
ABSTRACT
The fitness of multidrug-resistant tuberculosis (MDR-TB) is thought to be an important determinant of a strain’s ability to be transmitted. Studies in the laboratory have demonstrated that MDR-TB strains have reduced fitness but the relative transmissibility of MDR-TB versus drug-susceptible (DS) TB strains in human populations remains unresolved. We used data on genomic clustering from our previous molecular epidemiological study in Songjiang (2011-2020) and Wusheng (2009-2020), China, to compare the relative transmissibility of MDR-TB versus DS-TB. Genomic clusters were defined with a threshold distance of 12-single-nucleotide-polymorphisms and the risk for MDR-TB clustering was analyzed by logistic regression. In total, 2212 culture-positive pulmonary TB patients were enrolled in Songjiang and 1289 in Wusheng. The clustering rates of MDR-TB and DS-TB strains were 19.4% (20/103) and 26.3% (509/1936), respectively in Songjiang, and 43.9% (29/66) and 26.0% (293/1128) in Wusheng. The risk of MDR-TB clustering was 2.34 (95% CI 1.38-3.94) times higher than DS-TB clustering in Wusheng and 0.64 (95% CI 0.38-1.06) times lower in Songjiang. Neither lineage 2, compensatory mutations nor rpoB S450L were significantly associated with MDR-TB transmission, and katG S315 T increased MDR-TB transmission only in Wusheng (OR 5.28, 95% CI 1.42-19.21). MDR-TB was not more transmissible than DS-TB in either Songjiang or Wusheng. It appears that the different transmissibility of MDR-TB in Songjiang and Wusheng is likely due to differences in the quality of the local TB control programmes. Suggesting that the most effective way to control MDR-TB is by improving local TB control programmes.
Introduction
It was originally thought that drug-resistant (DR) mutations in Mycobacterium tuberculosis introduce fitness costs [Citation1, Citation2] that compromise the virulence of the DR strains and hinder their ability to be transmitted and cause secondary cases [Citation3, Citation4]. Accordingly, mathematical modelling predicted that this reduced fitness would limit the spread of multidrug-resistant tuberculosis (MDR-TB) and minimize its impact on global TB control [Citation5]. These predictions, however, appear to have grossly underestimated the growth and persistence of DR strains, as the MDR-TB epidemic has become a public health issue of global concern [Citation6]. In 2019, there were 464,000 global cases of rifampicin-resistant TB, 78% of which were MDR [Citation7]. Early in the MDR-TB epidemic, most MDR cases likely arose through the secondary acquisition of drug resistance as a result of inadequate therapy of drug-susceptible (DS) TB. Currently, though, the majority of MDR-TB cases are due to the transmission of MDR strains [Citation8–11]. The inconsistency between the theory of lower fitness in DR strains and the reality of the global MDR-TB epidemic has led researchers to explore the proposed fitness effects of DR mutations by comparing the relative transmissibility of MDR-TB versus DS-TB strains.
Although laboratory studies have demonstrated that DR strains have reduced fitness [Citation3, Citation4], in vitro fitness does not provide an accurate representation of bacterial transmissibility. The transmission of M. tuberculosis is a complex process influenced by many determinants, including host factors, bacterial factors and socioeconomic status. While it is difficult to control for the non-bacterial variables, population-based epidemiological studies nevertheless provide an in vivo measure of the transmissibility of M. tuberculosis.
Studies of the transmissibility of MDR-TB in human populations have approached the question in two ways. One approach compares the risk of infection or disease among household contacts of patients with DS-TB versus MDR-TB. The second approach compares the risk of genomic clustering in patients with DS-TB versus those with MDR-TB in the general population [Citation12, Citation13]. In a small study conducted in Brazil, Teixeira et al. found that the risk of infection and disease among household contacts of patients with DS-TB and MDR-TB was comparable [Citation14]. However, several studies found a greater risk of infection [Citation13, Citation15] and a lower risk of disease [Citation12] among household contacts of MDR-TB patients. Studies based on molecular epidemiology have also reached contradictory conclusions, with some studies finding that MDR-TB strains were more likely to belong to genomic-clusters than DS strains [Citation16–20], while other studies found less clustering of MDR strains [Citation21–24]. In addition to the discrepancy of the findings, the differences in study designs limit comparisons between these reports, and as a result, the relative transmissibility of MDR-TB versus DS-TB strains remains unresolved.
We previously conducted long-term prospective molecular epidemiological studies of TB in the Songjiang district of Shanghai and the Wusheng County of Sichuan, China [Citation25, Citation26]. In this report, we analyzed the data on genomic clustering from that study to compare the relative transmissibility of MDR-TB versus DS-TB.
Methods
Study design and population
Data for this work was obtained from our previous molecular epidemiological studies conducted in Songjiang and Wusheng. Songjiang is a suburban district of Shanghai, one of the most developed cities in China and a frequent destination for internal migrant workers. As 62% of the total Songjiang population are internal migrants, who contribute approximately 75% of the TB cases in the district, Songjiang is a representative site for epidemiological studies of TB in Chinese cities [Citation25]. Community physicians routinely identify individuals with TB-like symptoms or abnormal chest radiographs and refer them to the TB-designated hospital in Songjiang for diagnosis by sputum smear and culture. The study population was comprised of all culture-positive pulmonary TB patients 15 years or older who were diagnosed between Jan 1, 2011 and December 31, 2020. Whole-genome sequencing (WGS) was performed on all of the cultured isolates. At the time of TB diagnosis we collected demographic, clinical and laboratory information on each patient. The same study design used in Songjiang was also used to investigate TB patients diagnosed in Wusheng, a representative rural county in southwestern China, between July 1, 2009 and December 31, 2020 (Supplementary Figure S1) [Citation26]. The study was approved by the institutional review board of the Institutes of Biomedical Sciences, Fudan University and all enrolled patients provided written informed consent.
WGS
All clinical strains were re-cultured and sequenced as described [Citation27], and a previously validated pipeline was used to identify single nucleotide polymorphisms (SNPs) [Citation26]. In brief, raw sequence reads were trimmed with Sickle (version 1.33) and aligned to the inferred M. tuberculosis complex ancestor sequence [Citation28] using BWA-MEM. SAMtools (version 1.3.1) and Varscan (version 2.3.6) were then used to identify SNPs. Pairwise SNP distances were calculated based on the fixed SNPs with a frequency ≥75%. A genomic cluster was defined as strains differing by ≤12 SNPs, consistent with linkage through recent transmission [Citation11].
Based on the identified SNPs, a phylogeny tree was constructed with RAxML-NG (version 1.0.2) software, using the maximum likelihood method with 100 bootstraps and visualized with Interactive Tree of Life (https://itol.embl.de/). Drug-resistance profiles were predicted for the four first-line drugs based on the mutations reported to be associated with resistance [Citation29]. DS-TB was defined as susceptibility to the four first-line drugs and MDR-TB was defined as resistance to at least isoniazid and rifampicin. Compensatory mutations (CMs) were identified based on the mutations previously reported (Supplementary Table S4) [Citation30, Citation31].
Statistical analysis
Non-normal continuous data was expressed as medians and interquartile ranges (IQR), while categorical variables were described using proportions. Differences between groups were tested using the Wilcoxon rank sum test or the chi-square test. Logistic regression was used to calculate the risk of clustering of MDR-TB strains, given as odds ratios (OR) with 95% confidence intervals (CI). Variables with p-values less than 0.2 in the univariable analysis were included in the multivariable analysis to calculate the adjusted odd ratios (aOR). OR and 95% CI were also calculated to test the association between bacterial genetic background and increased MDR-TB transmission. Results with a p-value less than 0.05 were considered statistically significant. All analyses were performed in Stata version 14.0.
Results
General population characteristics
Between January 1, 2011 and December 31, 2020, there were 3157 bacteriologically confirmed pulmonary TB patients diagnosed in Songjiang, of which 2444 (77.4%) were culture-positive. After excluding patients whose isolates were non-tuberculous mycobacteria (NTM) and patients whose strains failed re-culture or alignment, a total of 2212 patients were included in the final analysis (A). Sample enrolment in Wusheng was described previously [Citation26]. In brief, between July 1, 2009 and December 31, 2020 there were 1473 culture-positive pulmonary TB patients diagnosed in Wusheng, and after excluding patients whose isolates were NTM or whose strains failed re-culture or alignment, a total of 1289 patients were included in the final analysis (B). The characteristics of patients are shown in . Compared with patients in Wusheng, patients in Songjiang were younger (30.0 years vs 50.0 years), had shorter diagnosis delays (3.7 weeks vs 3.9 weeks) and were less likely to be male (69.6% vs 79.0%).
Figure 1. Sample enrolment in Songjiang (A) and Wusheng (B).
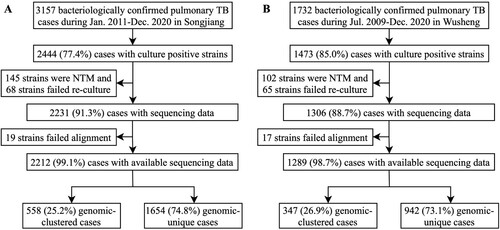
Table 1. Characteristics of pulmonary tuberculosis patients in Songjiang and Wusheng.
WGS for genotyping and drug resistance prediction
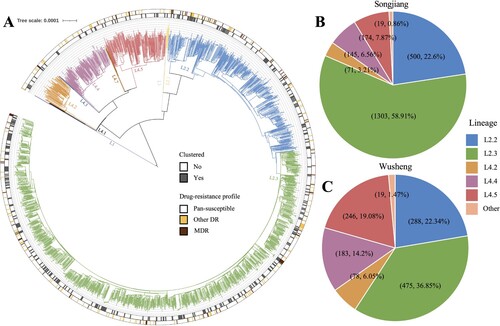
The whole genomes of 3501 strains were sequenced with a median depth of 134X (IQR, 101-158) and a median coverage of 99.3% (IQR, 99.2-99.4). Phylogenetic analysis revealed that most strains belonged to Lineage 2 (L2) (; ): 81.8% (1809/2212) in Songjiang and 59.4% (766/1289) in Wusheng. Almost all non-L2 strains belonged to L4, predominantly sublineages L4.2, L4.4 and L4.5 (). The drug-resistance profiles were similar between the two sites (). The proportions of DS-TB were 87.5% (1936/2212; 1128/1289) in both Songjiang and Wusheng, and the proportions of MDR-TB were 4.7% (103/2212) and 5.1% (66/1289) respectively.
Figure 2. (A) Phylogeny, clustering, and resistance profile of 2212 Mycobacterium tuberculosis strains isolated in Songjiang. (B) The sublineage composition of isolates from patients in Songjiang. (C) The sublineage composition of isolates from patients in Wusheng.
To identify risk factors associated with the notified MDR-TB patients, we compared the characteristics of MDR and non-MDR patients (Supplementary Table S1). There was a higher proportion of MDR in retreated patients in both sites. In Songjiang, 16.9% (21/124) of the retreated patients and 3.9% (82/2088) of the new patients had MDR-TB, while in Wusheng 16.3% (15/92) and 4.3% (51/1197) of retreated and new patients, respectively, had MDR-TB. In Songjiang there was also a higher proportion of MDR-TB in migrant patients (5.4%, 89/1652) than in patients whose home residence was registered (hukou) as Shanghai (2.5%, 14/560).
Transmission of MDR-TB and its risk factors
To assess the relative transmissibility, we calculated the clustering rates for MDR and DS strains. The clustering rates for MDR and DS strains were 19.4% (20/103) and 26.3% (509/1936), respectively in Songjiang, and 43.9% (29/66) and 26.0% (293/1128) respectively in Wusheng. The median cluster sizes of MDR and DS strains were similar in both Songjiang – 2 (IQR, 2-2) vs 2 (IQR, 2-3), and Wusheng – 2 (IQR, 2-3) vs 2 (IQR, 2-3) (Supplementary Figure S2). We then calculated the aOR for the risk of MDR clustering by adjusting for demographic, clinical and laboratory data (; Supplementary Table S3). This analysis estimated that in Wusheng the MDR strains were 2.34 (95% CI 1.38-3.94) times more likely to be clustered than DS strains, while in Songjiang the MDR strains were 0.64 (95% CI 0.38-1.06) times less likely to be clustered than DS strains, suggesting that the transmissibility of MDR-TB strains differed in the two sites.
Table 2. Multivariable logistic regression of risk factors for clustering in Songjiang and Wusheng.
To identify possible risk factors associated with MDR clustering, we compared the characteristics of clustered and non-clustered MDR patients (Supplementary Table S2). We found that in Wusheng none of the patient characteristics were associated with clustering, while in Songjiang, only gender and diagnostic delay were associated with MDR clustering. In Songjiang, the clustering rate was significantly higher (p = 0.019) in male MDR patients (25.7%, 18/70) than in female MDR patients (6.1%, 2/33) and the diagnosis delay was longer in clustered MDR patients (5.0 weeks vs. 2.7 weeks, p = 0.0298), but no other patient characteristics were associated with clustering.
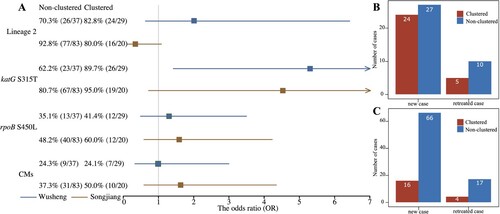
To determine the proportion of MDR-TB cases due to transmission, we separated the new and retreated MDR patients into those with clustered or non-clustered isolates (B/C). All clustered MDR patients and non-clustered new MDR patients presumably had primary infections with MDR strains. There were 20 and 29 clustered MDR-TB patients in Songjiang and Wusheng respectively, and 66 and 27, respectively, non-clustered new patients with MDR-TB. Therefore, at least 83.5% (86/103) of MDR patients in Songjiang and 84.8% (56/66) in Wusheng were likely caused by the transmission of MDR strains.
Figure 3. (A) Forest plot of the impact of lineage 2, katG S315 T, rpoB S450L and compensatory mutations (CMs) on MDR-TB transmission risk. (B) Number of new and retreated MDR-TB cases in Wusheng, stratified by clustered (red) and non-clustered (blue) cases. (C) Number of new and retreated MDR-TB cases in Songjiang, stratified by clustered (red) and non-clustered (blue) cases.
Impact of bacterial genetic background on MDR-TB transmission
Among the MDR-TB strains in Songjiang and Wusheng, 90.3% (93/103) and 75.8% (50/66), respectively, belonged to L2. Different M. tuberculosis lineages are thought to vary in their inherent fitness and it has been suggested that the genetic background of L2 strains can somehow mitigate the fitness costs of rifampicin-resistance mutations [Citation1]. Consistent with this notion, in Wusheng the 48.0% (24/50) clustering rate for L2 strains was higher than the 31.3% (5/16) rate for non-L2 strains. The opposite was found in Songjiang, however, where the 40.0% (4/10) clustering rate for non-L2 strains was more than twice the 17.2% (16/93) clustering rate for L2 strains. When we analyzed the overall association of L2 with MDR clustering, however (A), we found no significant correlation of lineage with clustering in either Wusheng (OR 2.03, 95% CI 0.63-6.43) or Songjiang (OR 0.31, 95% CI 0.08-1.15). Previous studies have suggested that L2 strains have an increased capacity for transmission [Citation32], and we also found that patients with an L2 strain had a greater risk of clustering (). L2 strains did not, however, show an increased rate of MDR clustering.
Compensatory mutations (CMs) are thought to alleviate the fitness costs of some drug-resistant mutations, especially the rpoB mutations confering resistance to rifampicin. If CMs restore the fitness MDR strains, they could perhaps augment the transmission success of MDR-TB [Citation33, Citation34]. We identified a total of 23 different CMs (Supplementary Table S4) that were present in 39.8% (41/103) of the MDR-TB strains in Songjiang and in 24.2% (16/66) of the MDR-TB strains in Wusheng. The clustering rates for MDR-TB strains with and without CMs were 24.4% (10/41) and 16.1% (10/62) in Songjiang, and 43.8% (7/16) and 44.0% (22/50) in Wusheng. Statistical analysis of these proportions (A), however, showed that CMs had no significant association with the risk of MDR clustering in either Songjiang (OR 1.68, 95% CI 0.64-4.40) or Wusheng (OR 0.99, 95% CI 0.33-3.00).
In clinical isolates, rpoB S450L is the most common mutation conferring rifampicin resistance and katG S315 T is the most common mutations conferring isoniazid resistance, and both mutations have been shown to have low in vitro fitness costs [Citation2, Citation35]. The rpoB S450L mutation was found in 50.5% (52/103) of MDR-TB strains in Songjiang and 37.9% (25/66) of MDR-TB strains in Wusheng. Although the clustering rates for strains with the rpoB S450L mutation were higher than for strains without this mutation in both Songjiang – 23.1% (12/52) vs 15.7% (8/51), and Wusheng – 48.0% (12/25) vs 41.5% (17/41) (A), the differences were not significant in either site: Songjiang (OR 1.61, 95% CI 0.61-4.25); Wusheng (OR 1.30, 95% CI 0.49-3.50). This suggests that if the putative low fitness cost of the rpoB S450L mutation promotes transmission, the effect is small and would require a larger data set to perhaps reach statistical significance.
A similar analysis was applied to the katG S315 T substitution that confers isoniazid resistance. This mutation was found in 83.5% (86/103) and 74.2% (49/66) of MDR-TB strains in Songjiang and Wusheng, respectively, and the clustering rates for strains with and without katG S315 T were 53.1% (26/49) and 17.6% (3/17) in Wusheng and 22.1% (19/86) and 5.9% (1/17) in Songjiang. Statistical analysis found that in Wusheng the MDR-TB strains with katG S315 T had a significantly increased risk of clustering (A) (OR 5.28, 95% CI 1.42-19.21), but there was no association of katG S315 T with clustering in Songjiang (OR 4.54, 95% CI 0.71-∞).
Discussion
In this report we compared the transmissibility of MDR-TB versus DS-TB strains using data from long-term molecular epidemiological studies conducted in Songjiang and Wusheng. We found that the clustering rates for MDR and DS strains were 43.9% and 26.0% in Wusheng and 19.4% and 26.3% in Songjiang, indicating that compared to DS-TB strains, the MDR-TB strains showed greater transmissibility in Wusheng (aOR = 2.34, 95% CI 1.38-3.94) but weaker transmissibility in Songjiang (aOR = 0.64, 95% CI 0.38-1.06). Further analysis showed that neither lineage 2, CMs nor the rpoB S450L substitution were significantly associated with MDR-TB transmission, and the katG S315 T substitution increased MDR-TB transmission only in Wusheng (OR 5.28, 95% CI 1.42-19.21).
Population-based molecular epidemiological studies have produced conflicting results on the relative transmissibility of MDR versus DS strains. Alland et al. collected 104 strains from patients diagnosed in a New York City hospital during 1989–1992 and found that the clustering risk for MDR-TB strains was 6.5 (95% CI 1.5-33.3) times higher than for DS-TB strains [Citation16]. Similarly, a small study in Archangel Oblast, Russia by Toungoussova et al. collected 119 strains in 1998 and 1999 and found that MDR-TB strains were much more likely to be clustered (OR 9.2, 95% CI 2.4-47.7) [Citation17]. Yang et al. conducted a molecular epidemiological study in five regions in China in 2009–2012 and also found that MDR-TB strains had a greater risk of clustering (OR 1.86, 95% CI 1.25-2.63) than DS-TB strains [Citation18]. In contrast, other studies reported opposite results. Garcia-Garcia et al. collected 326 strains in the Orizaba Health Jurisdiction of Mexico during 1995–1999 and found that MDR-TB strains had a lower risk of clustering (OR 0.31, 95% CI 0.12-0.81) [Citation22]. Godfrey-Faussett et al. conducted a molecular epidemiological study of 371 goldminers with culture-positive TB in South Africa and also found that the MDR-TB patients had a lower risk of clustering (OR 0.27, 95% CI 0.09-0.83) [Citation23]. In a molecular epidemiological study from 2014–2017 in Shenzhen, China, Yang et al. found that MDR-TB and DS-TB strains had similar risks of clustering (OR 0.85, 95% CI 0.46-1.57) [Citation24]. Many of these previous studies were of short duration and had small sample sizes, and the differences in study design and genotyping methods make it difficult to draw comparisons between them. We based our analysis on the results of a 10-year prospective molecular epidemiological study using the same design in both urban Songjiang and rural Wusheng and therefore believe our data faithfully represents the transmissibility of the TB strains and allow meaningful comparisons of the results from the two sites.
At present there is no model that faithfully simulates human TB transmission. This limits our understanding of the relative transmissibility of MDR-TB versus DS-TB and the role of different factors that influence transmission, but epidemiological data can provide useful information. There are many incompletely defined influences affecting MDR-TB transmission, including host factors, bacterial factors, and socioeconomic status. Socioeconomic status is especially complex and difficult to assess quantitatively, but is generally reflected in the quality or effectiveness of the local TB control programmes and its ability to find and cure local TB cases, thereby limiting transmission. We refer to this generically as the quality of the local TB control programmes, which is possible to improve. Because we cannot quantify the efficacy of TB prevention and control programme, we used the exclusion method to analyze the influence of other factors that may affect MDR-TB transmission. Our analysis found that both host (; Supplementary Table S3) and bacterial factors () had a limited effect on MDR-TB transmission, and therefore believe that the differences in the transmissibility of MDR-TB in Songjiang and Wusheng are mainly due to differences in the quality of the local TB control programmes. Although it is difficult to quantify and compare the effectiveness of local TB prevention and control programmes, our data, derived from two regions with distinct TB prevention and control programmes, provides an opportunity to assess their influence on the transmissibility of MDR-TB strains. Shanghai is one of the most developed cities in China with one of the best TB control programmes and a TB incidence of 26.5 per 100,000 population [Citation36]. Since 2011, Shanghai has implemented an integrated MDR-TB control model that includes MDR-TB patient detection, expert panel consultation on diagnosis, treatment in designated hospitals, case management and medical cost reimbursement [Citation37]. Wusheng is a poor rural area with a TB incidence of 95.8 per 100,000 population [Citation38]. Up until 2021, programmes for the diagnosis, treatment and management of MDR-TB patients were lacking in Wusheng and MDR-TB patients seeking treatment had to travel voluntarily to TB-designated provincial medical institutions far from their homes.
High quality TB prevention and control programmes that diagnose and effectively treat MDR-TB early in the course of the disease can minimize the time that MDR-TB patients are infectious and thereby interrupt MDR-TB transmission. As a result, the clustering rates of MDR and DS strains would be similar, as we found in Shanghai. In contrast, in areas with inadequate TB control programmes, the delay in diagnosing and appropriately treating MDR-TB would leave infectious MDR patients in their community for longer time periods, leading to more transmission. Without the ability to promptly detect MDR-TB, all patients would receive a first-line treatment regimen that would reduce DS-TB transmission but allow continued spread of MDR-TB strains, leading to higher MDR-TB clustering rates.
Although many drug-resistance mutations are thought to engender a vary extent of fitness cost [Citation1–4], other bacterial genetic factors, including belonging to Lineage 2, harbouring the katG S315 T and rpoB S450L substitutions and having CMs, have been associated with increased or restored bacterial fitness, manifested as an increased risk for genomic clustering [Citation1, Citation2, Citation33–35]. Several population-based molecular epidemiological studies have found that the bacterial genetic background was associated with increased MDR-TB transmission. Gygli et al. analyzed MDR-TB strains isolated in Georgia from 2011–2016 and found that strains carrying CMs had greater transmissibility (OR 1.34, 95% CI 1.05-1.71) [Citation31] than those without CMs. Nonghanphithak et al. analyzed DR strains isolated in Thailand from 2014–2017 and found that strains with the katG S315 T (OR 3.90, 95% CI 2.46-6.18) or rpoB S450L (OR 3.29, 95% CI 2.32-4.66) substitutions had greater transmissibility [Citation39]. These results, however, could reflect specific local circumstances and may not apply to all situations. We recently analyzed global data sets to assess the influence of these bacterial factors on MDR-TB transmission and found that only the katG S315 T substitution facilitates MDR-TB transmission, and this association was significant solely in developing countries and regions with high TB burdens [Citation40]. The current report again shows that the bacterial genetic background appears to have a limited effect on MDR-TB transmission, but also confirmed that katG S315 T can facilitate MDR-TB transmission in a high TB-burden rural setting with an inadequate control programme.
It is undeniable that the bacterial genetic background has a significant impact on TB transmission in regions where the specific environments and conditions lead large clusters of single clones [Citation31, Citation39]. For reasons that are undefined, however, no large single-clone clusters have been described in China [Citation41], suggesting that in China the bacterial genetic background is perhaps less important than in other settings.
While a favourable genomic background may increase the transmissibility of some MDR-TB strains, this advantage may not have the opportunity to be manifested if MDR-TB is promptly diagnosed and appropriately treated by an effective TB control programme. An inadequate TB control programme, however, may provide conditions in which the advantageous bacterial genetic factors can promote transmission, which would be lead to increased clustering rates. In our study this was seen with katG S315 T isoniazid resistance substitution, which increased MDR-TB transmission in the setting of inadequate TB control in Wusheng but not with the more effective TB control in Songjiang. Transmission is a complex process but the bacterial genetic background does not appear to be the main determinant of MDR-TB transmissibility and clustering, at least in China.
The major limitation of our study is that, despite its 10-year duration, the populations covered and the total number of MDR-TB patients are relatively small. In addition we did not comprehensively analyze and could not quantify the efficacies of the TB prevention and control programmes and their effects on MDR-TB transmission. Our characterization of TB control is based on the TB incidence rates at the two sites and the control measures that have been implemented. Because our study included both urban and rural settings, we believe our results are likely applicable to all of China, but may not be applicable to other settings, especially areas with a high TB incidence, large MDR outbreaks and poor TB control.
In conclusion, MDR-TB strains do not appear to be more transmissible than DS-TB strains in Songjiang and Wusheng. The effect of the host and bacterial factors on MDR-TB transmission appears to be limited and less important than the quality of the local TB control programmes. Our results imply that the most effective way to interrupt MDR-TB transmission and reduce the global incidence of MDR-TB is by improving the diagnosis, treatment and management of MDR-TB patients.
Authors’ contributions
M.L., Q.J., L.X., C.Y., X.S., C.C., and Q.G. conceived, designed and managed the study. L.L., M.G., Y.J., S.Z., Y.Q., J.H., and X.G. contributed the demographic, clinical and microbiological data. M.L., Q.J., and Y.C. did the data analyses. M.L., Q.J., C.Y., Y.C., H.T., and Q.G. drafted and revised the manuscript. All authors contributed to and gave input to the final article.
Ethical approval
The study was approved by the institutional review board of Biomedical Sciences, Fudan University and all enrolled patients provided written informed consent.
Supplemental Material
Download MS Word (1.6 MB)Acknowledgments
We thank the tuberculosis public health teams in the Songjiang District Centre for Disease Control and Prevention and the Wusheng County Centre for Disease Control and Prevention. We also thank Prof. Sebastien Gagneux for his valuable comments on this manuscript.
Data availability
The datasets used and analyzed during the current study are not accessible online, but may be made available upon written request to the corresponding author. Sequencing data were deposited in the Genome Sequence Archive (https://bigd.big.ac.cn/gsa) under BioProject PRJCA008815 and PRJCA010372.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Gagneux S, Long CD, Small PM, et al. The competitive cost of antibiotic resistance in mycobacterium tuberculosis. Science. 2006 Jun 30;312(5782):1944–1946.
- Gygli SM, Borrell S, Trauner A, et al. Antimicrobial resistance in mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol Rev. 2017 May 1;41(3):354–373.
- Cohen T, Sommers B, Murray M. The effect of drug resistance on the fitness of mycobacterium tuberculosis. Lancet Infect Dis. 2003 Jan;3(1):13–21.
- Luciani F, Sisson SA, Jiang H, et al. The epidemiological fitness cost of drug resistance in mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2009 Aug 25;106(34):14711–5.
- Dye C, Williams BG, Espinal MA, et al. Erasing the world’s slow stain: strategies to beat multidrug-resistant tuberculosis. Science. 2002 Mar 15;295(5562):2042–2046.
- Lange C, Dheda K, Chesov D, et al. Management of drug-resistant tuberculosis. Lancet. 2019 Sep 14;394(10202):953–966.
- WHO. Global tuberculosis report 2020. Geneva: World Health Organization; 2020.
- Casali N, Nikolayevskyy V, Balabanova Y, et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet. 2014 Mar;46(3):279–286.
- Eldholm V, Monteserin J, Rieux A, et al. Four decades of transmission of a multidrug-resistant mycobacterium tuberculosis outbreak strain. Nat Commun. 2015 May 11;6:7119.
- McBryde ES, Meehan MT, Doan TN, et al. The risk of global epidemic replacement with drug-resistant mycobacterium tuberculosis strains. Int J Infect Dis. 2017 Mar;56:14–20.
- Yang C, Luo T, Shen X, et al. Transmission of multidrug-resistant mycobacterium tuberculosis in Shanghai, China: a retrospective observational study using whole-genome sequencing and epidemiological investigation. Lancet Infect Dis. 2017 Mar;17(3):275–284.
- Grandjean L, Gilman RH, Martin L, et al. Transmission of multidrug-resistant and drug-susceptible tuberculosis within households: a prospective cohort study. PLoS Med. 2015 Jun;12(6):e1001843, discussion e1001843.
- Becerra MC, Huang CC, Lecca L, et al. Transmissibility and potential for disease progression of drug resistant mycobacterium tuberculosis: prospective cohort study. Br Med J. 2019 Oct 24;367:l5894.
- Teixeira L, Perkins MD, Johnson JL, et al. Infection and disease among household contacts of patients with multidrug-resistant tuberculosis. Int J Tuberc Lung Dis. 2001 Apr;5(4):321–328.
- Fox GJ, Anh NT, Nhung NV, et al. Latent tuberculous infection in household contacts of multidrug-resistant and newly diagnosed tuberculosis. Int J Tuberc Lung Dis. 2017 Mar 1;21(3):297–302.
- Alland D, Kalkut GE, Moss AR, et al. Transmission of tuberculosis in New York city. An analysis by DNA fingerprinting and conventional epidemiologic methods. N Engl J Med. 1994 Jun 16;330(24):1710–1716.
- Toungoussova OS, Sandven P, Mariandyshev AO, et al. Spread of drug-resistant mycobacterium tuberculosis strains of the Beijing genotype in the archangel oblast, Russia. J Clin Microbiol. 2002 Jun;40(6):1930–1937.
- Yang C, Shen X, Peng Y, et al. Transmission of mycobacterium tuberculosis in China: a population-based molecular epidemiologic study. Clin Infect Dis. 2015 Jul 15;61(2):219–227.
- Li M, Guo MC, Qiu Y, et al. Transmission characteristics of student tuberculosis: a 12-year prospective population-based genomic epidemiological study. Chin J Tuberc Respir Dis. 2023 Jan 12;46(1):19–26.
- Yang C, Sobkowiak B, Naidu V, et al. Phylogeography and transmission of M. tuberculosis in Moldova: a prospective genomic analysis. PLoS Med. 2022 Feb;19(2):e1003933.
- Saavedra Cervera B, Lopez MG, Chiner-Oms A, et al. Fine-grain population structure and transmission patterns of mycobacterium tuberculosis in southern Mozambique, a high TB/HIV burden area. Microb Genom. 2022 Jul;8(7):mgen000844.
- Garcia-Garcia ML, Jimenez-Corona ME, Ponce-de-Leon A, et al. Mycobacterium tuberculosis drug resistance in a suburban community in southern Mexico. Int J Tuberc Lung Dis. 2000 Dec;4(12 Suppl 2):S168–S170.
- Godfrey-Faussett P, Sonnenberg P, Shearer SC, et al. Tuberculosis control and molecular epidemiology in a South African gold-mining community. Lancet. 2000 Sep 23;356(9235):1066–1071.
- Yang T, Wang Y, Liu Q, et al. A population-based genomic epidemiological study of the source of tuberculosis infections in an emerging city: Shenzhen, China. Lancet Reg Health-W. 2021 Mar;8:100106.
- Yang C, Lu L, Warren JL, et al. Internal migration and transmission dynamics of tuberculosis in Shanghai, China: an epidemiological, spatial, genomic analysis. Lancet Infect Dis. 2018 Jul;18(7):788–795.
- Li M, Guo M, Peng Y, et al. High proportion of tuberculosis transmission among social contacts in rural China: a 12-year prospective population-based genomic epidemiological study. Emerg Microbes Infect. 2022 Dec;11(1):2102–2111.
- Jiang Q, Liu Q, Ji L, et al. Citywide transmission of multidrug-resistant tuberculosis under China’s rapid urbanization: a retrospective population-based genomic spatial epidemiological study. Clin Infect Dis. 2020 Jun 24;71(1):142–151.
- Comas I, Coscolla M, Luo T, et al. Out-of-Africa migration and neolithic coexpansion of mycobacterium tuberculosis with modern humans. Nat Genet. 2013 Oct;45(10):1176–1182.
- Papaventsis D, Casali N, Kontsevaya I, et al. Whole genome sequencing of mycobacterium tuberculosis for detection of drug resistance: a systematic review. Clin Microbiol Infect. 2017 Feb;23(2):61–68.
- Liu Q, Zuo T, Xu P, et al. Have compensatory mutations facilitated the current epidemic of multidrug-resistant tuberculosis? Emerg Microbes Infect. 2018 Jun 6;7(1):98.
- Gygli SM, Loiseau C, Jugheli L, et al. Prisons as ecological drivers of fitness-compensated multidrug-resistant mycobacterium tuberculosis. Nat Med. 2021 Jul;27(7):1171–1177.
- Yang C, Luo T, Sun G, et al. Mycobacterium tuberculosis Beijing strains favor transmission but not drug resistance in China. Clin Infect Dis. 2012 Nov;55(9):1179–1187.
- Handel A, Regoes RR, Antia R. The role of compensatory mutations in the emergence of drug resistance. PLoS Comput Biol. 2006 Oct;2(10):e137.
- Comas I, Borrell S, Roetzer A, et al. Whole-genome sequencing of rifampicin-resistant mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet. 2011 Dec 18;44(1):106–110.
- Alame Emane AK, Guo X, Takiff HE, et al. Highly transmitted M. tuberculosis strains are more likely to evolve MDR/XDR and cause outbreaks, but what makes them highly transmitted? Tuberculosis (Edinb). 2021 Jul;129:102092.
- Shen X, Yang C, Wu J, et al. Recurrent tuberculosis in an urban area in China: relapse or exogenous reinfection? Tuberculosis (Edinb). 2017 Mar;103:97–104.
- Wu Z, Zhang Q, Zhang Z, et al. Effectiveness of an integrated multi-drug resistant pulmonary tuberculosis control model in Shanghai, China. Chin J Antituberc. 2015;37(11):1118–1125.
- Li M, Qiu Y, Guo M, et al. Investigation on the cause of recurrent tuberculosis in a rural area in China using whole-genome sequencing: a retrospective cohort study. Tuberculosis (Edinb). 2022 Mar;133:102174.
- Nonghanphithak D, Chaiprasert A, Smithtikarn S, et al. Clusters of drug-resistant mycobacterium tuberculosis detected by whole-genome sequence analysis of nationwide sample, Thailand, 2014-2017. Emerg Infect Dis. 2021 Mar;27(3):813–822.
- Chen Y, Liu Q, Takiff HE, et al. Comprehensive genomic analysis of mycobacterium tuberculosis reveals limited impact of high-fitness genotypes on MDR-TB transmission. J Infect. 2022 Jul;85(1):49–56.
- Zhou Y, Anthony R, Wang S, et al. The epidemic of multidrug resistant tuberculosis in China in historical and phylogenetic perspectives. J Infect. 2020 Apr;80(4):444–453.