WB 3rd, et al. Structure validation by Cα geometry: φ, ψ, and Cβ deviation. Proteins. 2003;50:437–450.
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786.
- Wuerges J, Caputi L, Cianci M, et al. The crystal structure of Erwinia amylovora levansucrase provides a snapshot of the products of sucrose hydrolysis trapped into the active site. J Struct Biol. 2015;191:290–298.
- Meng G, Fütterer K. Structural framework of fructosyl transfer in Bacillus subtilis levansucrase. Nat Struct Mol Biol. 2003;10:935–941.
- Dale GE, Kostrewa D, Gsell B, et al. Crystal engineering: deletion mutagenesis of the 24 kDa fragment of the DNA gyrase B subunit from Staphylococcus aureus. Acta Crystallogr D Biol Crystallogr. 1999;55:1626–1629.
- Derewenda ZS. Application of protein engineering to enhance crystallizability and improve crystal properties. Acta Crystallogr D Biol Crystallogr. 2010;66:604–615.
- Caputi L, Nepogodiev SA, Malnoy M, et al. Biomolecular characterization of the levansucrase of Erwinia amylovora, a promising biocatalyst for the synthesis of fructooligosaccharides. J Agric Food Chem. 2013;61:12265–12273.
- Strube CP, Homann A, Gamer M, et al. Polysaccharide synthesis of the levansucrase SacB from Bacillus megaterium is controlled by distinct surface motifs. J Biol Chem. 2011;286:17593–17600.
- Yamamoto S, Iizuka M, Tanaka T, et al. The mode of synthesis of levan by Bacillus subtilis levansucrase. Agric Biol Chem. 1985;49:343–349.
- Holm L. DALI and the persistence of protein shape. Protein Sci. 2020;29:128–140.
- Naumoff DG. Furanosidase superfamily: search of homologues. Mol Biol. 2012;46:354–360.
- Pijning T, Anwar MA, Böger M, et al. Crystal structure of inulosucrase from Lactobacillus: insights into the substrate specificity and product specificity of GH68 fructansucrases. J Mol Biol. 2011;412:80–93.
- He C, Yang Y, Zhao R, et al. Rational designed mutagenesis of levansucrase from Bacillus licheniformis 8-37-0-1 for product specificity study. Appl Microbiol Biotechnol. 2018;102:3217–3228.
- Ni D, Xu W, Zhu Y, et al. Inulin and its enzymatic production by inulosucrase: characteristics, structural features, molecular modifications and applications. Biotechnol Adv. 2019;37:306–318.
- Porras-Domínguez JR, Ávila-Fernández Á, Miranda-Molina A, et al. Bacillus subtilis 168 levansucrase (SacB) activity affects average levan molecular weight. Carbohydr Polym. 2015;132:338–344.
- Fujita K, Hara K, Hashimoto H, et al. Purification and some properties of beta-fructofuranosidase I from Arthrobacter sp. K-1. Agric Biol Chem. 1990;54:913–919.
- Ortiz-Soto ME, Porras-Domínguez JR, Seibel J, et al. A close look at the structural features and reaction conditions that modulate the synthesis of low and high molecular weight fructans by levansucrases. Carbohydr Polym. 2019;219:130–142.
- Raga-Carbajal E, Carrillo-Nava E, Costas M, et al. Size product modulation by enzyme concentration reveals two distinct levan elongation mechanisms in Bacillus subtilis levansucrase. Glycobiology. 2016;26:377–385.
- Raga-Carbajal E, López-Munguía A, Alvarez L, et al. Understanding the transfer reaction network behind the non-processive synthesis of low molecular weight levan catalyzed by Bacillus subtilis levansucrase. Sci Rep. 2018;8:15035.
- Ortiz-Soto ME, Porras-Domínguez JR, Seibel J, et al. A close look at the structural features and reaction conditions that modulate the synthesis of low and high molecular weight fructans by levansucrases. Carbohydr Polym. 2019;219:130–142.
- Polsinelli I, Caliandro R, Demitri N, et al. The structure of sucrose-soaked levansucrase crystals from Erwinia tasmaniensis reveals a binding pocket for levanbiose. Int J Mol Sci. 2020;21:83.