
ABSTRACT
Discovery of small-molecule inducers of unique phenotypic changes combined with subsequent target identification often provides new insights into cellular functions. Here, we applied integrated profiling based on cellular morphological and proteomic changes to compound screening. We identified an indane derivative, NPD9055, which is mechanistically distinct from reference compounds with known modes of action. Employing a chemical proteomics approach, we then showed that NPD9055 binds subunits of heterotrimeric G-protein Gi. An in vitro [35S]GTPγS-binding assay revealed that NPD9055 inhibited GDP/GTP exchange on a Gαi subunit induced by a G-protein-coupled receptor agonist, but not on another G-protein from the Gαs family. In intact HeLa cells, NPD9055 induced an increase in intracellular Ca2+ levels and ERK/MAPK phosphorylation, both of which are regulated by Gβγ, following its dissociation from Gαi. Our observations suggest that NPD9055 targets Gαi and thus regulates Gβγ-dependent cellular processes, most likely by causing the dissociation of Gβγ from Gαi.
GRAPHICAL ABSTRACT
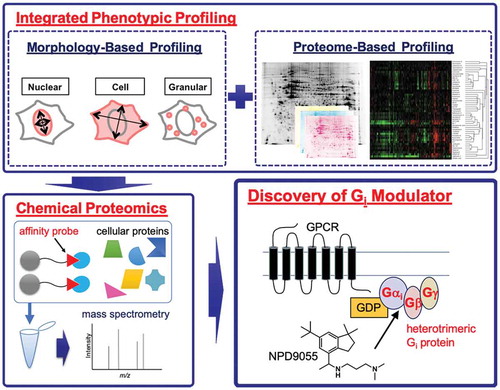
By combining integrated phenotypic profiling and chemical proteomics, a small-molecule modulator of heterotrimeric Gi-protein was discovered.
Discovery of small-molecule modulators of proteins has accelerated the understanding of cellular processes at the molecular level and is also important for drug discovery. However, available small-molecule probes for many proteins are still lacking[Citation1]. Therefore, exploring uncharacterized chemotypes with unique biological activity and connecting them with the target molecules is of great importance. Target-agnostic phenotypic screening, when compared to target-based screening, is a less biased approach that sometimes enables identification of unexpected targets necessary for achieving desired phenotypes[Citation2]. Indeed, phenotypic screening combined with subsequent target identification has led to the identification of unique bioactive compounds and their target molecules, which has contributed to uncovering cellular functions and developing new drugs for the treatment of diseases [Citation3,Citation4].
Generally, target identification is a time-consuming, but critical process of elucidating mechanisms of action of bioactive compounds discovered through phenotypic screening. Techniques for target identification are divided into direct and indirect approaches [Citation5,Citation6]. Chemical proteomics, i.e., affinity-based enrichment of proteins, is a direct approach that is widely used in the identification of cellular targets of bioactive compounds. Indirect approaches, such as profiling for mechanism elucidation, have also been developed. We have previously established two profiling techniques, MorphoBase and ChemProteoBase, to characterize bioactive compounds based on the induced cellular morphological and proteomic changes, respectively [Citation7,Citation8]. Based on comparisons with 207 and 120 reference compounds with known modes of action, respectively (see Table S1 in Ref. 7; and Table S1), both methods have been utilized for target prediction of uncharacterized bioactive compounds[Citation9]. More recently, a highly multiplexed morphology-based profiling method, termed Cell Painting assay, was established as a complementary approach based on similar logic[Citation10].
G-protein-coupled receptors (GPCRs), the largest family of cell surface receptors, regulate a wide range of physiological and pathological processes, and heterotrimeric G-proteins are key mediators of GPCR-regulated signaling [Citation11,Citation12]. Heterotrimeric G-proteins consist of Gα, Gβ, and Gγ subunits, and are regulated via an activation/inactivation cycle, as follows: (i) Gα usually exists in a GDP-bound state and forms a trimer with Gβ and Gγ; (ii) an agonist-activated GPCR stimulates the exchange of bound GDP for GTP on the Gα subunit; (iii) GTP binding to Gα leads to Gβγ dissociation, and the subsequent binding of Gα and Gβγ to downstream effectors; (iv) the signal is terminated by GTP hydrolysis, resulting in the reformation of an inactive heterotrimer. Based on sequence similarity of the α subunit, G-proteins are divided into four families, Gs, Gi/Go, Gq/G11, and G12/G13. Since GPCRs are major targets for therapeutic drugs, G-proteins have also attracted attention [Citation13,Citation14].
In the present study, we integrated MorphoBase and ChemProteoBase profiling systems for compound screening, and thereby identified NPD9055, an indane derivative that was mechanistically distinct from the reference compounds. In addition, our chemical proteomics approach and subsequent biochemical assays suggested that NPD9055 directly targets Gαi and modulates Gβγ-regulated cellular processes.
Results and discussion
The screening strategy for mechanistically unique bioactive compounds was to identify compounds that induced phenotypic changes distinct from those induced by well-characterized reference compounds in both MorphoBase and ChemProteoBase profiling systems. Since the throughput of both profiling methods is not high, we needed to narrow down test compounds prior to the profiling analyses. To this end, we first performed cytotoxicity assays using a panel of cancer cell lines, because morphology and the proteome are, in many cases, both affected during the process of cell death or cell-cycle arrest. In parallel, compound-induced morphological changes were observed under a microscope during the cytotoxicity assays. After testing 5472 compounds from the RIKEN Natural Products Depository (NPDepo) library, we found that an indane derivative, termed NPD9055 (1) (), was toxic against various cancer cell lines (IC50 values, 1.8–18 μM; Table S2), and notably induced morphological changes in v-src-transformed normal rat kidney (srcts-NRK) and human cervical cancer HeLa cells (Figure S1). Therefore, we next performed MorphoBase profiling for NPD9055. Using 71 parameters extracted from the nuclei, whole cells, and granules of NPD9055-treated srcts-NRK and HeLa cells, we quantified the morphological changes. The mechanistic uniqueness of NPD9055 was suggested by the probability scores for 15 well-validated target classes and Euclidean distances to the 207 reference compounds (Table S3). Next, we performed ChemProteoBase profiling as a complementary and more informative analysis based on changes at the molecular level. After the treatment of HeLa cells with NPD9055, changes in 296 protein levels were analyzed. We found increases in the levels of multiple proteins localized to the mitochondria or the endoplasmic reticulum (ER) (Figure S2; Table S4). When the patterns of changes in protein expression level are compared, two compounds with a similar mode of action usually have a cosine similarity score >0.7. To identify compounds that have a mode of action similar to that of NPD9055, we analyzed the pattern of changes in protein expression level between NPD9055 and our 120 reference compounds. However, we did not identify any compounds that have a cosine similarity score with NPD9055 > 0.7, indicating that the mode of action of NPD9055 is unique among our reference compounds ( and S3). This mechanistic uniqueness of NPD9055 suggested by the integrated phenotypic profiling prompted us to elucidate its mechanism of action.
Figure 1. Indane derivative NPD9055 has a unique mode of action

To identify the target molecule of NPD9055, we analyzed proteins binding to NPD9055 by using a chemical proteomics approach that employs affinity purification and subsequent mass spectrometric protein identification. To design a suitable affinity probe, we first synthesized and tested a small number of NPD9055 analogs. We found that the methyl group at the tertiary amino group can be extended to obtain compound 2 without the loss of cytotoxicity (Table S5). After testing compound 3, we synthesized a biotin-tagged pull-down probe 4; the probe exhibited a slight loss of cytotoxicity, most likely because of reduced cell membrane permeability (; Table S5). Using affinity probe 4 together with streptavidin beads, we performed pull-down assays using HeLa cell lysates in the presence or absence of unmodified NPD9055 as a competitor. Subsequent identification of the co-precipitated proteins by mass spectrometry and label-free quantification yielded six candidates whose intensity was significantly reduced in the presence of the competitor (): Gαi2 and Gβ2, which are components of a heterotrimeric G-proteinCitation[11]; nucleoporins Nup160, Nup37, and Nup85, which form the nuclear pore Nup107-160 complexCitation[15]; and dynamin-like 120 kDa protein. Therefore, the six identified proteins were divided into three groups: (i) components of a heterotrimeric G-protein, (ii) components of the nuclear pore, and (iii) dynamin-like 120 kDa protein (Figure S4a). We have previously established a method for compound immobilization onto a matrix by a photoaffinity reaction that is functional group-independent[Citation16]. Using this method, we prepared NPD9055-immobilized photoaffinity beads. To confirm the results of the experiment with the biotin-tagged pull-down probe (), we performed a competition pull-down assay with the photoaffinity beads. From the six potential binding proteins, only Gαi2 and Gβ2 (encoded by GNAI2 and GNB2, respectively) were identified in the photoaffinity bead experiment. Other G-protein subunits, Gαi1, Gαi3, Gβ1, Gβ4, and Gγ12 (encoded by GNAI1, GNAI3, GNB1, GNB4, and GNG12, respectively), were also identified as potential NPD9055-binding proteins in the same experiment (Figures S4b and S4c). To confirm the physical engagement of Gαi2 and Gβ2 by NPD9055, we then performed a cellular thermal shift assay, which is based on the changes in thermal stability of small-molecule protein targets[Citation17]. NPD9055 slightly disrupted the thermal stability of Gαi2 in a HeLa cell lysate with a shift in the melting temperature of – 1.1°C (,d). Therefore, we prioritized G-proteins, especially the ones from the Gi subfamily, for further in-depth investigation.
Figure 2. Gi subunits are NPD9055-binding proteins

During G-protein activation, GPCR agonist-stimulated GDP/GTP exchange occurs on Gα[Citation11]. To monitor this process, we previously established an in vitro [35S]GTPγS-binding assay, using a membrane fraction from transgenic silkworm expressing a human GPCR-Gα fusion protein to measure GPCR agonist-induced exchange of GDP to [35S]GTPγS on Gα [Citation18,Citation19]. Although this system lacks the Gβγ subunits, it enables examination of the direct effect of small molecules on Gα. Using the silkworm experimental system, we first investigated whether NPD9055 induced the GDP/GTP exchange on Gαi2 coupled to μ-opioid receptor in the absence of GPCR agonist. However, up to 100 µM NPD9055 did not induce GDP/GTP exchange on Gαi2 unlike morphine, an agonist of μ-opioid receptor (). Therefore, we next examined the influence of NPD9055 on GPCR agonist-stimulated GDP/GTP exchange on Gαi2. NPD9055 inhibited morphine-stimulated GDP/GTP exchange on Gαi2 coupled to μ-opioid receptor (), suggesting a direct interaction of NPD9055 with Gαi2. To assess the selectivity of inhibition, we also examined the effect of NPD9055 on Gαs, a protein from another G-protein family, using a membrane fraction from silkworms expressing human Gαs coupled to G-protein-coupled bile acid receptor (GPBA). NPD9055 did not affect taurolithocholic acid (TLC)-stimulated GDP/GTP exchange on Gαs (). Collectively, these observations suggest that NPD9055 directly and selectively targets Gαi, and inhibits GPCR agonist-stimulated GDP/GTP exchange on Gαi.
Figure 3. NPD9055 inhibits GPCR agonist-induced GDP/GTP exchange on Gαi in vitro.

Since NPD9055 inhibited the GPCR agonist-stimulated GDP/GTP exchange on Gαi2 in vitro (), we next investigated whether NPD9055 affected Gi-regulated cellular processes. Following the GPCR agonist-stimulated GDP/GTP exchange on Gαi and the subsequent dissociation of Gαi from Gβγ, both components separately modulate their effector molecules[Citation11]. One of the known effectors of Gαi is Rap1GAP2, which inactivates Rap1, resulting in the phosphorylation of ERK/MAPK[Citation20]. On the other hand, Gβγ regulates phospholipase Cβ, which hydrolyzes phosphatidylinositol 4,5-bisphosphate to the second messengers inositol 1,4,5-trisphosphate (IP3) and diacylglycerol[Citation21]. IP3 stimulates Ca2+ release from the ER via the IP3 receptor on the ER membrane[Citation22]. Furthermore, both IP3 and diacylglycerol activate protein kinases C, resulting in the phosphorylation of ERK/MAPK[Citation23]. We first examined the influence of NPD9055 on Ca2+ homeostasis, and found that NPD9055 increased the intracellular Ca2+ levels directly after its addition to HeLa cells (). To investigate the source of the increased Ca2+ levels, we performed another Ca2+ assay in the presence or absence of extracellular Ca2+. NPD9055 increased the intracellular Ca2+ levels even in the absence of extracellular Ca2+ (), suggesting that the increased Ca2+ levels are at least partly derived from intracellular organelle(s), such as the ER. We next investigated whether NPD9055 affected the phosphorylation level of ERK/MAPK, and found that NPD9055 induced ERK/MAPK phosphorylation 10 min after its addition to HeLa cells (). These observations suggest that NPD9055 modulates Gβγ-regulated cellular processes, such as Ca2+ release and ERK/MAPK phosphorylation.
Figure 4. NPD9055 affects Gβγ-regulated cellular processes

To date, several G-protein modulators targeting Gαi or Gβγ have been reported [Citation13,Citation14]. One of the most recognized Gαi modulators is pertussis toxin produced by the bacterium Bordetella pertussis. Pertussis toxin catalyzes ADP-ribosylation of the key C-terminal cysteine residue in the Gαi subfamily, and thereby interferes with the activation of Gi mediated by an agonist-bound GPCR[Citation24]. Pertussis toxin inhibits Ca2+ level increase induced by a chemotactic peptide formyl-methionyl-leucyl-phenylalanine in differentiated HL-60 cells[Citation25]. Since NPD9055 increased the intracellular Ca2+ levels (), NPD9055 seems to be mechanistically distinct from pertussis toxin. Another related G-protein modulator is Surfen/NSC12155. Gβγ binds to many effectors at a common “hot spot” that is obscured by GDP-bound Gα in the inactive state, and Surfen was identified as binding to the hot spot[Citation26]. Surfen caused the dissociation of Gβγ from GDP-bound Gα without inducing a nucleotide exchange on Gα in vitro, and induced Ca2+ release and ERK/MAPK phosphorylation in differentiated HL-60 cells[Citation26]. Based on these previous findings and on data presented in the current study (-), we hypothesized that the Gαi-binder NPD9055 was similar to Surfen in terms of the mechanism of action. However, unlike NPD9055, Surfen did not increase the intracellular Ca2+ levels, induce ERK/MAPK phosphorylation, or was not cytotoxic up to 30 µM, at least in HeLa cells (Figure S5), suggesting a different mechanism of action. In addition, neither Gallein (a direct inhibitor of Gβγ) nor UK14304 (an agonist of Gi-coupled α2-adrenoceptor expressed in HeLa cells) [Citation27] showed any effect in the same assays (Figure S5).
Nevertheless, it is widely recognized that Gβγ can dissociate from Gα and regulate Gβγ effectors without an agonist-activated GPCR or nucleotide exchange. Some proteins, such as activator of G-protein signaling 3 (AGS3), share the G-protein regulatory (GPR)/GoLoco motif consisting of 19 amino acid residues, and interact with GDP-bound Gαi/o via that motif. Proteins belonging to the GoLoco family act as GDP dissociation inhibitors that selectively interfere with the GDP/GTP exchange on Gαi/o [Citation28–30]. Importantly, despite the inhibitory activity of GDP dissociation, the GoLoco peptide derived from AGS3 causes the dissociation of Gβ1γ2 from Gαi1[Citation31]. A crystal structure analysis revealed that the GoLoco motif interacts with the nucleotide-binding pocket of Gαi1, suggesting that Gβγ- and GoLoco-binding are mutually exclusive[Citation32]. Based on the above reports on the GoLoco motif, we propose a mechanism of action for NPD9055, namely, that is (i) targeting GDP-bound Gαi, (ii) dissociation of Gβγ from Gαi, and (iii) activation of Gβγ-regulated signaling.
Further, to the best of our knowledge, neither the nuclear pore Nup107-160 complex nor dynamin-like 120 kDa protein indicated by chemical proteomics () (neither of them was identified as NPD9055-binding in the pull-down assay using the photoaffinity beads) has been reported as a Ca2+ level modulator. Therefore, these cellular components do not appear to be the targets responsible for the NPD9055-associated phenotype. Taken together, our observations suggest that Gi is the primary target of NPD9055. Further experimental comparison with known G-protein modulators will provide additional insight into the mechanism of action of NPD9055.
Conclusion
By integrating morphology- and proteome-based profiling systems, we successfully identified the biologically uncharacterized indane derivative NPD9055. In addition, employing a chemical proteomics technique, we connected the indane chemotype with its cellular target Gαi. Since the dysregulation of G-protein-mediated signaling is involved in various diseases including cancer, exploration of the available small-molecule modulators that directly target G-proteins is of great interest. NPD9055 may serve as a chemical tool to uncover the functions of Gαi, Gβγ, and related molecules. Furthermore, by combining integrated phenotypic profiling and chemical proteomics, the developed strategy may open up new opportunities for drug discovery targeting unexplored molecules.
Author contributions
T.K., Y.F., M.M., P.J., M.U., J.W., and S.T. performed the biological experiments and analyzed the data. E.S. synthesized NPD9055 analogs. Y.K. prepared photoaffinity beads. T.K., Y.F., H.W., and H.O. conceived and designed the experiments. H.O., H.W., N.W., and S.Z. supervised the research project. T.K., Y.F., and H.O. wrote the manuscript with the support of other authors.
Supporting information
The Supporting Information is available free of charge on the journal website. Materials and methods; supporting tables and figures. (PDF and Excel files)
Table_S8._Proteomic_results_of_the_competition_pull-down_using_NPD9055-immobilized_photoaffinity_beads.xlsx
Download MS Excel (581.3 KB)Table_S7._Proteomic_results_of_the_competition_pull-down_using_affinity_probe_4__biological_replicate_2_.xlsx
Download MS Excel (1.9 MB)Table_S6._Proteomic_results_of_the_competition_pull-down_using_affinity_probe_4__biological_replicate_1_.xlsx
Download MS Excel (1.8 MB)Table_S4.NPD9055-induced_expression_change_and_subcellular_localization_of_each_protein_spot.xlsx
Download MS Excel (29.1 KB)Final_Kawamura_SI_BBB.docx
Download MS Word (2.8 MB)Acknowledgments
We thank M. Tanaka, H. Aono, K. Honda, and T. Nogawa for technical support, T. Shimizu and M. Kawatani for suggestions (Chem. Biol., RIKEN CSRS), and the RIKEN NPDepo for providing compounds. We also thank Y. Uehara (Iwate Medical University) for providing srcts-NRK cells.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Edfeldt FN, Folmer RH, Breeze AL. Fragment screening to predict druggability (ligandability) and lead discovery success. Drug Discov Today. 2011;16(7–8):284–287.
- Eggert US. The why and how of phenotypic small-molecule screens. Nat Chem Biol. 2013;9(4):206–209.
- Moffat JG, Rudolph J, Bailey D. Phenotypic screening in cancer drug discovery - past, present and future. Nat Rev Drug Discov. 2014;13(8):588–602.
- Wagner BK, Schreiber SL. The power of sophisticated phenotypic screening and modern mechanism-of-action methods. Cell Chem Biol. 2016;23(1):3–9.
- Kapoor S, Waldmann H, Ziegler S. Novel approaches to map small molecule-target interactions. Bioorg Med Chem. 2016;24(15):3232–3245.
- Ziegler S, Pries V, Hedberg C, et al. Target identification for small bioactive molecules: finding the needle in the haystack. Angew Chem Int Ed Engl. 2013;52(10):2744–2792.
- Futamura Y, Kawatani M, Kazami S, et al. Morphobase, an encyclopedic cell morphology database, and its use for drug target identification. Chem Biol. 2012;19(12):1620–1630.
- Muroi M, Kazami S, Noda K, et al. Application of proteomic profiling based on 2D-DIGE for classification of compounds according to the mechanism of action. Chem Biol. 2010;17(5):460–470.
- Muroi M, Futamura Y, Osada H. Integrated profiling methods for identifying the targets of bioactive compounds: morphoBase and ChemProteoBase. Nat Prod Rep. 2016;33(5):621–625.
- Bray MA, Singh S, Han H, et al. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat Protoc. 2016;11(9):1757–1774.
- Milligan G, Kostenis E. Heterotrimeric G-proteins: a short history. Br J Pharmacol. 2006;147(Suppl 1):S46–55.
- Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005;85(4):1159–1204.
- Smrcka AV. Molecular targeting of Gα and Gβγ subunits: a potential approach for cancer therapeutics. Trends Pharmacol Sci. 2013;34(5):290–298.
- Ayoub MA. Small molecules targeting heterotrimeric G proteins. Eur J Pharmacol. 2018;826:169–178.
- Loïodice I, Alves A, Rabut G, et al. The entire Nup107-160 complex, including three new members, is targeted as one entity to kinetochores in mitosis. Mol Biol Cell. 2004;15(7):3333–3344.
- Kanoh N, Honda K, Simizu S, et al. Photo-cross-linked small-molecule affinity matrix for facilitating forward and reverse chemical genetics. Angew Chem Int Ed Engl. 2005;44(23):3559–3562.
- Martinez Molina D, Jafari R, Ignatushchenko M, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341(6141):84–87.
- Enomoto R, Kurosawa A, Nikaido Y, et al. A novel partial agonist of GPBA reduces blood glucose level in a murine glucose tolerance test. Eur J Pharmacol. 2017;814:130–137.
- Nikaido Y, Kurosawa A, Saikawa H, et al. In vivo and in vitro evaluation of novel μ-opioid receptor agonist compounds. Eur J Pharmacol. 2015;767:193–200.
- Mochizuki N, Ohba Y, Kiyokawa E, et al. Activation of the ERK/MAPK pathway by an isoform of rap1GAP associated with G alpha(i). Nature. 1999;400(6747):891–894.
- Camps M, Carozzi A, Schnabel P, et al. Isozyme-selective stimulation of phospholipase C-beta 2 by G protein beta gamma-subunits. Nature. 1992;360(6405):684–686.
- Hanson CJ, Bootman MD, Roderick HL. Cell signalling: IP3 receptors channel calcium into cell death. Curr Biol. 2004;14(21):R933–935.
- Kolch W, Heidecker G, Kochs G, et al. Protein kinase C alpha activates RAF-1 by direct phosphorylation. Nature. 1993;364(6434):249–252.
- Katada T. The inhibitory G protein G(i) identified as pertussis toxin-catalyzed ADP-ribosylation. Biol Pharm Bull. 2012;35(12):2103–2111.
- Krause KH, Schlegel W, Wollheim CB, et al. Chemotactic peptide activation of human neutrophils and HL-60 cells. Pertussis toxin reveals correlation between inositol trisphosphate generation, calcium ion transients, and cellular activation. J Clin Invest. 1985;76(4):1348–1354.
- Surve CR, Lehmann D, Smrcka AV. A chemical biology approach demonstrates G protein βγ subunits are sufficient to mediate directional neutrophil chemotaxis. J Biol Chem. 2014;289(25):17791–17801.
- Chan AS, Lau WW, Szeto AC, et al. Differential Regulation of CXCL8 Production by Different G Protein Subunits with Synergistic Stimulation by Gi- and Gq-Regulated Pathways. J Mol Biol. 2016;428(19):3869–3884.
- De Vries L, Fischer T, Tronchère H, et al. Activator of G protein signaling 3 is a guanine dissociation inhibitor for Galpha i subunits. Proc Natl Acad Sci U S A. 2000;97(26):14364–14369.
- Natochin M, Lester B, Peterson YK, et al. AGS3 inhibits GDP dissociation from galpha subunits of the Gi family and rhodopsin-dependent activation of transducin. J Biol Chem. 2000;275(52):40981–40985.
- Natochin M, Gasimov KG, Artemyev NO. Inhibition of GDP/GTP exchange on G alpha subunits by proteins containing G-protein regulatory motifs. Biochemistry. 2001;40(17):5322–5328.
- Ghosh M, Peterson YK, Lanier SM, et al. Receptor- and nucleotide exchange-independent mechanisms for promoting G protein subunit dissociation. J Biol Chem. 2003;278(37):34747–34750.
- Kimple RJ, Kimple ME, Betts L, et al. Structural determinants for GoLoco-induced inhibition of nucleotide release by Galpha subunits. Nature. 2002;416(6883):878–881.