
ABSTRACT
Previous studies have shown that the increased prevalent ST764 clone in China, Japan, and other Asian areas. However, the knowledge of the genetic features and virulence characteristics of methicillin-resistant Staphylococcus aureus (MRSA) ST764 in China is still limited. In this study, we identified 52 ST764-SCCmec type II isolates collected from five cities in China between 2014 and 2021. Whole genome sequencing showed that the most common staphylococcal protein A (spa) types of ST764 in China were t002 (55.78%) and t1084 (40.38%). Virulence assays showed that ST764-t1084 isolates had high haemolytic activity and α-toxin levels. Of the critical regulatory factors affecting α-toxin production, only the SaeRS was highly expressed in ST764-t1084 isolates. Mouse abscess model indicated that the virulence of ST764-t1084 isolates was comparable to that of S. aureus USA300-LAC famous for its hypervirulence. Interestingly, ST764-t002 isolates exhibited stronger biofilm formation and cell adhesion capacities than ST764-t1084 isolates. This seems to explain why ST764-t002 subclone has become more prevalent in China in recent years. Phylogenetic analysis suggested that all ST764 isolates from China in Clade III were closely related to KUN1163 (an isolate from Japan). Notably, genomic analysis revealed that the 52 ST764 isolates did not carry arginine catabolic mobile element (ACME), which differed from ST764 isolates in Japan. Additionally, most ST764 isolates (69.23%) harboured an obvious deletion of approximately 5 kb in the SCCmec II cassette region compared to KUN1163. Our findings shed light on the potential global transmission and genotypic as well as phenotypic characteristics of ST764 lineage.
Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) is a major pathogen associated with high rates of morbidity and mortality since these pathogenic bacteria are resistant to multiple classes of antibiotics [Citation1]. During the last few decades, MRSA strains spread all over the world being one of the most important nosocomial pathogens [Citation2]. Among these epidemic MRSA strains, ST5 and ST239 are the typical hospital-associated MRSA (HA-MRSA) clones prevalent in Asia countries [Citation3–6]. ST5-MRSA-II is one of the global HA-MRSA clones (New York/Japan clone) [Citation3].
As a single-locus variant of the ST5 HA-MRSA lineage with the features of community-associated MRSA, ST764 isolate was first identified in 2006 in Japan [Citation7]. Later, it was found that the ST764-SCCmec II genotype is increasingly prevalent in various environments in Japan, including long-term care institutions [Citation8], outpatient clinics, and hospitals [Citation9–13]. These epidemiological findings showed that ST764-SCCmec II is highly prevalent during nosocomial MRSA infections. Despite the HA-MRSA clones, ST764 isolates had been reported frequently carried Arginine catabolic mobile element (ACME) type II′ adjacent to its type II SCCmec element in Japan [Citation8,Citation11,Citation14–16]. ACME, a genomic island of the staphylococcus, was previously thought to enhance bacterial survival and growth in the host [Citation8]. The truncated form ACME II, namely ACME II′, which is also assumed to improve the existence of MRSA within the host, has been detected in different lineages of MRSA [Citation17–19]. However, whether the ST764-SCCmec II isolates in China carry ACME II′ remains to be elucidated.
The potential for infiltration of ST764 into the nosocomial settings and resistance to topical antibiotic mupirocin have made it an alarming public health concern [Citation20,Citation21]. Our previously published multicentre longitudinal study indicates that ST5 and its variant ST764 displayed a jagged growth trend in China over the preceding seven years [Citation22]. A recent study reported that ST5/ST764-SCCmec II was the most prevalent type of HA-MRSA isolate in southern China [Citation23]. Furthermore, there are studies that reported S. aureus isolated from the environmental surface in hospitals were predominantly ST764-t002-II isolates [Citation24]. ST764-MRSA has established pandemic in China, especially prevalent in coastal cities such as Shanghai and Guangzhou [Citation22–25]. Therefore, the ST764 clone may form a dominant clone in coastal cities in the future, and this emerging clone may pose a serious threat to hospitals. Additionally, several staphylococcal enterotoxin genes, such as seb, seg, seh, sem, sen, seo, and sei, have been frequently found in ST764 isolates [Citation25,Citation26]. Although ST764 isolates are Panton-Valentine leukocidin (PVL)-negative, they can cause bloodstream infection and invasive infections (e.g. necrotizing fasciitis and keratitis) [Citation11,Citation14,Citation27]. However, the virulence of ST764-MRSA has not been fully investigated to date, and there is limited information on their epidemiological trends. It is crucial to promptly detect MRSA new clones and effectively stop clone spread. This prompted us to study ST764-MRSA isolates to better understand the cause and prospect of the rise of ST764-MRSA in China.
In the present study, we sequenced 52 ST764-MRSA isolates from five provinces in China utilizing the Illumina NovaSeq platform. The virulence potential of ST764 isolates was analysed in vivo and in vitro. Phylogenetic analysis was carried out to investigate the evolutionary dynamics of Chinese ST764 clonotype. We aimed to reveal the genetic features and virulence characteristics of ST764 clonotype and thus will be useful for the development of infection prevention and control measure.
Materials and methods
Ethics approval
This study was approved by the Ethics Committee of Shanghai Pulmonary Hospital, School of Medicine, Tongji University, Shanghai, China. All animal experiments were carried out under the guidelines approved by the Ethics Committee of the Shanghai Pulmonary Hospital of Tongji University School, Tongji University, Shanghai (Project number: K22-183Y).
Bacterial isolates and growth conditions
We collected and analysed a total of 643 non-duplicated clinical MRSA isolates from 7 provinces/municipalities in China between 2014 and 2021, from which the epidemiological data of 565 clinical MRSA isolates (including 46 ST764 isolates) between 2014 and 2020 have been published in our previous study [Citation22]. Among 643 MRSA isolates, 52 ST764 MRSA isolates were identified by WGS. The Illumina sequences of the 52 ST764 isolates are available in NCBI (Accession number: PRJNA902152). The full details of 52 ST764 isolates were shown in Table S1. In this study, S. aureus isolates were cultivated in tryptic soy broth (TSB) (Oxoid) and incubated with shaking at 220 rpm in a 37°C incubator.
Whole genome sequencing and analysis
Whole genome sequencing (WGS) of 52 ST764-MRSA isolates was performed on the Illumina NovaSeq platform. Raw reads were quality filtered with fastp v0.20.1 [Citation28]. De novo genome assembly of WGS data was performed using Unicycler v0.4.8 [Citation29]. The programme mlst in GitHub (https://github.com/tseemann/mlst) was utilized to perform multi-locus sequence typing of the S. aureus assemblies. Staphylococcal cassette chromosome mec (SCCmec) and staphylococcal protein A (spa) typing of strains was performed using SCCmecFinder and SpaFinder, respectively [Citation30,Citation31]. The virulence genes and antibiotic resistance genes were identified by ABRicate v1.0.0 (https://github.com/tseemann/abricate) based on VFDB [Citation32] and CARD [Citation33] databases. To explore the phylogenetic relationships among Chinese ST764 and ST764 isolates from other countries, S. aureus genome assemblies were downloaded from NCBI RefSeq (data cutoff was October 2022). The resulting 97 ST764 assemblies were then extracted. The phylogenetic relationship of 149 ST764 isolates was constructed by ParSNP using S. aureus SARJ15 (GenBank accession number: NZ_JAOBNV010000010.1) as the reference [Citation34], followed by visualization and annotation using the interactive tree of life (iTOL) [Citation35]. To infer transmission, the pairwise SNP distances between all isolates were assessed. Furthermore, the SCCmec in ST764 was compared with a reference SCCmec II sequence (S. aureus N315, GenBank accession number: D86934.2) using Blastn. An alignment of the USA300 (GenBank accession number: CP000255.1) acquired-ACME related genes (acrA and opp3) was carried out using Blastn. BRIG software (http://brig.sourceforge.net/) was used to generate comparisons [Citation36].
Antimicrobial susceptibility testing
The antimicrobial susceptibility of a total of 10 antimicrobial agents was evaluated. For erythromycin (ERY), clindamycin (CLI), ciprofloxacin (CIP), and tetracycline (TET), susceptibility testing was performed using the disc diffusion method on Mueller-Hinton agar plates (Oxoid, UK). The microdilution broth method was used to evaluate the MICs of cefoxitin (FOX), gentamicin (GEN), oxacillin (OXA), linezolid (LNZ), mupirocin (MUP), and vancomycin (VAN). All antimicrobial susceptibility testing and interpretative criteria were according to break points mentioned in the Clinical Laboratory Standards Institute guidelines (CLSI, 2020). S. aureus ATCC 25923 and ATCC 29213 served as controls for disk diffusion method and MIC testing, respectively.
Haemolytic activity assay
Bacterial cultures were incubated in TSB medium for 24 h and supernatants were collected by centrifugation at 8000 rpm for 2 min. Then 100 µl of bacterial supernatant was collected and mixed with 900 μL PBS containing 3% defibrinated rabbit blood cells, and the mixtures were incubated at 37°C for 1 h. Triton X-100 served as the positive control, and 0.9% (w/v) NaCl was set as the negative control. After centrifugation, the supernatant was collected, and its absorbance was measured at 600 nm. Experiments were repeated three times.
Western blot analysis
Western blots were performed as previously described [Citation37]. Briefly, ST764 isolates (six randomly selected isolates each in t002 isolates and t1084 isolates, and these isolates same apply to the experiment that follows) were grown in TSB with shaking for 24 h and bacterial culture supernatants were collected. The protein concentration of each sample was determined by Bradford assays to keep the total amount of protein loaded for SDS-PAGE consistent in the twelve isolates. Proteins were mixed with Omni-EasyTM Protein Sample Loading Buffer (EpiZyme Biotechnology Co., Ltd., Shanghai, China) and heated for 10 min at 95°C. Samples were separated by 12.5% SDS-PAGE and were transferred to the PVDF membrane. After blocking with 5% bovine serum albumin (BSA) at 4°C for overnight, the membrane was incubated with a rabbit anti-Hla IgG antibody (Sigma) at a 1/2500 dilution. The membrane was then incubated with Goat Anti-Rabbit IgG HRP (Biosharp) at a 1/5000 dilution. Omni-ECLTM Pico Light Chemiluminescence Kit (EpiZyme Biotechnology Co., Ltd., Shanghai, China) was used to develop image.
Growth curve
To compare the growth curve between ST764-t002 and ST764-t1084 isolates, ST764 isolates (randomly selected isolates as illustrated above) were grown overnight. Overnight cultures of S. aureus were diluted 1:200 in fresh TSB medium and incubated at 37°C with shaking (220 rpm) for 24 h. OD600 was measured with Microplate Reader at 1–12 and 24 h. All experiments were repeated in triplicate.
Biofilm semi-quantitative assay
The 52 ST764 isolates were grown in TSB at 37°C for 16 h with shaking (220 rpm), diluted with TSBG (containing 0.5% glucose) 100 times, and then added to sterile 96-well plates. The plates were incubated for 24 h at 37°C. Subsequently, the supernatant was aspirated and discarded, then gently washed three times with sterile PBS. The biofilms were fixed with 99% methanol and then stained with 2% crystal violet for 10 min, then gently washed with flowing water, and dried at 37°C. Optical density value measured at 600 nm. S. epidermidis ATCC 35984 was used as a positive control. Sterile TSBG and S. epidermidis ATCC 12228 served as the negative control. Experiments were conducted in triplicates and repeated three times.
Cell adhesion assay
The A549 lung epithelial cells were grown in DMEM medium after supplementing with 10% foetal bovine serum at 37°C and 5% CO2. About 5 × 105 cells were seeded into 12-well plates. Before use, the plates were washed twice with PBS. ST764 isolates (ten randomly selected isolates each in t002 isolates and t1084 isolates) were incubated for 6 h at 37°C to the logarithmic growth phase and resuspended in DMEM medium without serum. Then the cells were infected with bacteria (MOI = 10:1) and co-incubated at 37°C for 2 h. Subsequently, the supernatant was aspirated and discarded, and the plates were washed three times with sterile PBS to remove loosely adherent bacteria. Then cells were dissociated with 200 µl trypsin-EDTA for 3 min at 37°C. A549 cells were lysed with 0.05% Triton X-100. Bacterial CFU was determined by plating serial dilutions of epithelial cell lysates onto trypticase soy agar (TSA) plates.
Real-time fluorescence quantitative PCR
The ST764 isolates were grown for 24 h in TSB with shaking (220 rpm) at 37°C. Total RNA was isolated using a Total RNA purification Kit (Sangon Biotech, Shanghai, China) following to the manufacturer’s protocol. Total RNA was reverse transcribed into cDNA using the PrimeScript RT reagent kit with gDNA Eraser (Takara Bio, Inc.). S. aureus N315 was used as a control (relative expression = 1), and gyrB was used as the internal reference gene. Real-time quantitative PCR (RT-qPCR) was performed using TB Green™ Premix Ex Taq™ II (Takara) on QuantStudio™ 5 Real-Time PCR System (Applied Biosystems). RNA expression levels of hla, saeR, saeS, agrA, RNAIII, sbi and nuc genes were calculated by the formula 2−ΔΔCt [Citation37]. The primer pairs were shown in Table S2. Each reaction was performed trice.
Mouse skin abscess model
BALB/C mice (six weeks old, female) were randomly allocated into eight groups (n = 5 per group). ST764 isolates, HA-MRSA clone N315, and CA-MRSA clone USA300-LAC were grown overnight in TSB. A 200-fold dilution of the overnight cultures was reinoculated into TSB media for 9 h (the post-exponential phase) and washed three times with normal saline solution. Then, mice were injected subcutaneously in the shaved flank with 100 μL PBS containing 1 × 107 bacterial cells suspended. Lesion and abscess sizes were monitored daily with calliper using the formula: (A = π × (L × W)). After measuring and recording the skin abscess for five days, the mice were euthanized and the skin was dissected. Bacterial load was determined in abscess homogenates by serial dilution and culture on TSA plates.
Galleria mellonella larvae model
Prepare for isolates was described as above. Groups of 15 larvae of G. mellonella weighing 250–300 mg (Tianjin huiyude Biotech, Tianjin, China) were used in all assays. G. mellonella was injected with 10 μL of bacterial suspension in saline (3.0 × 106 CFU/larvae) in the last left posterior leg using Hamilton syringe. Larvae with injected with 10 μL sterile 0.9% (w/v) NaCl and un-injected larvae as control. Then the treated larvae were placed in sterile petri plates and kept at 37°C for observation. The survival rate was assessed every 12 h for three days. The experiment was repeated at least three times.
Statistical analysis
All analyses were performed using GraphPad Prism 8 (GraphPad Software Inc. San Diego, CA, USA). Results derived from samples between two groups were treated with unpaired two-tailed Student’s t-test and Chi-square test. P < .05 was statistically considered to be significant. Error bars in the figures represented the standard deviation of a data set (mean ± standard). *P < .05, **P < .01, ***P < .001, ****P < .0001.
Results
Demographic characteristics of the selected ST764 clonotype isolates
Of the 643 MRSA isolates collected from seven cities in China between 2014 and 2021, 52 ST764-MRSA SCCmec type II isolates were identified. The most common staphylococcal protein A (spa) types of ST764 in China were t002 (55.77%, 29/52) and t1084 (40.38%, 21/52). The remaining two spa types were t045 (1.92%, 1/52) and t042 (1.92%, 1/52). Relative information of t002 and t1084 isolates is listed in . There were no significant differences in age or gender between t002 and t1084 isolates. The ST764-t002 isolates were significantly (P = .003) recovered as the predominant type from sputum (65.52%, 19/29) and blood (20.69%, 6/29), while the ST764-t1084 isolates were mostly sputum (57.14%, 12/21) and pus (33.3%, 7/21). The clinical relevance of infectious disease types caused by t002 and t1084 isolates may be left determined. The full details of 52 ST764 isolates were shown in Table S1. These isolates were mostly isolated from Shanghai (n = 28), followed by Guangdong (n = 19), Zhejiang (n = 2), Hubei (n = 2), and one isolate from Jiangxi. Isolates were primarily recovered from sputum (n = 32), followed by blood (n = 9) and pus (n = 7) samples.
Table 1. Constitution in gender, age group, and specimen sources of the ST764-t002 and ST764-t1084 genotype S. aureus isolates.
Resistance gene analysis and antibiotic susceptibility test
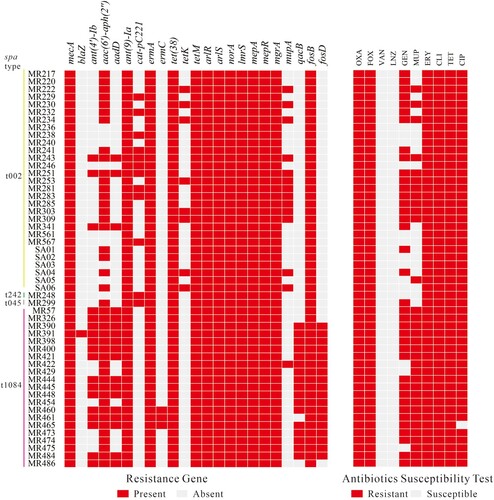
The antimicrobial resistance gene and antibiotic susceptibility testing of 52 ST764 isolates are shown in . Aminoglycoside resistance genes including ant(4′)-Ib, aac(6′)-aph(2″), and aadD exhibited differing profiles in ST764-t1084 and ST764-t002 isolates (71.43% versus 10.35%, P < .001; 100% versus 72.41%, P = .015; 71.43% versus 10.35%, P < .001). Mupirocin resistance gene mupA was detected in more ST764-t002 isolates than ST764-t1084 isolates (44.83% versus 4.76%, P < .001). Notably, antiseptic resistance gene qacB and fosfomycin resistance gene fosD were only found to be present in most ST764-t1084 isolates (80.95% and 85.71%, respectively) but not in ST764-t002 isolates. Subsequently, antibiotic susceptibility testing was performed on 52 ST764 isolates. As shown in , 52 ST764 isolates were resistant to oxacillin, cefoxitin, erythromycin, clindamycin and tetracycline, while were all susceptible to vancomycin and linezolid.
Figure 1. Antibiotic resistance genes (left) and antibiotics susceptibility profiles (right) of 52 ST764 S. aureus isolates collected in this study.
Virulence gene analysis
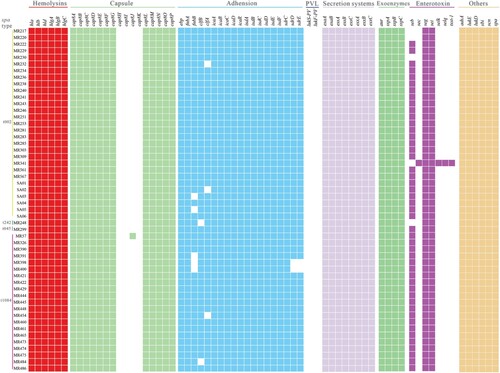
Genes encoding virulence factors in the 52 ST764 isolates are described in . The Panton-Valentine leucocidin genes (lukS-PV and lukF-PV) were lacking in all ST764 isolates. The enterotoxin B gene (seb) was found to be present in most isolates (90.38%, 47/52). In addition, the bicomponent toxin leucocidin genes (lukD and lukE) and enterotoxin genes (seg, sei, sem, sen, seo, and set6-15) existed in all isolates. Almost all ST764 isolates (98.08%, 51/52) did not have sec, selk, selq and tsst-1.
Figure 2. Virulence gene profiles of the 52 ST764 S. aureus isolates. Coloured blocks represent the presence of genes and white blocks represent absence. The horizontal colour bar (from left to right) represents genes associated with haemolysins, capsule, adhesion, PVL, secretion systems, exoenzymes, enterotoxins, and other virulence genes.
ST764-t1084 isolates exhibited higher haemolytic activity but lower adhesion abilities in comparison to ST764-t002 isolates
Although virulence gene analysis showed that ST764-t1084 and ST764-t002 isolates showed similar patterns, the different spa types have diverse degrees of virulence [Citation38]. To analyse the virulence characteristic of ST764 isolates collected in this study, we separated them according to spa types and compared the haemolytic activity, hla mRNA and α-toxin expression levels, biofilm formation ability, cell adhesion capacity between ST764-t002 and ST764-t1084 genotype isolates. Haemolytic activity assay was performed on the 52 ST764 isolates. As shown in (A), ST764-t1084 isolates displayed a high level of lysis capacity against erythrocytes in comparison with ST764-t002 isolates (OD600: 0.1245 ± 0.0617 versus 0.0836 ± 0.0379, P < .001). We next tested whether ST764-t1084 isolates exhibited high haemolysis activity due to high hla expression by RT-qPCR and western blot. Six randomly selected isolates belonging to each t002 and t1084 to test the level of hla expression. A significant difference in the expression level of hla between ST764-t002 and ST764-t1084 isolates was observed (P < .05) ((B)). The western blot assay results of ST764-t1084 and ST764-t002 isolates showed that there were significant differences in α-toxin production (P < .001) ((C,D)). To investigate whether the observed phenotypical differences were due to growth ability, growth curves of these isolates (six isolates each in t002 and t1084 as above) were assessed. However, there was no difference between ST764-t1084 and ST764-t002 isolates (Figure S1).
Figure 3. Phenotypic tests of the ST764 S. aureus isolates. (A) Erythrocyte lysis capacity (separated by spa types) of the 52 ST764 S. aureus isolates. (B) The transcriptional level of hla (six randomly selected isolates each in t002 isolates and t1084 isolates). Transcript levels were normalized to gyrB transcript level. S. aureus N315 was used as a control (relative expression = 1). (C) Western blot assay was used to assess the secretion of α-toxin in the twelve ST764 S. aureus isolates (randomly selected isolates as illustrated above). (D) Evaluation of grey values of the secretion α-toxin using Image J software. (E) Semi-quantitative biofilm formation ability (separated by spa types) of the 52 ST764 S. aureus isolates. (F) Cell adhesion assays in A549 lung epithelial cells of randomly selected ST764 S. aureus isolates (10 randomly selected isolates each in t002 isolates and t1084 isolates).
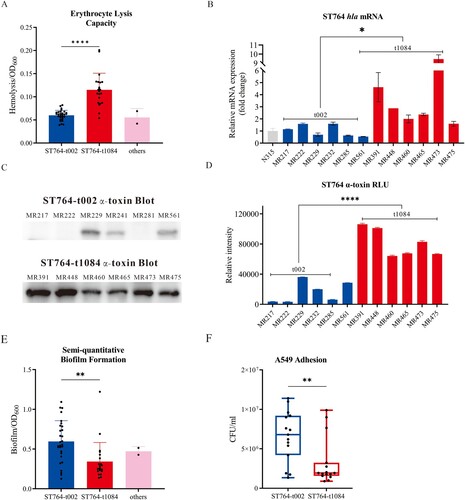
MRSA infections continue to be a challenge in hospitals environment. Previous studies showed that the majority of S. aureus isolated from environmental surfaces in several hospitals were ST764-t002-II isolates [Citation24]. This led us to examine the adhesion capabilities of ST764 isolates. We therefore performed a semi-quantitative biofilm test on 52 ST764 isolates. The biofilm-forming capabilities of each isolate are shown in (E). Although ST764-t002 isolates exhibited lower virulence than ST764-t1084 isolates, ST764-t002 isolates had stronger biofilm formation capability than ST764-t1084 isolates (OD600: 0.5956 ± 0.2617 versus 0.3432 ± 0.2400, P = .0011). To further explore that the adhesion capabilities of ST764 isolates, we performed the cell adhesion assay using 10 randomly selected isolates each in t002 and t1084. As shown in (F), the A549 epithelial cell adhesion ability of ST764-t002 isolates was stronger than that of ST764-t1084 isolates (CFU/106·mL−1: 6.572 ± 3.240 versus 3.249 ± 2.987, P = .0068).
The SaeRS two-component system contributes to the haemolytic activity of ST764-t1084 isolates
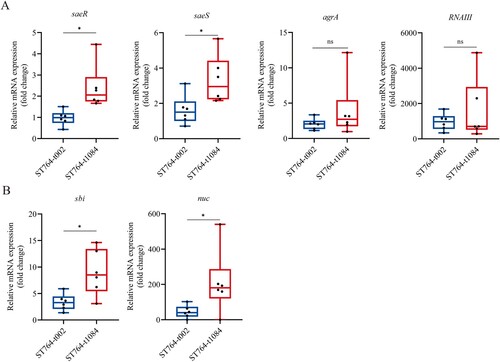
The expression of α-toxin in S. aureus is affected by several key regulators, including agr, RNAIII, saeR, and saeS [Citation39]. To further understand the differences in haemolytic activity, hla mRNA and α-toxin levels between the included ST764 isolates, we further examined the transcription levels of the four regulators. As shown in (A), both saeS and saeR displayed high transcript levels in the ST764-t1084 isolates. This trend, however, was not observed in agr and RNAIII. To confirm the impact of saeR and saeS on SaeRS Two-Component System (TCS) activity, we examined the transcript levels of well-studied saeR/S target genes: IgG binding protein (sbi) and nuclease (nuc) [Citation40,Citation41]. Similar to saeRS, ST764-t1084 isolates displayed higher transcript levels for both sbi and nuc when compared to ST764-t002 isolates ((B)). In combination, these results suggest that ST764-t1084 isolates have a higher Sae activity.
Figure 4. RT-qPCR detection of the expression levels of virulence factors in ST764-t002 and ST764-t1084 isolates. (A) The levels of mRNA expression for the α-toxin regulators. (B) The levels of mRNA expression for the Sae target genes. Transcript levels were normalized to gyrB transcript level.
To explore whether the higher SaeRS activity among ST764-t1084 isolates linked to the genome mutations, we analysed the genome sequence of ST764 isolates using N10CSA27 as the reference (ST764-t002, GenBank accession number: CP094443). After excluding intergenic and synonymous mutations, we observed 28 general SNPs in ST764-t1084 isolates (Table S3). However, no general mutations exist in SaePQRS region of ST764-t1084 isolates. Our data therefore suggest that genomic mutations are unlikely to be the result of the higher Sae-activity in ST764-t1084 isolates.
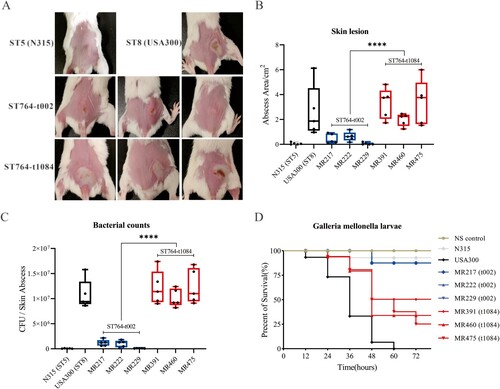
Virulence of ST764 isolates in vivo
After observing the differences of ST764-t002 and ST764-t1084 isolates on the activity of haemolytic, and the expression levels of hla and α-toxin, we sought to examine its virulence in vivo using mouse skin abscess model and G. mellonella larvae model. The closely related to ST764 HA-MRSA clone N315, and CA-MRSA clone USA300-LAC famous for its hypervirulence, were selected and compared with ST764 to evaluate differences in virulence. After subcutaneously inoculated with 100 μL PBS containing 1 × 107 bacterial cells for 48 h, the abscess area was shown in (A). The abscess size of ST764-t1084 isolates was significantly larger than ST764-t002 isolates (2.89 ± 1.38 in ST764-t1084 isolates versus 0.37 ± 0.38 in ST764-t002 isolates, P < .0001), but at levels comparable to USA300 ((B)). As shown in (C), the mice infected with ST764-t1084 isolates had significantly higher CFU counts compared to ST764-t002 isolates (P < .0001). In G. mellonella larvae models, G. mellonella infected with USA300 all died within 60 h (n = 15), while mostly infected with ST764-t002 and ST764-t1084 isolates were still alive after 72 h ((D)). However, the percentage of surviving larvae was significantly lower after infection with ST764-t1084 isolates compared to ST764-t002 isolates and N315 isolate (G. mellonella survival ≤60% at 72 h). Taken together, our data indicated that the Chinese MRSA ST764-t1084 isolates were highly virulent.
Figure 5. Comparison of the virulence of the ST764 S. aureus isolates in vivo. (A) Representative mouse back skin abscess after 48 h inoculation of isolates. Randomly selected three isolates in each spa type were used for infections in mouse back skin. N315 and USA300 were selected and compared with ST764. (B) Skin abscess areas of 40 mice (five mice for each group) using the formula π × (L × W) after 48 h infection. (C) Bacterial load in abscess homogenates was determined by serial dilution and culture on TSA plates. (D) The survival rates of Galleria mellonella larvae infected with ST764 S. aureus isolates.
Phylogenetic construction of ST764 clonotype S. aureus
To explore the evolution of ST764 isolates, we constructed a phylogenetic tree with our 52 ST764 isolates and the 97 ST764 isolates from NCBI RefSeq during the time of the study (October 2022). As shown in (A–C), three clades could be distinguished. In the largest clade, Clade III, phylogenetic analysis showed that 110 ST764 isolates derived from China clustered together with the former one ACME-negative isolate (KUN1163, AP020324) from Kyoto, Japan, making it the main clade out of Chinese ST764 clones ((B)). This indicated that the Chinese ST764 may have evolved from Japan, followed by spread in the Chinese population. Clade II can be divided into two subclades (IIa and IIb). Noteworthy, all ST764-t1084 isolates (n = 27) were found solely from China and formed one distinct monophyletic group (Clade IIb) within the Clade II. Mapping of the isolates revealed the independent evolution of t1084 and other spa types in ST764 clonotype, demonstrating that the ST764-t1084 was a novel spa type different from other ST764 isolates. Accordingly, based on the above analysis, we can speculate that the ST764-t1084 subclone might be a variant of ST764 formed in China. However, the Chinese ST764-t002 subclone may have evolved from the Japan ST764 linage, then formed pandemic in China.
Figure 6. Phylogenetic analysis of ST764 S. aureus isolates. (A) ParSNP phylogenetic tree based on the 2301 core-SNPs of the 149 ST764 S. aureus isolates. Six groups of independent epidemics (paired SNPs of isolates ≤4 were defined as the same transmission) were identified. (B) Classification of Country, SCCmec type, spa type, and isolated year of the 149 isolates. The colours are the same as shown above. (C) Unrooted tree of ST764 S. aureus isolates. Branches in this tree could be classified into three clades, including Clade I, Clade II and Clade III. EICU: Emergency Intensive Care Unit; Burn Unit: The Department of burn unit; RICU: The Respiratory Intensive Care Unit; MICU: The Medical Intensive Care Unit; Cardiology: The Department of Cardiology; Pancreatic surgery: The Department of Pancreatic surgery.
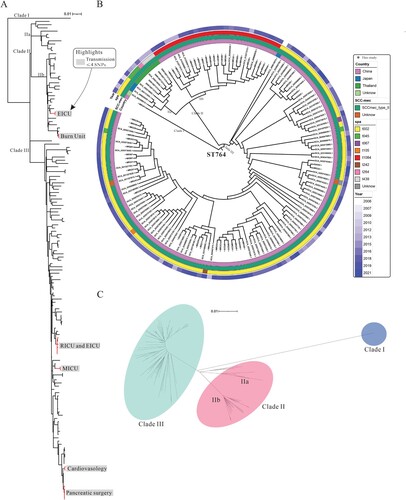
Paired distance between isolates was critical in determining the genetic relatedness of paired isolates. Therefore, we calculated all-against-all pairwise distances of 149 isolates and interpreted them with epidemiological information to identify possible transmission pairs. As shown in Figure S2, paired distances for 149 ST764 isolates were from 0 to 315. Isolates with paired distances of less than or equal to four SNPs were considered to be from the same transmission [Citation42]. Six independent small-scale transmissions were identified among the 52 ST764 isolates collected in this study were shown in (A). Notably, the isolates in each pair were solely collected from the same hospital, and the dates of collection were relatively close together, indicating that these ST764 isolates in each pair were either recently infected from the hospital environment or were transmitted from patient to patient. Hence, this is suggestive of frequent within-hospital transmission of ST764 clones.
The genetic structure of ST764 isolates
Phylogenetic analysis showed that 27 ST764-t1084 isolates from China in Clade IIb and three ST764 ACME-positive isolates from Japan in Clade IIa belonged to the same subclade. To further investigate the variations between the ST764 isolates from China and Japan, we identified the previously described ACME-SCCmec genetic elements. The ACME-related arcA and opp3 genes were not detected in the 52 ST764 isolates collected in this study, which differed from the ACME-positive ST764 isolates in Japan [Citation8,Citation11,Citation14,Citation15].
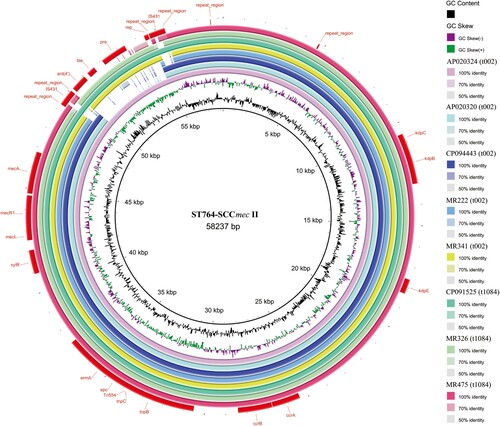
According to the results of phylogenetic analysis in this study, 110 ST764 isolates from China in Clade III showed the closest relationship to KUN1163 (AP020324) isolate from Japan. Therefore, we chose KUN1163 for further investigation. Notably, in the SCCmec II cassette region, we observed an obvious deletion of approximately 5 kb in nearly all ST764-t002 isolates (28/29, 96.55%) compared to KUN1163 (), except for one isolate in Clade I. Among the 21 ST764-t1084 isolates collected in this study, six isolates (MR454, MR474, MR473, MR429, MR422, and MR475) also harbour this deletion in SCCmec II region, and the six isolates were gathered in the same subclade in Clade IIb. The deletion region contains the plasmid recombinase enzyme gene pre, the bleomycin resistance gene ble, and an ant(4′) aminoglycoside resistance gene that has been described to provide high-level resistance to amikacin, tobramycin, and kanamycin. The ST764-t002 isolates with this deletion formed a large group in Clade III and have been spreading in China over the last five years, which indicated that the shorter SCCmec II variant could be a factor accounting for the fitness of MRSA ST764 isolates in healthcare settings.
Figure 7. Structural comparison of SCCmec. Comparative structural analyses of SCCmec from eight isolates. The SCCmec in ST764 was compared with a reference SCCmec II sequence (S. aureus strain N315, GenBank accession number: D86934.2). Each circle represents an isolate. Isolates MR222, MR341, MR326 and MR475 were included in our data set, while genomic information for the remaining four isolates was retrieved from the NCBI database (GenBank accession number: AP020324, AP020320, CP094443 and CP091525). AP020320, CP094443 and CP091525 were used as reference strains. Isolates are grouped in different colours, and the blank places indicate differences.
Discussion
Previous studies have reported that ST764 isolates have become the common hospital-associated S. aureus clone in Shanghai, China [Citation22–25]. However, reports on the characteristics of ST764 isolates and their evolutionary histories are still insufficient. To this end, we described here the genomic context, virulence characteristic, and phylogenetic analysis of 52 ST764 isolates collected in China from 2017 to 2021, combined with the globally available ST764 genomes, with which we were able to shed light on the emergence of ST764 clonotype in China. To the best of our knowledge, this is the first study focusing on the genetic feature and virulence potential of epidemiologically emerging clone ST764 in China.
ST764-t002 isolates have been reported from human infections in China, Japan and Thailand, but reports of ST764-t1084 isolates are rare. Therefore, we compared the genotype and phenotypic characteristics between ST764-t002 and ST764-t1084 isolates. Indeed, there are clear differences in genotype and phenotype in ST764-t1084 compared with ST764-t002 isolates. The antimicrobial resistance genes of ant (4′) – IIb, aadD, qacB and fosD were mainly present in ST764-t1084 isolates, while tetK and mupA were mainly present in ST764-t002 isolates. This indicated that ST764-1084 isolates might have potential resistance to chlorhexidine and fosfomycin. Chlorhexidine is a chemical disinfectant that is commonly used in hospital settings [Citation43]. The potential resistance to chlorhexidine may reflect the establishment and spread of ST764-1084 isolates in hospitals. Moreover, we compared the virulence of ST764-t1084 isolates relative to ST764-t002 isolates both in vitro and in vivo. Of note, phenotypic tests and animal experiments showed that ST764-t1084 isolates had higher haemolytic activity, hla mRNA and α-toxin expression levels in comparison to ST764-t002 isolates. Mouse abscess model indicated that ST764-t1084 isolates with virulence similar to that of S. aureus USA300-LAC famous for its hypervirulence. All these findings indicated that ST764-t1084 isolates with higher virulence, both in vitro and in vivo. Therefore, we next explored what factors contribute to the enhanced virulence of the ST764-t1084 isolates.
α-toxin is one of the critical virulence determinants implicated in the pathogenesis of S. aureus, associated with pneumonia[Citation44], skin and soft tissue infections (SSTI) [Citation45], and sepsis [Citation46]. The production of α-toxin is mainly controlled by the SaeRS two-component system and the global toxin accessory gene regulator (agr) [Citation47,Citation48]. In S. aureus, SaeR and SaeS are a part of the two-component system that control the expression of numerous important virulence factors such as coagulase, nuclease, IgG binding protein, toxic shock syndrome toxin, and α-toxin via response regulators and histidine kinases [Citation49]. Agr is a well-defined quorum sensing system in S. aureus, controlled by the regulatory effector molecule RNAIII, which plays an important role in the transcription regulation of virulence genes [Citation50]. Genetic annotation showed that both ST764-t002 and ST764-t1084 isolates were hla-positive and PVL-negative. On the basis of our data, we postulated that the increased expression levels of hla and α-toxin may be attributed to the high activity of sae and/or agr. The upregulated expression of sae activity on ST764-t1084 isolates might explain the highly haemolytic activity of the strain. The upregulated expression of target genes (i.e. ncu and sbi) regulated by saeRS TCS further supports our above speculation ((B)). Future molecular studies are needed to elucidate the mechanisms underlying the hyper-virulence in ST764-t1084 isolates.
In this study, higher biofilm-forming and cell adhesion capabilities were found for ST764-t002 isolates. This could in part explain the prevalence of ST764-t002 in hospital settings. These isolates could persist in the hospital environment and were difficult to eradicate due to their strong adherence to the surfaces of medical equipment and human lung epithelial cells. Additionally, the low pathogenicity of ST764-t002 isolates may allowed it to coexist with the host for a long time, contributing the dissemination of this clone [Citation51]. The further spread of ST764 isolates in China should be closely monitored.
Unlike those of other countries, the Chinese ST764 isolates mainly formed two large clades (Clade IIb and Clade III). Although ST764 MRSA was previously mostly assigned to spa type t002, other types t242 and t045, which are closely related to t002, were detected in the present study. Meanwhile, in Clade IIb, a novel spa type (ST764-t1084) we identified were found solely from China, suggesting a restricted epidemic of ST764-t1084 clone in China. These findings suggested the expansion of genetic diversity in ST764-MRSA, probably due to the increased frequency of transmission among the population in China. Notably, in the SCCmec II cassette region, we observed a larger deletion of approximately 5 kb had occurred in nearly all ST764-t002 isolates and a part of ST764-t1084 isolates. The large size of SCCmec type II probably limits their horizontal transfer [Citation52]. We speculate that this may be explained by the concept of fitness cost. Shorter variants (a deletion in SCCmec II in China ST764-t002 isolates) are increased in number in the last five years, which may indicate that these variants have a lower biological cost and better fitness [Citation53]. In addition, we cannot rule out the importance of genetic background. The genotype ST764 spa-t002 was the most prevalent (29/52, 55.77%) in our collected isolates. This may indicate that this deletion in SCCmec II in Chinese ST764 isolates is more functional and beneficial to host MRSA. Future studies are warranted to examine whether this deletion in SCCmec II correlates with the decreased fitness cost in ST764 isolates.
In conclusion, this study provided the first detailed characterization of ST764 isolates. Our experimental data indicated that ST764-t1084 isolates had high levels of haemolytic activity, α-toxin production and abscesses formation. Mouse abscess model indicated that the virulence of ST764-t1084 isolates was comparable to that of S. aureus USA300-LAC famous for its hypervirulence. The mapping of ST764 isolates showed that the independent evolution of ST764-t1084 and other spa types, demonstrating that the ST764-t1084 was a novel spa type that differed from other ST764 subclones. Additionally, the ST764-t002 isolates exhibited higher biofilm formation and adhesion ability than ST764-t1084 isolates. Approximately 5 kb deletion was found in SCCmec II in mostly ST764 isolates (69.23%) collected in this study. These variants may carry a compensated biological cost, resulting in further spread of already successful ST764-SCCmec II isolates. This also seems to explain why ST764-t002 subclone has formed pandemic in China, in recent years. Thus, dynamic monitoring of the spread of the ST764-SCCmec II clone in China should be implemented.
Supplementary_Table_3.xls
Download MS Excel (2.9 KB)Supplementary_Table_2.xlsx
Download MS Excel (9.6 KB)Supplementary_Table_1.xlsx
Download MS Excel (13.4 KB)Figure_S2.tif
Download TIFF Image (3 MB)Figure_S1.tif
Download TIFF Image (457.6 KB)Acknowledgements
The authors thank all the staff members at our institution.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Turner NA, Sharma-Kuinkel BK, Maskarinec SA, et al. Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat Rev Microbiol. 2019;17:203–218.
- Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–532.
- Lakhundi S, Zhang K. Methicillin-resistant Staphylococcus aureus: molecular characterization, evolution, and epidemiology. Clin Microbiol Rev. 2018;31:e00020-18.
- Monecke S, Coombs G, Shore AC, et al. A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS One. 2011;6:e17936.
- Jian Y, Zhao L, Zhao N, et al. Increasing prevalence of hypervirulent ST5 methicillin susceptible Staphylococcus aureus subtype poses a serious clinical threat. Emerg Microbes Infect. 2021;10:109–122.
- Jian Y, Lv H, Liu J, et al. Dynamic changes of Staphylococcus aureus susceptibility to vancomycin, teicoplanin, and linezolid in a central teaching hospital in Shanghai, China, 2008–2018. Front Microbiol. 2020;11:908.
- Zaraket H, Otsuka T, Saito K, et al. Molecular characterization of methicillin-resistant Staphylococcus aureus in hospitals in Niigata, Japan: divergence and transmission. Microbiol Immunol. 2007;51:171–176.
- Aung MS, Kawaguchiya M, Urushibara N, et al. Molecular characterization of methicillin-resistant Staphylococcus aureus from outpatients in northern Japan: increasing tendency of ST5/ST764 MRSA-IIa with arginine catabolic mobile element. Microb Drug Resist Larchmt N. 2017;23:616–625.
- Nakaminami H, Noguchi N, Ito A, et al. Characterization of methicillin-resistant Staphylococcus aureus isolated from tertiary care hospitals in Tokyo, Japan. J Infect Chemother Off J Jpn Soc Chemother. 2014;20:512–515.
- Aung MS, Urushibara N, Kawaguchiya M, et al. Clonal diversity and genetic characteristics of methicillin-resistant Staphylococcus aureus isolates from a tertiary care hospital in Japan. Microb Drug Resist Larchmt N. 2019;25:1164–1175.
- Kawamura K, Kitaoka K, Kimura K, et al. Spread of seb-positive methicillin-resistant Staphylococcus aureus SCCmec Type II-ST764 among elderly Japanese in nonacute care settings. Microb Drug Resist Larchmt N. 2019;25:915–924.
- Yamasaki F, Takeuchi S, Uehara Y, et al. Prevalence and characteristics of methicillin-resistant Staphylococcus aureus colonization among healthcare professionals in a university hospital in Japan. J Gen Fam Med. 2019;20:190–192.
- Ogura K, Kaji D, Sasaki M, et al. Predominance of ST8 and CC1/spa-t1784 methicillin-resistant Staphylococcus aureus isolates in Japan and their genomic characteristics. J Glob Antimicrob Resist. 2022;28:195–202.
- Takano T, Hung W-C, Shibuya M, et al. A new local variant (ST764) of the globally disseminated ST5 lineage of hospital-associated methicillin-resistant Staphylococcus aureus (MRSA) carrying the virulence determinants of community-associated MRSA. Antimicrob Agents Chemother. 2013;57:1589–1595.
- Urushibara N, Kawaguchiya M, Onishi M, et al. Novel structures and temporal changes of arginine catabolic mobile elements in methicillin-resistant Staphylococcus aureus genotypes ST5-MRSA-II and ST764-MRSA-II in Japan. Antimicrob Agents Chemother. 2016;60:3119–3122.
- Kawaguchiya M, Urushibara N, Ghosh S, et al. Genetic diversity of emerging Panton–Valentine leukocidine/arginine catabolic mobile element (ACME)-positive ST8 SCCmec-IVa meticillin-resistant Staphylococcus aureus (MRSA) strains and ACME-positive CC5 (ST5/ST764) MRSA strains in northern Japan. J Med Microbiol. 2013;62:1852–1863.
- Shore AC, Rossney AS, Brennan OM, et al. Characterization of a novel arginine catabolic mobile element (ACME) and staphylococcal chromosomal cassette mec composite island with significant homology to Staphylococcus epidermidis ACME Type II in methicillin-resistant Staphylococcus aureus Genotype ST22-MRSA-IV▿. Antimicrob Agents Chemother. 2011;55:1896–1905.
- Espedido BA, Steen JA, Barbagiannakos T, et al. Carriage of an ACME II variant may have contributed to methicillin-resistant Staphylococcus aureus sequence type 239-like strain replacement in Liverpool Hospital, Sydney, Australia. Antimicrob Agents Chemother. 2012;56:3380–3383.
- Thurlow LR, Joshi GS, Clark JR, et al. Functional modularity of the arginine catabolic mobile element contributes to the success of usa300 methicillin-resistant Staphylococcus aureus. Cell Host Microbe. 2013;13:100–107.
- Chen S, Jin Y, Lin C, et al. Low prevalence of mupirocin resistance among Staphylococcus aureus clinical isolates from a Chinese tertiary hospital. J Med Microbiol. 2019;68:201–205.
- Chen W, He C, Yang H, et al. Prevalence and molecular characterization of methicillin-resistant Staphylococcus aureus with mupirocin, fusidic acid and/or retapamulin resistance. BMC Microbiol. 2020;20:183.
- Wang B, Xu Y, Zhao H, et al. Methicillin-resistant Staphylococcus aureus in China: a multicentre longitudinal study and whole-genome sequencing. Emerg Microbes Infect. 2022;11:532–542.
- Zhao R, Wang X, Wang X, et al. Molecular characterization and virulence gene profiling of methicillin-resistant Staphylococcus aureus associated with bloodstream infections in southern China. Front Microbiol. 2022;13:1008052.
- Zhang H, Tian L, Chen T, et al. Prevalence and WGS-based characteristics of MRSA isolates in hospitals in Shanghai, China. Front Microbiol. 2022;13. Available from: https://www.frontiersin.org/articles/10.3389fmicb.2022.1002691.
- Gu F, He W, Xiao S, et al. Antimicrobial resistance and molecular epidemiology of Staphylococcus aureus causing bloodstream infections at Ruijin Hospital in Shanghai from 2013 to 2018. Sci Rep. 2020;10:6019.
- He C, Xu S, Zhao H, et al. Leukotoxin and pyrogenic toxin superantigen gene backgrounds in bloodstream and wound Staphylococcus aureus isolates from eastern region of China. BMC Infect Dis. 2018;18:395.
- Aung MS, Urushibara N, Kawaguchiya M, et al. Clonal diversity of methicillin-resistant Staphylococcus aureus (MRSA) from bloodstream infections in northern Japan: identification of spermidine N-acetyltransferase gene (speG) in staphylococcal cassette chromosomes (SCCs) associated with type II and IV SCCmec. J Glob Antimicrob Resist. 2021;24:207–214.
- Chen S, Zhou Y, Chen Y, et al. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinforma Oxf Engl. 2018;34:i884–i890.
- Wick RR, Judd LM, Gorrie CL, et al. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13:e1005595.
- Kaya H, Hasman H, Larsen J, et al. SCCmecFinder, a web-based tool for typing of staphylococcal cassette chromosome mec in Staphylococcus aureus using whole-genome sequence data. mSphere. 2018;3:e00612–17.
- Bartels MD, Petersen A, Worning P, et al. Comparing whole-genome sequencing with Sanger sequencing for spa typing of methicillin-resistant Staphylococcus aureus. J Clin Microbiol. 2014;52:4305–4308.
- Liu B, Zheng D, Jin Q, et al. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019;47:D687–D692.
- Alcock BP, Raphenya AR, Lau TTY, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020;48:D517–D525.
- Treangen TJ, Ondov BD, Koren S, et al. The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15:524.
- Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–W296.
- Alikhan N-F, Petty NK, Ben Zakour NL, et al. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402.
- Wang X, Zhao H, Wang B, et al. Identification of methicillin-resistant Staphylococcus aureus ST8 isolates in China with potential high virulence. Emerg Microbes Infect. 2022;11:507–518.
- Shang W, Hu Q, Yuan W, et al. Comparative fitness and determinants for the characteristic drug resistance of ST239-MRSA-III-t030 and ST239-MRSA-III-t037 strains isolated in China. Microb Drug Resist Larchmt N. 2016;22:185–192.
- Bronner S, Monteil H, Prévost G. Regulation of virulence determinants in Staphylococcus aureus: complexity and applications. FEMS Microbiol Rev. 2004;28:183–200.
- Olson ME, Nygaard TK, Ackermann L, et al. Staphylococcus aureus nuclease is an SaeRS-dependent virulence factor. Infect Immun. 2013;81:1316–1324.
- Mainiero M, Goerke C, Geiger T, et al. Differential target gene activation by the Staphylococcus aureus two-component system saeRS. J Bacteriol. 2010;192:613–623.
- Steinig EJ, Duchene S, Robinson DA, et al. Evolution and global transmission of a multidrug-resistant, community-associated methicillin-resistant Staphylococcus aureus lineage from the Indian subcontinent. mBio. 2019;10:e01105–19.
- Kim H, Park S, Seo H, et al. Clinical impact of and microbiological risk factors for qacA/B positivity in ICU-acquired ST5-methicillin-resistant SCCmec type II Staphylococcus aureus bacteremia. Sci Rep. 2022;12:11413.
- Bubeck Wardenburg J, Bae T, Otto M, et al. Poring over pores: α-hemolysin and Panton–Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med. 2007;13:1405–1406.
- Kobayashi SD, Malachowa N, Whitney AR, et al. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J Infect Dis. 2011;204:937–941.
- Berube BJ, Wardenburg JB. Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins. 2013;5:1140–1166.
- Montgomery CP, Boyle-Vavra S, Daum RS. Importance of the global regulators Agr and SaeRS in the pathogenesis of CA-MRSA USA300 infection. PLoS One. 2010;5:e15177.
- Novick RP, Ross HF, Projan SJ, et al. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 1993;12:3967–3975.
- Jenul C, Horswill AR. Regulation of Staphylococcus aureus virulence. Microbiol Spectr. 2019;7(2).
- Le KY, Otto M. Quorum-sensing regulation in staphylococci – an overview. Front Microbiol. 2015;6:1174.
- Knight GM, Budd EL, Whitney L, et al. Shift in dominant hospital-associated methicillin-resistant Staphylococcus aureus (HA-MRSA) clones over time. J Antimicrob Chemother. 2012;67:2514–2522.
- Mongkolrattanothai K, Boyle S, Kahana MD, et al. Severe Staphylococcus aureus infections caused by clonally related community-acquired methicillin-susceptible and methicillin-resistant isolates. Clin Infect Dis Off Publ Infect Dis Soc Am. 2003;37:1050–1058.
- Ender M, McCallum N, Adhikari R, et al. Fitness cost of SCCmec and methicillin resistance levels in Staphylococcus aureus. Antimicrob Agents Chemother. 2004;48:2295–2297.