
ABSTRACT
There is experimental and clinical data to indicate the contribution of immune-escape mechanisms in relapsed/refractory pediatric leukemia. Studies have shown the accumulation of mutations that translate to peptides containing tumor-specific epitopes (neoantigens). The effectiveness of neoantigen-based vaccines has been shown in several clinical trials in adults. Though the initial results are encouraging, this knowledge must be developed to account for the uniqueness of pediatric cancer biology. We have completed the initial proof-of-concept analysis on a high-risk pediatric leukemia specimen and identified usable neoantigen sequences. We describe this approach, including the bioinformatics method and experimental model to verify their function that can be further broadened for personalized neoantigen prediction and testing for the generation of anticancer vaccines against high-risk pediatric leukemias.
Immunotherapy has emerged as an effective therapy due to low toxicity, fewer side effects and effective clinical responses in several adult cancers. Neoantigens that are created by tumor-specific non-synonymous mutations carry the potential to serve as effective targets for immunotherapy.Citation1 As neoantigens are expressed only in tumor cells, they are exempted from central tolerance and can elicit a robust immune response which is specific to tumor cells.Citation2 Next-generation sequencing and development of algorithms for the prediction of MHC-I binding epitopes have enabled the detection of tumor-specific mutations which can be used for the generation of personalized therapeutic cancer vaccines.Citation3 These neoantigen-based vaccines activate cytotoxic T cells that could specifically recognize and kill cancer cells and provide immunological memory that can lead to long-term protection against the recurrence of cancer.Citation4 Clinical trials testing personalized neoantigen-based vaccines have been conducted in adult melanoma, glioblastoma, and breast cancer patients.Citation3 These trials demonstrated that neoantigen vaccines were able to induce tumor-specific CD4+ and CD8+ T cell response, depicting their potential for further development into a mainstream therapy.Citation5–8
Despite the promising results in adult cancers, the antitumor potential of neoantigen vaccines has not been well studied in pediatric cancers. In this study, we report personalized neoantigen peptides generated for a pediatric patient diagnosed with high-risk T cell acute lymphocytic leukemia (T-ALL) showing effective in vitro CD8+ T cell activity against the leukemic blasts, providing a practical treatment backbone for future novel immune therapeutic approaches for children with high-risk leukemias.
This study was approved by the local Institutional Review Board [#HREBA.CC-160286_REN5] and all patient samples were collected following parental informed consent. Bone marrow specimen from a 13-year-old who was diagnosed with T-ALL and presented with a mediastinal mass was collected during the diagnostic procedure and a leukemic blast enriched population was prepared by Ficoll (GE Healthcare) density gradient centrifugation. Peripheral blood cells were obtained from blood collected one year following completion of treatment when the child had normal blood cell counts and no evidence of disease. These cells were used to generate normal RNA sequence and to perform ELISPOT assays as described below.
For the whole exome analysis, RNA was extracted from the patient’s leukemic blasts and normal blood cells using the Qiagen RNeasy kit (Qiagen). The sequencing and bioinformatics analysis to identify neoantigen peptides were performed by MedGenome as described previously.Citation9 Briefly, the Agilent SureSelect Human All Exome kit (50 Mb panel size) was used to generate libraries and sequence them on the HiSeq 2500 platform (Illumina Inc.) to generate 2 × 75 bp paired-end data. Illumina Stranded mRNA library preparation kit with PolyA selection was used for RNA sequencing (Illumina Inc.). RNA-seq reads aligned to the human genome version GRCh38 using STAR v2.4.1 were used to compute the gene level expression counts. This involved counting the number of reads aligning concordantly within a pair and uniquely to each gene locus using gene models defined by NCBI and Ensembl gene annotations, and RefSeq mRNA database. The gene expression was estimated using Cufflinks v2.2.1.
Raw reads were assessed by FastQC for Phred score quality. Adapter trimming was performed by the cutadapt program (v2.5) (https://cutadapt.readthedocs.io/en/stable/index.html). Adapter-trimmed reads were then analyzed using the GATK good-practice workflow (Genome Analysis Toolkit, Broad Institute). Briefly, trimmed reads were aligned against human genome hg19 using BWA (Burrows-Wheeler Aligner)-MEM (Maximal Exact Matches) (v0.7.12).Citation10 Aligned reads were further sorted and indexed using Samtools (v1.2).Citation11 These binary alignment map (BAM) files were assessed for PCR duplicates, and likely duplicates were flagged using the MarkDuplicates tool of the Picard package (v1.140) in the GATK tool suite (Genome Analysis Toolkit, Broad Institute). The sequences were further realigned around known indels from population frequency databases and the base quality score was recalibrated. The somatic variants were identified using the Strelka2 program (v2.0.17) and annotated using the variant annotation pipeline VariMAT (v2.4.1), which were further filtered, and only on-target variants were considered to determine the Tumor Mutational Burden (TMB). Known germline variants reported in the population frequency databases such as dbSNP, 1,000 G and ExAC at ≥0.1% were removed.Citation12–14 The total number of exonic variants and Indels are shown in
Potential immunogenic neoepitopes were predicted from somatic mutations by generating overlapping 9-mer peptides with the mutant amino acid occupying each of the nine positions of the peptide. Using patient’s HLA, the binding affinity of each of the 9-mer peptide was determined using NetMHCpan4.0.Citation15 Peptides with binding affinities ≤1000 nM were selected for the further prioritization of epitopes for immune-proteasomal processing using NetChop 3.1 and transporter associated with antigen processing (TAP) binding using the TAP processing module of IEDB. Peptides having proteasomal cleavage score >10 and TAP binding affinity <0.5 were selected. The expression of the mutant transcript was assessed and expression >1 FPKM was used as a cutoff and the resultant peptides were rank ordered based on the composite immunogenicity score calculated from MHC class I binding affinities, expression of the somatic variants at the RNA level and TCR binding predictions.
The prioritized neoepitope peptides were assayed for T cell response using a primed CD8+ T cell and leukemia co-culture assay. Peripheral blood mononuclear cells (PBMCs) were isolated from blood at one year post completion of treatment, using a standard Ficoll gradient. CD8+ T cells were isolated from PBMCs (RPMI 1640 + 10% FBS supplemented with IL-7 10 ng/ml) using the MACS cell separation method (Miltenyi). Monocytes from PBMCs were isolated by the adherence method and differentiated into dendritic cells (DCs) in RPMI 1640 + 10% FBS supplemented with GM-CSF 100 ng/ml and IL-4 25 ng/ml. DCs were pulsed overnight with the neoantigen peptides synthesized in a GMP facility (Shanghai Royobiotech Ltd.) at a concentration of 50 μg/ml. CD8+ T cells were co-cultured with the pulsed DCs for fivedays (1:10 DC, CD8+ ratio) in RPMI 1640 + 10% FBS supplemented with IL-2 10 ng/ml for priming. The primed CD8+T-cells were then co-cultured with thawed leukemia cells from the patient in a 1:1 ratio (25,000 total cells/well) in RPMI 1640 + 10% FBS supplemented with IL-7 and IL-2 and interferon gamma (IFN-γ) production was analyzed by the ELISPOT assay (R&D systems) following the manufacturer’s protocol.
The RNA seq analysis identified the class I HLAs for the patient to be HLA-B07:02, HLA-C07:02, HLA-B58:01, HLA-A02:01, HLAC03:02, HLA-A01:01. The whole exome sequencing of the patient’s leukemia cells and PBMCs identified somatic mutations which were orthogonally validated and assessed for expression of the mutated alleles by RNA-seq (). Our analysis showed that >99% of the total reads aligned to the reference genome for each sample. The average insert size of the aligned reads after applying filters such as for mapping quality, duplicate %, and cross mapping was ~160bp. summarizes the analysis workflow for exomes and RNA sequencing data.
Table 1. Data summary of exome and RNA sequencing. Tables summarizing the (A) whole exome and (B) RNA sequencing data from the patient’s leukemia cells (Initial tumor) and normal PBMCs (Final normal)
Table 2. Read alignment summary for exomes and RNA sequencing. Tables summarizing alignment data from exome sequencing (A) and RNA sequencing (B) of patient’s leukemia cell (Initial tumor) and normal PBMCs (Final normal)
About 80% of exome and RNA sequencing reads mapped to target genes with an average depth of sequencing ranging from 117–250X (). Somatic variant calling identified variants that were considered for further analysis to calculate the TMB. shows a summary of somatic variants identified in this study. All the non-synonymous coding mutations (Frameshift, Missense, Nonsense, In-frame, Start loss, and Stop loss) were used to estimate the TMB of 0.34 mut/Mb for this patient.
Table 3. Summary of the target region for exome sequencing. Table summarizing coverage, depth of sequencing and on-target reads from patient’s leukemia cells (Initial tumor) and normal PBMCs (Final normal). A targeted mean coverage of 117x for normal and 251x for tumor with 95.43% and 98.27%% bases covered at ≥10x for normal and tumor respectively
Table 4. Summary of passed and on-target somatic variants. Table summarizing the variants from selected reads that were reported from the whole exome sequencing of the patient’s leukemia cells (initial tumor). The on-target is calculated based on the coordinate of the target regions provided by the vendor. SNP = Single nucleotide polymorphism; Ts/Tv = Transitions/Transversions
For neoepitope prioritization, 9-mer overlapping peptides covering each somatic variants were rank-ordered based on their composite immunogenicity score based on MHC class I binding affinities, mutant expressions and TCR binding predictions. shows a list of the peptides from the mutated genes that were tested for in vitro CD8+ T cell activation.
Table 5. Top prioritized epitopes from the tumor sample. Table enlisting mutant peptides representing the top prioritized epitopes predicted from missense mutations reported in the patient’s leukemia cells
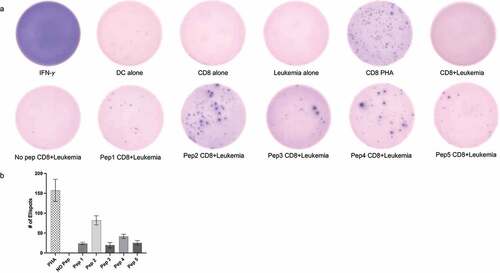
We next performed IFN-γ ELISPOT assay using autologous PBMCs and leukemia cells to determine the immunogenicity of the neoantigen peptides. indicates that all five peptides prioritized by the pipeline were able to induce a CD8+ T cell-specific immune response. A blinded manual evaluation of the ELISPOTs from three independent counts showed that peptide 2 (RMFWHLMPF) representing a missense mutation in STAT5B protein was most immunogenic. Peptide 4 (FPTNAFGDI) representing a missense mutation in TRPM8 also showed significant activation of CD8+ T cells when co-cultured with patient leukemia cells. Peptides 1, 3 and 5 showed low-moderate CD8+ T cell activation as indicated by the number of IFN-γ ELISPOTs compared to cells alone and no peptide controls. Overall, these results indicate that the neoantigen peptides generated by the whole exome sequencing and bioinformatics pipeline were able to activate CD8+ T cells when co-cultured with the patient’s leukemia cells, indicating that these peptides have the potential to be used as neoantigen vaccines in the future.
Figure 1. Neoantigen peptides can induce tumor specific CD8+ T cell mediated response in vitro. (a) IFN-γ ELISPOT results showing CD8+ T cell activation when co-cultured with patient leukemia cells. All five peptides representing neoantigens (Pep1-5) showed higher number of IFN-γ spots as compared to cells alone or no peptide controls. CD8+ T cells treated with 0.5 μg/ml phytohemagglutinin (PHA) for 24 hrs were used as a positive control. Representative images from three separate experiments are displayed (b) Quantitation of ELISPOTS from three separate individual manual counts displaying the number of spots in different conditions.
Recently, there have been several clinical studies involving the use of neoantigen peptide-based vaccines for different types of cancers in adults, alone or in combination with immune checkpoint inhibitors.Citation1,Citation5,Citation9 Although there are few active clinical trials that specifically aim at developing peptide-based vaccines for children,Citation16,Citation17 these studies have been limited largely to CNS tumors. Our study establishes a feasible experimental method to identify effective therapeutic neoantigen vaccines in high-risk pediatric leukemia patients. The aim is to generate effective personalized neoantigen-derived peptide vaccines using a combination of bioinformatics and cell-based assays, potentially in combination with other therapeutic agents and protocols.
The identification of highly immunogenic peptide sequences that are expressed exclusively on cancer cells is the most important step in designing a cancer vaccine. While very few antigens meet these criteria, this is also impacted by the observation that pediatric malignancies in general carry fewer mutations and potential neoantigens in comparison to adult patients.Citation18–20 This has been considered a critical limitation in the development of pediatric cancer-focused neoantigen vaccines in the past. Nevertheless, a recent report showed the potential of the approach to identify putative targetable neoepitopes based on somatic missense mutations and gene fusions using whole-genome sequencing.Citation15 The evaluation of the neoepitope landscape of somatic alterations, missense mutations, and oncogenic gene fusions in 540 childhood cancer genomes and transcriptomes showed that >80% pediatric cancers (leukemias, brain tumors and solid tumors) had at least one predicted neoepitope, indicating the potential of neoantigen vaccine therapies in pediatric oncology.Citation21
The bioinformatics pipeline utilized in our report was efficient in identifying mutations and predicting neoantigens that were unique to the patient’s leukemia cells. The resultant 9-mer neoantigen peptides were tested in an in vitro T cell activation assay to demonstrate that the predicted neoantigens were capable to induce an immune response specific to the leukemia cells. Limitations of this pre-clinical study include the potential exclusion of the complex accessory immune cells and soluble components that may participate and contribute to the vaccine activity in patients. In addition, we have also not examined the utility of other agents and combinations that may enhance the vaccine response in the clinical setting, such as checkpoint inhibitors. Finally, additional studies are required to determine the most favorable dose and immunization regimens that may be necessary to optimize the efficacy of such neoantigen vaccines in patients. Despite these limitations, this report provides important proof-of-concept data to generate expanded studies using neoantigen peptide vaccines for the management of high-risk leukemias in children in the future.
Acknowledgments
The authors acknowledge the contribution of Dr Amit Chaudhury from Medgenome Inc. for this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Fang Y, Mo F, Shou J, Wang H, Luo K, Zhang S, Han N, Li H, Ye S, Zhou Z, et al. A pan-cancer clinical study of personalized neoantigen vaccine monotherapy in treating patients with various types of advanced solid tumors. Clin Cancer Res. 2020;26:4511–20.
- Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135–46. doi:10.1038/nrc3670.
- Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. 2021;18(4):215–29. doi:10.1038/s41571-020-00460-2.
- Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol. 2018;18(3):168–82. doi:10.1038/nri.2017.131.
- Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie W-R, Hildebrand WH, Mardis ER, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–08. doi:10.1126/science.aaa3828.
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–21. doi:10.1038/nature22991.
- Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–26. doi:10.1038/nature23003.
- Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, Platten M, Tabatabai G, Dutoit V, van der Burg SH, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565(7738):240–45. doi:10.1038/s41586-018-0810-y.
- Majumder S, Shah R, Elias J, Manoharan M, Shah P, Kumari A, Chakraborty P, Kode V, Mistry Y, Coral K, et al. A cancer vaccine approach for personalized treatment of Lynch Syndrome. Sci Rep. 2018;8(1):12122. doi:10.1038/s41598-018-30466-x.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi:10.1093/bioinformatics/btp324.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–79. doi:10.1093/bioinformatics/btp352.
- Clarke L, Fairley S, Zheng-Bradley X, Streeter I, Perry E, Lowy E, Tassé A-M, Flicek P. The international Genome sample resource (IGSR): a worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017;45(D1):D854–D9. doi:10.1093/nar/gkw829.
- Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, Hamamsy T, Lek M, Samocha KE, Cummings BB, et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017;45(D1):D840–D5. doi:10.1093/nar/gkw971.
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Ward MH, Kholodov M, Baker J, Phan L, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11. doi:10.1093/nar/29.1.308.
- Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: improved Peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol. 2017;199(9):3360–68. doi:10.4049/jimmunol.1700893.
- NCT02960230. H3.3K27M peptide vaccine with nivolumab for children with newly diagnosed DIPG and other gliomas.
- NCT03299309. PEP-CMV in recurrent MEdulloblastoma/MALIGNANT GLIOMA.
- Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, Davidson S, Edwards M, Elvin JA, Hodel KP, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017;171(5):1042–56 e10. doi:10.1016/j.cell.2017.09.048.
- Majzner RG, Heitzeneder S, Mackall CL. Harnessing the immunotherapy revolution for the treatment of childhood cancers. Cancer Cell. 2017;31(4):476–85. doi:10.1016/j.ccell.2017.03.002.
- Hutzen B, Paudel SN, Naeimi Kararoudi M, Cassady KA, Lee DA, Cripe TP. Immunotherapies for pediatric cancer: current landscape and future perspectives. Cancer Metastasis Rev. 2019;38(4):573–94. doi:10.1007/s10555-019-09819-z.
- Chang T-C, Carter RA, Li Y, Li Y, Wang H, Edmonson MN, Chen X, Arnold P, Geiger TL, Wu G, et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017;9(1):78. doi:10.1186/s13073-017-0468-3.