
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
SARS-CoV-2 has caused a global pandemic with significant humanity and economic loss since 2020. Currently, only limited options are available to treat SARS-CoV-2 infections for vulnerable populations. In this study, we report a universal fluorescence polarization (FP)-based high throughput screening (HTS) assay for SAM-dependent viral methyltransferases (MTases), using a fluorescent SAM-analogue, FL-NAH. We performed the assay against a reference MTase, NSP14, an essential enzyme for SARS-CoV-2 to methylate the N7 position of viral 5’-RNA guanine cap. The assay is universal and suitable for any SAM-dependent viral MTases such as the SARS-CoV-2 NSP16/NSP10 MTase complex and the NS5 MTase of Zika virus (ZIKV). Pilot screening demonstrated that the HTS assay was very robust and identified two candidate inhibitors, NSC 111552 and 288387. The two compounds inhibited the FL-NAH binding to the NSP14 MTase with low micromolar IC50. We used three functional MTase assays to unambiguously verified the inhibitory potency of these molecules for the NSP14 N7-MTase function. Binding studies indicated that these molecules are bound directly to the NSP14 MTase with similar low micromolar affinity. Moreover, we further demonstrated that these molecules significantly inhibited the SARS-CoV-2 replication in cell-based assays at concentrations not causing cytotoxicity. Furthermore, NSC111552 significantly synergized with known SARS-CoV-2 drugs including nirmatrelvir and remdesivir. Finally, docking suggested that these molecules bind specifically to the SAM-binding site on the NSP14 MTase. Overall, these molecules represent novel and promising candidates to further develop broad-spectrum inhibitors for the management of viral infections.
Introduction
COVID-19 pandemic has lasted for more than two years. The aetiological coronavirus (CoV), SARS-CoV-2, was first reported in Wuhan, China in December 2019 and has spread to more than 200 countries and territories. It has resulted in more than 500 million infections and 6 million deaths according to the World Health Organization. Many SARS-CoV-2 variants with varying infection rates and lethality have evolved during this period. So far, only a few small molecule drugs, including remdesivir, Paxlovid and molnupiravir, have been approved by U.S. Food and Drug Administration (FDA) for the treatment or Emergency Use Authorization of COVID-19 [Citation1–3]. Both remdesivir and molnupiravir target the viral RNA-dependent RNA polymerase NSP12 [Citation1,Citation3], whereas Paxlovid inhibits the viral main protease NSP5 [Citation2]. In addition to drugs, COVID-19 vaccines have been developed at amazing pace, which have helped reduce the death rate substantially [Citation4]. However, vaccination does not prevent people from infection or reinfection of rapidly developing variants of SARS-CoV-2. There is an urgent need for the development of small molecule drugs for unvaccinated individuals and people who are infected despite being vaccinated.
The SARS-CoV-2 enzymes are potential targets for small molecule drug discovery. SARS-CoV-2 is single-stranded positive sense RNA virus with large genome size (∼30 kb) [Citation5]. It encodes 4 structural, 16 non-structural (NSP) and 6 accessory proteins. SARS-CoV-2 has a type I cap at the 5’ end [Citation6]. A crucial step in the viral replication pathway is the RNA 5’-capping, a conserved biochemical mechanism in CoVs [Citation7] and in many other viruses such as (+)ssRNA flaviviruses [Citation8–12], dsDNA poxviruses [Citation13], dsRNA reoviruses [Citation14], and (-)RNA viruses such as vesicular stomatitis virus [Citation15]. For CoVs, this capping process involves four steps [Citation16,Citation17]. In step (1) the γ phosphate is removed from the nascent 5′-triphosphorylated RNA (5′-pppRNA) to produce a 5′-diphosphorylated RNA (5′-ppRNA) by an RNA triphosphatase (RTPase). In step (2), a GMP moiety is transferred from GTP to the 5′-ppRNA by a guanylyltransferase (GTase) to form the core cap structure GpppN-RNA. In step (3), a guanine-N7-methyltransferase (N7-MTase) methylates the cap guanine at the N7 position. In the final step (4), a nucleoside-2′-O-MTase (2′-O-MTase) methylates the ribose-2′-OH position of the first nucleotide of the RNA.
All viral MTases use S-adenosylmethionine (SAM) as methyl donor and generate S-adenosyl-L-homocysteine (SAH) as a by-product. SARS-CoV-2 encodes two different proteins – NSP14 and NSP16 – to perform the N7- and 2’-O MTase activities in steps (3) and (4), respectively [Citation7,Citation18–22]. NSP14 is a bifunctional enzyme with an N-terminal 3’→5’ exoribonuclease (ExoN) domain [Citation23] and a C-terminal N7 MTase domain, whereas NSP16 only has 2’-O MTase function [Citation24]. Both NSP14 and NSP16 form complexes with co-factor protein NSP10 [Citation25–27]. However, NSP10 is only essential for the 2’-O MTase activity of NSP16 and the ExoN activity of NSP14 [Citation7,Citation22,Citation25,Citation28] but not for the N7 MTase activity of NSP14 [Citation7,Citation25,Citation27]. The NSP14 MTase plays an essential role in viral RNA 5’-capping by methylating viral RNA at the N7 position of the 5’ guanine cap [Citation18]. The N7-MTase activity facilitates viral mRNA stability, viral translation initiation and is essential for viral replication [Citation18,Citation29,Citation30].
MTases are validated drug targets [Citation31,Citation32]. Identification of potent inhibitors against viral MTases has been hampered by a lack of proper chemical probes to develop high throughput screen (HTS) assays [Citation33–39]. Most existing MTase function assays rely on radioactive materials such as 32P-labelled RNA substrate, which is not suitable for HTS. In addition, several HTS assays were developed using non-radioactive RNA substrates [Citation35]. However, they have various limitations. Pearson et al. reported a mass spectrometry (MS)-based HTS assay to identify inhibitors for the NSP14 MTase [Citation35]. The assay requires a specific RapidFire MS instrument which is not widely available for many academic and industrial laboratories. HTS assays were also developed based on homogenous time-resolved fluorescence (HTRF®) [Citation33,Citation37]. Although the HTRF assay does not use radioactive materials, the high cost of the commercial EPIgeneous Methyltransferase Kit (Cisbio, MA) prohibits large-scale HTS for academic institutions. Another limitation is that any assay based on the MTase enzyme activity will require generation of large quantity of capped viral RNA substrates. However, generation of large quantity enough for large-scale primary HTS screen is prohibitive because available assays will use micromolar concentration of the RNA substrates in a reaction [Citation37]. Interestingly, Kasprzyk et al. reported a functional Py-FLINT MTase HTS assay based on a pyrene-labelled GpppA fluorescent substrate probe [Citation39]. Although the Py-FLINT assay is suitable for large-scale HTS, the assay may only be limited to a few viral MTases that can use the GpppA dinucleotide as a substrate. It is reported that many viral MTases including flavivirus and SARS-CoV-2 NSP16/10 MTases cannot methylate the GpppA dinucleotide [Citation7,Citation40–42].
In this study, we have developed a fluorescence polarization (FP)-based HTS assay to identify inhibitors targeting the co-factor SAM-binding site of SARS-CoV-2 NSP14. In our assay, FP originated from an organic molecule, FL-NAH that targets the SAM binding site on NSP14. FL-NAH is fluorescent and structurally analogous to SAM and can be replaced by high-affinity compounds [Citation43]. Therefore, the assay could be applied for any SAM-dependent MTases such as SARS-CoV-2 NSP14 and NSP16, and flavivirus NS5 MTases. Using this assay, we performed a small scale HTS and identified two compounds, NSC 111552 and 288387, as candidate inhibitors of the NSP14 MTase. Inhibition of the MTase activity, binding affinity, antiviral efficacy and combination with known SARS-CoV-2 drugs were further investigated. These compounds are potent inhibitors of SARS-CoV-2 replication and can be further optimized for the development of structure-based inhibitors.
Results
FL-NAH binds to the NSP14 MTase and NSP14/10 complex with similar binding affinity
Using FL-NAH, we developed an FP-based assay to identify and characterize inhibitors targeting the co-factor SAM-binding site of the SARS-CoV-2 NSP14 MTase. FL-NAH is a fluorescent non-hydrolyzable SAM analog that can mimic SAM to bind SAM-dependent MTases ((A)) [Citation43]. It was previously used to develop an HTS SAM-displacement assay for a histone MTase MLL1 [Citation43]. The fluorescent ligand containing fluorescein linked to aza-adenosylhomocysteine – namely, fluorescein N-adenosylhomocysteine (FL-NAH) – is built based on the backbone structure of the SAM cofactor ((A)).
Figure 1. FL-NAH binding to the NSP14 MTase. (A) Chemical structure of FL-NAH. (B) SDS-PAGE analysis of purified NSP14, NSP14-NS10, and NSP16 of SARS-CoV-2, and ZIKV NS5. St, molecular weight standard; lane 1, NSP14; lane 2, SARS-CoV-2 NSP14-NSP10 complex; lane 3, SARS-CoV-2 NSP16; lane 4, ZIKV NS5. (C) Dose-dependent FL-NAH FP assay. FL-NAH (50 nM) was applied to 2-fold diluted concentration series of NSP14 and NSP14-NSP10 complex. FP was calculated by measuring the parallel and perpendicular fluorescence with excitation and emission wavelengths of 485 nm and 528 nm, respectively. N = 3. (D) Dose-dependent inhibition of FL-NAH binding to the NSP14 MTase by SAM and SAH. The NSP14 MTase was incubated with concentration series of SAM and SAH for 30 min. FL-NAH (50 nM) was added and further incubated for 30 min before fluorescence measurement. N = 3. FP values in the presence of SAM or SAH were normalized to that of the DMSO control (100%).
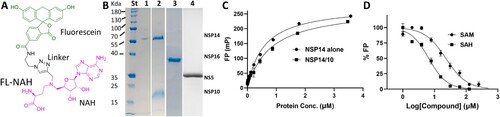
To develop a SAM-displacement assay for the SARS-CoV-2 NSP14 MTase, we individually expressed and purified NSP14 and NSP10 ((B). We also co-expressed and purified the NSP14-NSP10 complex ((B), Supplemental Figure 1). Both affinity and size exclusion chromatography indicated that NSP10 and NSP14 formed a stable complex (Supplemental Figure 1(A)), which is consistent with previous findings [Citation26,Citation44–46]. Using native gel electrophoresis, we confirmed that individually purified NSP10 and NSP14 formed a complex (Supplemental Figure 1(B)). We further showed that NSP10 bound to NSP14 with high affinity of 0.56 µM, determined by microscale thermophoresis (MST) (Supplemental Figure 1(C)). Overall, we have generated functional NSP14 and NSP14-NSP10 complex.
Using these purified proteins, we investigated whether binding of FL-NAH to the NSP14 MTase could be monitored by FP. As shown ((C)), FL-NAH could bind to the NSP14 MTase, leading to FP changes in a manner depending on the concentration of NSP14. By fitting the experimental data, we determined that FL-NAH bound to the NSP14 MTase with a binding affinity KD of 0.56 µM. In addition, we investigated if FL-NAH is also bound to the NSP14/NSP10 complex. Our results showed that that FL-NAH also bound to the NSP14-NSP10 complex dose-dependently, with a slightly weaker binding affinity of 0.81 µM than that for NSP14 alone ((C)). The results confirmed that NSP10 was not required for the NSP14 MTase function. Since NSP10 had only minimum effects on the NSP14 MTase activity, all the following experiments were performed using the NSP14 MTase alone.
SAM and SAH displace FL-NAH from the NSP14 MTase
As FL-NAH is a SAM analogue, it is expected to bind at the MTase SAM-binding site. To investigate this, we tested if SAM and its by-product SAH could displace FL-NAH from binding to the NSP14 MTase. Our results showed that SAM and SAH inhibited the FL-NAH binding to the NSP14 MTases dose-dependently ((D)). We determined the IC50-disp, defined as compound concentration at which 50% inhibition of the binding of FL-NAH to NSP14 was achieved, by non-linear fitting experimental curve. Our results showed that SAH and SAM inhibited the binding of FL-NAH to NSP14 with IC50-disp of 7.2 and 25 µM, respectively. Interestingly, SAM is less efficient in inhibiting FL-NAH binding to the MTases than SAH, as has been reported for other MTases [Citation43].
FP-based FL-NAH-displacement assay is universal for SAM-dependent MTases
To test if the SAM-displacement assay is suitable for HTS, we used SAH (25 µM) as a control inhibitor. Our results showed that the SAM-displacement FP assay is very robust in a 96-well plate format with satisfactory signal/background (S/B) ratio (4.7), Z-factor (0.7), and coefficient variation (CV, 4.5%) against the NSP14 MTase ((A)). The optimized HTS assay contained 50 nM FL-NAH and 0.5 µM NSP14 in a 25 µL reaction mixture.
Figure 2. FP-based FL-NAH displacement HTS. (A) SAH (25, 31.25, or 50 µM) inhibited FL-NAH (50 nM or 30 nM) binding to the SARS-CoV-2 NSP14 (0.5 µM), ZIKV NS5 (2.5 µM), or SARS-CoV-2 NSP16 (0.5 µM) MTases in 96-well plate. ****, p < 0.0001. (B) FL-NAH FP HTS assay statistics against the NCI Diversity Set VI compound library. Scales for Z-factor and CV were on the left axis; scale for S/B was on the right axis. (C) Structures of NSC 111552 and 288387. (D) Dose-dependent inhibition of FL-NAH binding to the NSP14 MTase by NSC 111552 and 288387. N = 3. Assay was performed similarly as described in (D). FP values in the presence of compounds were normalized to that of the DMSO control (100%).
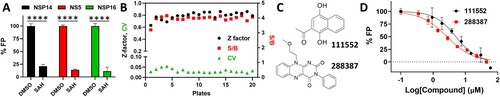
Because FL-NAH is a SAM analogue, we hypothesize that it is suitable for other viral MTase as well. To test this hypothesis, we tested if this assay could be applied to the other SARS-CoV-2 MTase, the NSP16 2’-O MTase, and an additional MTase of other virus, the NS5 MTase of ZIKV, a devasting flavivirus recently causing global epidemic [Citation47,Citation48]. We expressed and purified the NSP16 and NS5 MTases ((B)) and performed FL-NAH FP-assay in the presence and absence of SAH. Clearly, FL-NAH binding to NSP16 and the ZIKV NS5 MTases led to significant FP, which could be quenched by SAH or SAM ((A)). These data demonstrated that the FL-NAH assay could be universally applicable to any viral MTases that use SAM as a methyl donor.
HTS screening
To identify inhibitors targeting the SAM-binding site of the SARS-CoV-2 NSP14 MTase, we performed a small-scale HTS against the NCI diversity set VI compound library, containing 1584 compounds dissolved in DMSO in twenty 96-well plates. A single concentration of 15 µM of each compound was used in HTS. For each plate screening, DMSO was used as a negative control, whereas SAH at 25 µM was used as a positive control for inhibition. The quality of the screening assay was assessed by calculating Z-factor, S/B ratio, and CV for each plate, average of which are 0.8%, 4.0% and 3%, respectively ((B)). The results indicated a high-quality screen.
The FL-NAH-displacement primary HTS screen identified 12 compounds showing inhibition larger than 50% for the binding of FL-NAH to the NSP14 MTase. Ten compounds were eliminated from further investigation due to compound autofluorescence and upon cheminformatics analysis of the chemical structures of the hit compounds. Two compounds, NSC 111552 and 288387 were chosen for further dose-response confirmation ((C)). Our results showed that NSC 111552 and 288387 inhibited the FL-NAH binding to the SARS-CoV-2 NSP14 MTase dose-dependently with IC50-disp values of 5.1 and 2.7 µM, respectively ((D), ).
Table 1. Inhibition of FL-NAH binding to the SARS-CoV-2 NSP14 MTases (IC50-disp), inhibition of MTase activity (IC50-HTRF, IC50-TLC, IC50-MS), antiviral efficacy (EC50), and cytotoxicity (CC50) in Vero cells of compounds.
Inhibition of the MTase activity
Candidate inhibitors were further evaluated for their efficacy in inhibition of the NSP14 MTase activity by using multiple methods. We first used an HTRF assay, based on the quantification of released SAH from the MTase enzymatic reaction. The HTRF assay uses an anti-SAH antibody conjugated with Tb cryptate and a SAH labelled with d2 fluorophore (SAH-d2) as a tracer. In the absence of SAH, the anti-SAH Tb cryptate can form a FRET pair with the d2 fluorophore present in the bound SAH-d2 ((A)). SAH produced from a successful MTase reaction will compete with and displace SAH-d2 from the Tb-cryptate anti-SAH antibody, resulting in a reduction in signal. Using a GpppA dinucleotide as a substrate as described previously [Citation33], we performed the HTRF functional MTase assay, using the EPIgeneous Methyltransferase kit (Cisbio, MA). Our result showed that compounds NSC 111552 and 288387 nearly equally inhibited the NSP14 N7-MTase activity with IC50-HTRF values of 2.2 µM ((B), ).
Figure 3. Functional NSP14 MTase activity assays. (A) Schematic figure to show the HTRF-based assay for MTase activity. SAM-dependent MTase produces SAH after transferring methyl group to its substrate. The released SAH displaces SAH-d2 from the variable region of an α-SAH Tb cryptate-conjugated antibody, leading to a decreased HTRF signal through the disruption of the Tb cryptate – d2 FRET pair. (B) HTRF analyses of dose-dependent inhibition of FL-NAH binding to the NSP14 MTase by compounds NSC 111552 and 288387. N = 3. TR-FRET values in the presence of compounds were reverse normalized to that of the (-) MTase control (0%) and that of the DMSO control (100%). (C) TLC analyses of dose–response inhibition of the N7 methylation activity of the SARS-CoV-2 NSP14 MTase by compound NSC 111552 and 288387. Upper panels, TLC analyses of inhibition of the SARS-CoV-2 NSP14 MTase by of NSC111552 (left panel) and NSC288387 (right panel). The migration positions of the G*pppA and m7G*pppA molecules are labeled on the side of the TLC images. Lower panels, curve fitting to determine the IC50 values for each compound on the N7 activities of the SARS-CoV-2 NSP14. The percentage of activity was determined after quantification of G*pppA and m7G*pppA. The IC50 value was determined by fitting of the dose−response curve. (D) MS analyses of dose−response inhibition of the N7 methylation activity of the SARS-CoV-2 NSP14 MTase by compound NSC 111552 and 288387.
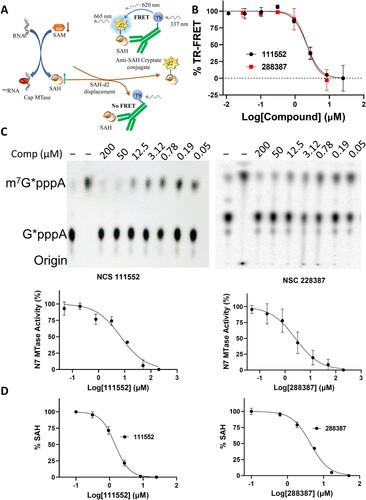
To further verify the inhibition, we carried out an MTase inhibition experiment to directly visualize and quantify N7-methylation of the RNA guanine cap using a traditional thin layer chromatography (TLC) method and radiolabeled G*pppA-capped RNA substrate, where the * indicates that the following phosphate was 32P labelled, as we described previously [Citation12,Citation40]. We first in vitro transcribed both unmethylated and N7-methylated RNAs representing the first 90 nucleotides of the West Nile virus genome as we described previously [Citation49,Citation50]. As shown ((C), ), NSP14 could efficiently N7-methylate the G*pppA-RNA to generate the cap-0 structure (m7G*pppA). Using this assay, we quantified the anti-MTase activity of NSC 111552 and 288387. Our data showed that the N7-MTase activity of NSP14 was efficiently inhibited by NSC 111552 and 288387 with IC50-TLC values of 7.0 and 2.5 µM, respectively.
To unambiguously evaluate the MTase inhibition efficacy, we developed a mass spectrometry (MS)-based assay to directly detect and measure the reaction by-product SAH, which will significantly minimize the risk of identifying false-positive hits. Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) successfully detected the by-product SAH, allowing us to quantify the SAH yield to evaluate the inhibitory potency of NSC 111552 and 288387 for the NSP14 MTase ((D), ). The LC-MS/MS results confirmed that NSC 111552 and 288387 can inhibit the production of SAH by the NSP14 MTase reaction dose-dependently with IC50-MS of 1.5 and 4.3 µM, respectively ().
Overall, these three independent and complementary approaches unambiguously verified the inhibitory efficacy of compounds NSC 111552 and 288387 against the SARS-CoV-2 NSP14 MTase. The IC50 values are slightly different between assays, likely due to differences in assay conditions, substrates, and detection methods.
Cytotoxicity and antiviral analyses
To confirm the inhibitory effects of viral replication in mammalian cells, we first performed a cell proliferation assay to measure the cytotoxicity of these compounds in Vero cells. A WST-8 cell proliferation assay showed that the two compounds, NSC 111552 and 288387, are not toxic to mammalian cells with CC50 of 61.8 and 64.4 µM, respectively ((A), ).
Figure 4. Analysis of Cytotoxicity and antiviral activity of compounds NSC 111552 and 288387. (A) Cytotoxicity of NSC 111552 (left panel) and 288387 (right panel). Vero cells were treated with various concentrations of NSC 111552 and 288387, followed by cell viability assay at 42 h post-incubation. N = 3. (B) Inhibition of SARS-CoV-2 replication by NSC 111552 (left panel) and 288387 (right panel). Vero cells were seeded in 96 well plated. After 24 h, media was replaced with fresh media containing indicated concentrations of NSC 111552 (left panel) and 288387 (right panel), followed by infection with SARS-CoV-2. At 72 h post-infection, wells were stained with crystal violet; and viral plaque were counted.
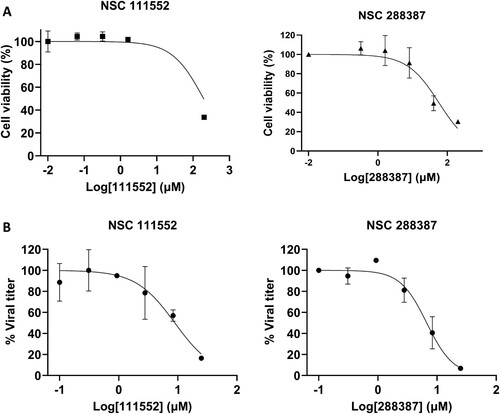
Viral titer reduction assay was performed to evaluate the compounds’ antiviral efficacy. The SARS-CoV-2 viral titre was reduced in a dose-dependent manner by compounds NSC 111552 and 288387 ((B), ). NSC 111552 and 288387 showed an antiviral efficacy EC50 of 8.5 and 5.7 µM, respectively ().
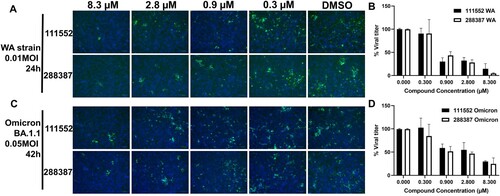
We also performed the immunofluorescence assay (IFA) in both SARS-CoV-2 wild-type (WT) strain (Washington strain, 0.01 MOI) or Omicron strain (BA.1.1, 0.05 MOI). Our results suggested that the compounds NSC 111552 and 288387 dose-dependently reduced the viral titre ((A–D)). Overall, these results indicated that the compounds we identified have antiviral activity in mammalian cells with limited cytotoxic effects.
Figure 5. Immunofluorescence assay for detection of SARS-CoV-2 WT and Omicron-infected cells treated with NSC 111552 and 288387. (A,C) IFA images of dose-dependent inhibition of SARS-CoV-2 WT Washington Strain (WA) (A) and Omicron BA.1.1 strain (B) by compounds NSC 111552 and 288387. Vero E6 cells were infected with the SARS-CoV-2 WA strain (A and B) and the Omicron strain (C and D), treated with compounds at indicated concentrations for 24 h for the WA strain or 42 h for the Omicron strain, fixed and immunolabeled with a primary SARS-CoV-2 nucleocapsid monoclonal antibody and a goat anti-mouse secondary Alexa-488 antibody. Blue, DAPI staining. (B,D) Normalized IFA data shown in panels A and C. The intensities of Alexa-488 positive cells for the DMSO control were set as 100%. N = 3.
Combination with known drugs
The strategy of combining treatments for improved efficacy has been clinically successful for infectious diseases, most notably HIV and HCV [Citation51,Citation52]; indeed, in HIV pharmacotherapy combinations include more than two antivirals [Citation53]. There has been considerable interest in combination approaches within the SARS-CoV-2 community, given the rapid development of VOCs and VOIs. In addition, persistent SARS-CoV-2 infection have been reported with an increasing frequency [Citation54–63].
Therefore, we explored the potential of our NSP14 MTase inhibitor NSC111552 in combination with known SARS-CoV-2 drugs, including nirmatrelvir and remdesivir, targeting the SARS-CoV-2 main protease (Mpro) and RNA-dependent RNA polymerase (RdRp), respectively. We conducted a checkerboard combination assay as we and other described previously [Citation64,Citation65]. Our results showed that these inhibitors targeting different SARS-CoV-2 enzymes have significant synergistic effects, with maximal ZIP-scores [Citation65] of 21 and 24 between MTase inhibitor NSC111552 and Mpro inhibitor nirmatrelvir, and between NSC111552 and RdRp inhibitor remdesivir, respectively ((A,C)). In the presence of NSC111552 (1.67 µM), the EC50 for nirmatrelvir was shifted ∼ 1.8-fold, from 56 nM to about 31 nM ((B)). Similarly, NSC111552 (0.56 µM) decreased EC50 for remdesivir about 2.4-fold, from 40 nM to 17 nM ((D)). It is noted that NSC111552 at these concentrations only minimally impacted SARS-CoV-2 replication (<10% inhibition) ((C)). Therefore, these results suggest that the NSP14 MTase inhibitor synergizes with Mpro and RdRp inhibitors.
Figure 6. Synergy between inhibitors of NSP14 MTase, Mpro, and RdRp. (A,C) Topographic two-dimensional map of synergy scores for dose-response interaction matrix between NSC111552 and nirmatrelvir (A) and between NSC111552 and remdesivir (C), using Vero E6 cells against the SARS-CoV-2 WA strain in the presence of P-gp efflux inhibitor CP-100356 (2 µM) [Citation67–72]. The synergy scores were determined in SynergyFinder [Citation66] from the above checkerboard combination assay. (B,D) Dose-response of compounds alone and in combination with fixed concentration of one compound and varying that of the other. N = 3.
![Figure 6. Synergy between inhibitors of NSP14 MTase, Mpro, and RdRp. (A,C) Topographic two-dimensional map of synergy scores for dose-response interaction matrix between NSC111552 and nirmatrelvir (A) and between NSC111552 and remdesivir (C), using Vero E6 cells against the SARS-CoV-2 WA strain in the presence of P-gp efflux inhibitor CP-100356 (2 µM) [Citation67–72]. The synergy scores were determined in SynergyFinder [Citation66] from the above checkerboard combination assay. (B,D) Dose-response of compounds alone and in combination with fixed concentration of one compound and varying that of the other. N = 3.](/cms/asset/e9a47dee-badc-42e4-80f6-5a5acdbf9a1c/temi_a_2204164_f0006_oc.jpg)
Direct binding of NSC 111552 and 288387
We used microscale thermophoresis (MST) to confirm the direct binding of NSC 111552 and 288387 with the NSP14 protein. We labelled the target protein NSP14 with a fluorescent dye according to the manufactory manual. NSC 111552 and 288387 bound to NSP14 with a binding constant (KD) of 2.1 and 1.5 µM, respectively. These data confirmed direct binding of NSC 111552 and 288387 to the NSP14 MTase ().
Figure 7. Analysis of binding of NSC 111552 and 288387 to the SARS-CoV-2 NSP14 protein using MST. Dose-response curve was generated by fitting experimental data by titrating NSC 111552 and 288387 from 0.6 mM to 9.1 nM against NSP14 (40 nM). N = 3.
Docking of inhibitors with NSP14
Docking analysis was performed on NSC 111552 and 288387 to assess their binding with NSP14. The SAM binding site of NSP14 was determined using structural information provided in the literature [Citation20] as well as docking of SAH. Docking showed that SAH forms extensive interactions in the active site (most notably with R289, D352, and F426) and results in an MMGBSA score of −57.74.
Each inhibitor was docked into the cofactor binding site using the Glide module. The resulting poses were subsequently minimized using the Prime MM-GBSA program in Maestro, allowing for residue flexibility within 4 Å of the ligand. Binding affinities were predicted using the resulting MM-GBSA ΔG binding score. For NSC111552, the best pose was revealed to have a MM-GBSA score of –42.42 ((A)). It was observed to bind in the cofactor binding pocket nicely with the naphthalene ring overlaying with the purine ring of SAH forming an interaction with F426, and the carbonyl oxygen of NSC111552 aligning well with the in-ring oxygen of the furanose portion of SAH ((A)). In addition, the hydroxyl group at the 4-position was observed to act as a hydrogen bond acceptor with the side chain of N388 and as a hydrogen bond doner with the backbone carbonyl of H427.
Figure 8. Binding interaction of NSC 111552 and 288387 with NSP14. (A) 3D overlay of NSC111552 (orange) with SAH (purple) (left) with the 2D interaction diagram of 111552 (right). (B) 3D overlay of NSC288387 (orange) with SAH (purple) (left) with the 2D interaction diagram of 288387 (right).
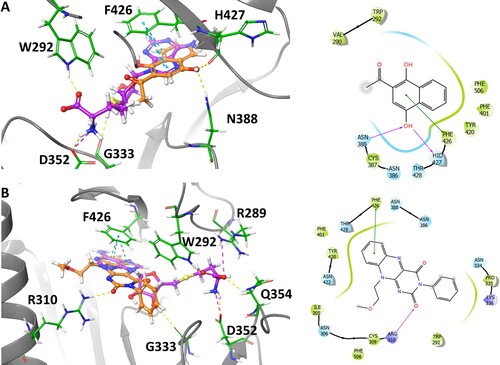
The hit NSC288387 was docked to the NSP14 structure resulting in a MM-GBSA score of −64.63 ((B)). The resulting pose was observed to align the flavin ring with the purine ring of SAH with the methoxyethyl substituent extending into a hydrophobic pocket. The N-phenyl imide portion extends out behind the furanose of SAH towards R310 ((B)). This ligand also picked up some interactions with the flavin ring forming a pi-stacking interaction with F426, and one of the carbonyls acted as an H-bond acceptor with the guanidine portion of R310. Overall, both NSC 111552 and 288387 showed significant overlapping with SAH in the NSP14 catalytic pocket, corroborating that they are SAM/SAH competitive inhibitors.
Discussion
RNA capping is an essential biochemical process in viral life cycle and is thus recognized as an attractive target for antiviral drug discovery [Citation31,Citation73]. Although nearly all MTases use SAM as a methyl donor, drugs specific to a particular MTase have been developed and approved by FDA to treat various human diseases [Citation74,Citation75], including tazemetostat for EZH2 histone MTase [Citation75], and 5-azacytidine and decitabine for DNA MTase (DNMT) [Citation74,Citation76]. Previously our lab have identified a flavivirus-specific pocket near the co-factor SAM-binding pocket [Citation77], leading to identification of inhibitors specific to viral MTase but not the human counterpart [Citation78]. Overall, our result and other published research suggest that inhibitors specific to a viral MTase can be developed.
The SARS-CoV-2 RNA MTase is a potential therapeutic target to develop therapeutics to combat the COVID-19 pandemic [Citation29]. It has been shown that inhibition of N7 methylation can inhibit the overall virus replication [Citation29,Citation79]. Although several other groups identified potential inhibitors of the SARS-CoV-2 NSP14 MTase [Citation34,Citation35,Citation37–39,Citation80–82]. It is currently unclear if these identified compounds possess antiviral activity because cellular antiviral activities for the majority of the identified compounds were not evaluated, except a few compounds in two manuscripts [Citation37,Citation39]. Basu et al. showed that compounds PF-03882845, Inauhzin and Trifluperidol had moderate anti-MTase and antiviral activities, with IC50 values of 1.1, 12.9, and 23 µM and EC50 values of 11.0, 13.0 and 59.8 µM, respectively [Citation37]. Kasprzyk et al. reported that three compounds, Pyridostatin, Reactive blue 2, and Evans Blue had appreciable anti-MTase and antiviral activities, with IC50 values of 4.1, 4.8, and 4.9 µM and EC50 values of 3.6, 16.3 and 31.0 µM, respectively [Citation39].
In this manuscript, we report the development of an FP-based HTS platform against a critical enzyme, the SARS-CoV-2 NSP14, involved in generating RNA cap structures essential for RNA translation and masking viral RNA from the host immune response. Moreover, we describe the screening and identification of potential inhibitors against the NSP14 protein of SARS-CoV-2.
We initially screened the NCI Diversity compound libraries using an FP-based assay. The top hit molecules were further characterized with a commercially available HTRF MTase functional assay (Cisbio, MA). Furthermore, we evaluated their antiviral efficacy and potential cytotoxicity using cell-based assays. Through this process, two potent inhibitors of the SARS-CoV-2 NSP14 MTase were identified. NSC 111552 and 288387 were found to inhibit the binding of FL-NAH, a fluorescent SAM analogue, with low micromolar IC50-disp. These molecules were further evaluated for their anti-MTase activity using three different approaches, HTRF, TLC with 32P-labelled substrate, and MS. All three methods generated similar results showing that these two molecules efficiently inhibited the NSP14 MTase activity with low micromolar IC50. Moreover, using MST, we showed the direct binding of NCS 111552 and 288387 to the NSP14 MTase with binding affinities in low micromolar range as well. Furthermore, we evaluated their antiviral potency and cytotoxicity using cell-based assays. Our results showed that these molecules inhibited the SARS-CoV-2 replication with single-digit micromolar EC50 values and therapeutic index around 10. Lastly, we showed that NSC111552 significantly synergized with known SARS-CoV-2 drugs targeting other viral enzymes, including nirmatrelvir targeting the viral main protease and remdesivir targeting the viral RdRp.
In summary, these molecules provide a starting point for further medicinal modification to develop more potent and selective inhibitors against the SARS-CoV-2 NSP14 MTase.
Materials and methods
Expression and purification of NSP14 and NSP14/NSP10 complex
Plasmid pET28-6His-TEV-NSP14 encoding full-length SARS-CoV-2 NSP14 was purchased from Addgene (Cat#: 154954) (Watertown, MA), which is a gift from Justin Kollman. E coli BL21(DE3) cells were transformed with the NSP14 plasmid. Next day, a transformed single colony was used to inoculate 50 ml LB media containing 50 µg/ml of kanamycin and grown in a shaker with 250 rpm at 37°C for overnight. A culture medium (1 L) was inoculated with the overnight grown pre-culture and further grown at 37 °C with 250 rpm until OD600 reached 0.6–0.8. Protein expression was induced by the addition of Isopropyl β-D-thiogalactoside (IPTG) to a final concentration of 0.1 mM; and the cells were further grown at 16°C for 16–18 h.
The cells were harvested by centrifugation at 5000g for 10 min. The bacterial pellet was resuspended in a lysis buffer containing 20 mM HEPES, pH 8.5, 500 mM NaCl, 10% glycerol and 1 mM DTT. Protease inhibitor cocktail was added to the lysis buffer. The cells were lysed by sonication. The lysate was centrifuged at 15,000g for 30 min. The supernatant containing the soluble protein was loaded onto Ni-NTA resin equilibrated with the lysis buffer. The resin was washed with the lysis buffer containing 20 mM imidazole. The bound NSP14 was eluted with lysis buffer containing 250 mM imidazole. The eluted fractions were analysed by SDS-PAGE. The fractions containing the protein of interest were combined, concentrated to 5 ml and loaded onto a Superdex 200 column equilibrated with a protein storage buffer containing 20 mM HEPES, pH 8.5, 150 mM NaCl, 10% glycerol and 1 mM DTT. The fractions containing purified NSP14 were pooled, concentrated, aliquoted, flash-frozen in liquid nitrogen and stored at −80°C until further use.
To generate the NSP14/NSP10 complex, the NSP10 gene sequence was codon optimized, synthesized, and inserted into the plasmid pGEX-6P-1 by GeneUniversal. The NSP14/NSP10 complex was obtained through co-expression of NSP14 and NSP10. Briefly, E coli BL21(DE3) cells were co-transformed with the pET28-6His-TEV-NSP14 and GST-NSP10 plasmids. Protein expression was carried out similarly as described above, except that a final concentration of IPTG at 0.2 mM was used. Cell lysate was loaded onto a glutathione Sepharose-4B column, pre-equilibrated with the cell lysis buffer. Upon extensive washing to remove non-specific binders with the lysis buffer, home-made GST-3C protease was added to the column for on-column digestion at 4°C overnight to remove the GST-tag. Flow-through and two-column washing fractions were collected, combined, and concentrated before further purification using size exclusion chromatography with a Superdex 200 column equilibrated with 20 mM HEPES, pH 8.5, 150 mM NaCl, 10% glycerol and 1 mM DTT on an AKTA Pure 25M system. The fractions containing purified NSP14/NSP10 complex were pooled, concentrated, aliquoted, flash-frozen in liquid nitrogen and stored at −80°C until further use.
To generate NSP16, the NSP16 gene sequence was codon optimized, synthesized, and inserted into the plasmid pET28a by GeneUniversal. NSP16 was expressed and purified, similarly as described above for NSP14.
To generate the ZIKV NS5 MTase, the gene sequence encoding the ZIKV NS5 MTase domain (amino acids 1-274) was codon-optimized, synthesized and cloned into the pET28a vector using the Nde1 and EcoR1 restriction sites by Bio Basic, Inc. Protein expression and purification were carried out similarly as described above for the NSP14 protein.
FL-NAH FP assay
For inhibitor screening against SARS-CoV-2 NSP14, an FP-based assay was performed using FL-NAH, a fluorescent analogue of the methyl donor SAM, as described earlier [Citation43]. The screening assay was performed in a reaction buffer consisting of 20 mM HEPES, pH 8.5, 150 mM NaCl, 10% glycerol, 1 mM DTT, 0.01% triton X-100, 50 nM FL-NAH and 0.5 µM NSP14 or NS5. The assay was performed in a 25 µL reaction volume in 96-well black polypropylene plates, against the NCI diversity set VI library (1584 compounds, 20 plates). NSP14 was initially incubated with DMSO or the inhibitor for 30 min at ambient temperature. FL-NAH was added to the reaction and FP was measured after 30 min using excitation and emission wavelengths of 485 and 528 nm, respectively. The FP was indicated in millipolarization units (mP). For IC50-disp measurement, the assay was carried out in the presence of 50 nM FL-NAH and the increasing concentration of the inhibitor or DMSO.
Z-factor was calculated from standard deviations and the FP values of samples in the absence and presence of SAH (25 µM) using the following equation, as previously described [Citation83].
where
and
are the standard deviations of samples in the absence and presence of SAH, respectively. The
and
present the mean FP with or without SAH, respectively.
Substrate generation for NSP14
GpppA-capped RNA is a substrate for NSP14. To synthesize the GpppA capped RNA, DNA templates were synthesized using a pA12-WNV-replicon. Primers WNV190T7F and WNV-90-rev-HDV were used to synthesize the template DNA using Q5 High-Fidelity DNA Polymerase as per manufacture protocol. Zymo DNA Clean & Concentrator-5 Kit was used to purify the PCR products per manufacturer protocol. In vitro WNV RNA (short, < 0.3 kb) transcription was performed using the HiScribe T7 Quick High Yield RNA Synthesis kit (New England Biolabs) overnight at 37°C. DNase I digestion was performed to remove the template DNA. The RNA was further purified following the kit instructions using Monarch RNA Cleanup Kit (NEB, 500ug). We verify the integrity of the purified WNV RNA in formaldehyde-denatured 2% agarose gel.
Capping of RNA was done with the Vaccinia capping system (New Egland Biolabs) as per the manufacturer's protocol. In brief, 70 µg of GpppRNA was denatured by heating on 65°C for 10 min, followed by cooling for 5 min on ice. The denatured GpppRNA was mixed with 14 µl of 10X capping buffer, 7 µl of 10 mM GTP, 7 µl of RNAase free water, and 7 µl of vaccinia capping enzyme in a total volume of 140 µl. Reactions were incubated at 37°C for 2 h. Capped GpppRNA was checked in 2% denatured agarose gel with formaldehyde.
HTRF MTase functional assay
The NSP14 MTase activity was measured by detecting the release of SAH from MTase activity. The MTase reaction product SAH was measured using a commercially available MTase kit, EPIgeneous Methyltransferase Assay 1000 Tests (CisBio Bioassays). The MTase reaction was conducted at 30°C in a 10 µl reaction volume with 100 nM NSP14 protein, 2 µM SAM (CisBio), 2 µM capped GpppA RNA in a P7 reaction buffer composed of 50 mM Tris-HCl, pH 7.0, 2 mM DTT, 20 mM NaCl as described previously [Citation40,Citation84]. The reaction was started with the the addition of NSP14 and was allowed to proceed for 20 min before quenching by adding 2 µl of 5 M NaCl to a final concentration of 1 M.
After quenching, 2 µl of detection Buffer 1 (CisBio) was added to the reaction mixture and incubated for 10 min. Following 10 min incubation, 4 µl of diluted SAH d2 reagent (CisBio) was added. 50x SAH-d2 was prepared as suggested by the manufacturer, by adding one-part SAH-d2 to 49 parts Detection Buffer 2 (CisBio). After 5 min, 4 µl of diluted SAH Tb Crypate Antibody solution was added to the reaction mixture. 1× SAH Tb Crypate Antibody solution was prepared by adding one-part α-SAH Tb Cryptate antibody (CisBio) to 49 parts Detection Buffer 2 (CisBio).
The reaction mixture was incubated for 1 hour at room temperature on a shaker at 500 rpm. After 1 h incubation, Homogenous Time-Resolved Fluorescence (HTRF) measurements were taken on Bioteck Cytation 5 as described earlier [Citation37]. Lag time of 100 µs was used to take a reading after excitation at λ = 330 nm. Emission wavelengths of λ = 665 nm and λ = 620 nm were used to record the reading. Further the experimental HTRF ratio (HTRFexp) was calculated as a ratio of emission intensities: I665/I620. To calculate the normalized HTRF ratio, HTRF ratio were also calculated using wells without enzyme (E0) and without SAH-d2 (d20), representing the maximum and minimum possible HTRF values, respectively. The normalized HTRF ratio was then calculated as a linear transformation of the experimental HTRF ratio, the E0 ratio, and the d20 ratio:
Inhibitors were pre-incubated with the enzyme for 30 min at RT to calculate the HTRF for inhibitors.
Quantification of SAH production by LC-MS/MS analysis
MTase reactions were performed as described above, except that after 20 min, the reaction was quenched by addition of 50 µl methanol (MeOH). Each sample (10 µl reaction mixture and 50 µl MeOH) was mixed with 10 µl AP-GSH (internal standard, IS, 100 ng/ml). The mixture was vortexed for 30s and centrifuged at 15000 rpm for 10 min in an Eppendorf 5424R centrifuge. The supernatant was collected and diluted with 450 µl Milli Q water for solid-phase extraction. In brief, the diluted supernatant was loaded onto an ISOLUTE® C18 SPE Column (1 ml/100 mg, Biotage, Salem, NH) pre-conditioned with 1 ml methanol followed by 1 ml water; the unbound fraction was collected and 5 µl was used for LC-MS/MS analysis.
SAH was detected using LC-MS/MS. The LC-MS system consisted of an Agilent 1290 UPLC system (Agilent Technologies, Santa Clara, CA) and a Sciex Qtrap6500+ Mass Spectrometer (AB SCIEX, Framingham, MA). Analytes were separated on an EclipsePlus C8 column (2.1 × 100 mm, 1.8 µm, Agilent) at a temperature of 35°C, with mobile phase A containing 0.1% formic acid (v/v) in water and mobile phase B containing 0.1% formic acid (v/v) in methanol. Elution was at a flow rate of 0.2 ml/min as follows: 1% B (0-0.5 min), 1% B→95% B (0.5-4 min), 95% B (4–5 min), 95% B→1% B (5–5.1 min), 1% B (5.1–8 min). The MS was operated in the positive ion mode, using electrospray ionization. The ion spray voltage and temperature were set at 5000 V and 500°C, respectively. Curtain gas, ion source gas1 and ion source gas2 were set at 25, 50, 50 psi, respectively. SAH and IS were detected using Multiple Reaction Monitoring (MRM), with a dwell time of 200 msec per transition, at m/z 385.0/136.1 and 457.0/328.0, respectively. Retention times for SAH and IS were 3.6 and 4.04 min, respectively. For quantitative analysis of SAH, standards (5 to 2000 nM in 10 µl methanol), along with 10 µl IS (at 100 ng/ml in methanol), were added to 10 µl of blank matrix to construct the calibration curve.
TLC-based MTase inhibition assay
Substrates G*pppA-RNA representing the first 190 nucleotides of the WNV genome were labelled at 5′-end with 32P using the vaccinia virus capping enzyme as per manufacturer instruction (New England Biolabs) and as described previously [Citation49]. We used two Sephadex G-25 spin columns (GE Healthcare) to purify the RNA. Further RNA was extracted using phenol-chloroform, washed with 70% ethanol, and precipitated using 100% ethanol. Our previous studies have shown that the optimal pH values for the N7 and 2′-O methylations are 7 and 10, respectively [Citation40,Citation84]. Different pH requirements allowed us to selectively perform the N7 methylation of GpppA-RNA to m7GpppA-RNA without subsequent 2′-O methylation. The N7 methylation (20-μl total volume) was performed in the presence of 3 pmol G*pppA-RNA substrate, 80 μM SAM, 1 μg (27 pmol) MTase in the P7 buffer at RT for 1 hr. Inhibition of methylation was also performed as described above. The methylation reaction mixtures were digested with 1 U nuclease P1 in 50 mM sodium acetate (pH 5.2) at 37°C for 6 h or overnight. They were further analysed on polyethyleneimine cellulose thin-layer chromatography (TLC) plates with 0.3 M (NH4)2SO4 as a solvent. After the TLC plates were dried, spots representing different cap structures (GpppA, m7GpppA) were quantified with a PhosphorImager. All experiments were performed in duplicate. The percentage of activity was determined after quantification of m7G*pppA and G*pppA. The IC50 values were determined by fitting the dose-response curve of the averaged values using the GraphPad Prism 9 software package.
Cytotoxicity assay
Cytotoxicity for led compounds NSC 111552 and 288387 was measured by a CCK-8 cell proliferation assay kit using WST-8 as a substrate (GLPBio), as we described previously [Citation85]. Briefly, ∼1 × 105 cells in 100 μl of media were seeded into a 96-well plate. Plates were incubated for 20–24 h at 37°C with 5% CO2. The media was removed, and 100 μl of media containing decreasing concentrations of NSC 111552 and 288387 were added to the wells. All determinations were performed in triplicates. After 42 h incubation at 37°C, WST-8 assays were performed according to manufacturers’ protocols. A Synergy H1 microtiter plate reader (BioTek Instruments) was used to record absorbance at 450 nm. After adjusting the absorbance for background and comparing it to the DMSO controls, the cytotoxic concentration CC50 was calculated using a sigmoidal nonlinear regression function to fit the dose-response curve using the GraphPad Prism 9.
Viral inhibition assay
To confirm the in vitro inhibitory assay of identified compounds NSC 111552 and 288387 on SARS-CoV-2 and ZIKV replication, we used Vero cells (ATCC #CCL-81) that are a model cell line for SARS-CoV-2 and ZIKV viral infection assay as previously described [Citation37,Citation86]. SARS-Related Coronavirus 2 (SARS-Cov-2), Isolate USA-WA1/2020 (BEI NR-52281), was obtained from Dr. Janko Nikolich-Žugich’s laboratory. Vero cells were suspended in the DMEM medium containing 10% FBS and seeded 3.125 × 104 cells into each well of 96 wells plates and cultured overnight with 5% CO2 at 37°C. The next day with 90% of cell confluency, a 2× solution was generated by dispensing 10 mM stocks of compounds into a V-bottom 96-well plate and DMSO control. The spent media were removed from assay plate culturing Vero cells. 100 µl of fresh medium with compound and SARS-CoV-2 virus stock with proper dilution to reach about 137 virus plaques per well was added to each well. The plates were further incubated for 3 h with 5% CO2 at 37°C, then overlayed with 100 µl per well of 1% methylcellulose in media. Plates were further incubated with 5% CO2 at 37°C for 72 h. After 72 h post-infection, plates were fixed with 10% Neutral Buffered Formalin for 30 min and stained for SARS-CoV-2 viral plaque using 0.9% crystal violet. All SARS-CoV-2-related work was conducted in a biological safety cabinet in a biosafety level 3 laboratory at the University of Arizona. The plaque was counted, and EC50 was calculated using GraphPad Prism 9.
Immunofluorescence assays
Vero E6 cells were seeded at a density of 3 × 104 cells per well in a 96-well flat bottom plate and incubated at 37 °C for 24 h. After the addition of compounds at different concentrations, the cells were infected with the SARS-CoV-2 WT strain (Washington strain, 0.01 MOI) or the Omicron strain (BA.1.1, 0.05 MOI), the cells were incubated for 24 h for the Washington strain or 42 h for the Omicron strain.
Cells were fixed with 10% 10% Formalin Solution for 30 min. Cells were detected using 1:100-diluted SARS-CoV-2 nucleocapsid monoclonal antibody (Invitrogen, MA17403) at 4°C for overnight. After washing with double-distilled water, cells were incubated with a 1:2000 dilution of goat anti-mouse IgG (H + L) highly cross-adsorbed secondary antibody, Alexa Fluor™ Plus 488 (Thermo Scientific, A32723TR) and counterstained with 1 µg of 4′,6-diamidino-2-phenylindole (DAPI) per ml. Cells were visualized using a microscope.
Compound combination
The 2D compound-compound interactions were determined by checkerboard titration in a 96-well plate. Vero- E6 cells were seeded one day before combination test at 3 × 104cells/each well. The first compound (A) was serially diluted along the x axis (columns 3 to 8); and the second compound (B) was serially diluted along the y axis (from row A to F) in the 96-well plate. The last two columns (columns 9 and 10) contained compound B-alone controls and the last two rows (row G and H) contained compound A-alone controls. After the cells grew into monolayer, the medium was discard, followed by addition of 50 ul compound mixture. Then 50 ul SARS-CoV-2 WA strain was added at MOI of 0.01. The plate was incubated at 37°C, 5% CO2 for 1 h. Then 100 ul overlay medium (DMEM + 2% FBS + 1% Methylcellulose) was added and the plate was further incubated at 37°C, 5% CO2. After 3 days, cells were fixed with 10% neutral formalin for 30 min, wash with water for more than 6 times and then stained with 50 ul Crystal Violet (0.5% in methanol/water) for 4 min. The staining solution was removed by aspiration and the plate was rinsed with water and dried thoroughly. Plaques were counted and numbers were recorded to calculate the percentage of inhibition. SynergyFinder was used to analyse the synergistic effect of the two compounds (https://synergyfinder.fimm.fi/synergy/20230202071113839465/). Each combination testing experiment has three replicates.
Microscale thermophoresis (MST)
Monolith NT.115 Microscale Thermophoresis (MST) instrument (NanoTemper Technologies) was used for this assay. Monolith protein labelling kit RED-NHS was purchased from NanoTemper Technologies. Briefly, the NSP14 protein was labelled using a RED-NHS labelling kit (NanoTemper) following the manufacturer’s instructions. A serial dilution of ligands NSC 111552 and 288387 (0.6 mM to 9.1 nM) was prepared and titrated against 40 nM labelled NSP14. The assays were read in 20% excitation power and medium MST power.
For binding of NSP10 with NSP14, NSP10 was diluted serially from 1 µM to 0.003 nM and titrated against 50 nM labeled NSP14 protein. The assays were read at medium MST power and 20% excitation power.
Molecular docking
The Schrodinger Maestro modelling platform was used to predict binding poses of NSC 111552 and 288387 into the SAM-binding pocket of NSP14. The protein structure was obtained from the Protein Data Bank (PDB ID: 7EGQ). Ligands were prepared using the LigPrep module; and ionization states were generated at pH 7.0 ± 2 using the OPLS4 force field. Protein structure was prepared and minimized in a similar fashion as the ligands using the OPLS4 force field. The binding site of NSP14 was determined using structural information provided in literature as well as docking of the endogenous ligand SAH. Docking was performed using the Ligand Docking module with the Glide docking program. A series of conformations of the ligands were generated by using molecular dynamics with various random seeds. Each of the conformations was translated through the active site using flexible ligand docking. The top poses were then subjected to Prime MM-GBSA minimization using the VGSB solvation model and OPLS4 forcefield. Residues within 4 Å of ligand were allowed to be flexible, while other residues remained rigid. Binding affinities were predicted and scored using the resulting MM-GBSA ΔG binding value.
Supplemental Material
Download TIFF Image (3.8 MB)Acknowledgment
The authors thank Drs Jennifer L Uhrlaub and Janko Nikolich-Žugich at the University of Arizona for the gift Omicron BA.1.1 SARS-CoV-2 strain
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- de Wit E, Feldmann F, Cronin J, et al. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc Natl Acad Sci U S A. 2020 Mar 24;117(12):6771–6776.
- Owen DR, Allerton CMN, Anderson AS, et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science. 2021 Dec 24;374(6575):1586–1593.
- Wahl A, Gralinski LE, Johnson CE, et al. SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature. 2021 Mar;591(7850):451–457.
- Samrat SK, Tharappel AM, Li Z, et al. Prospect of SARS-CoV-2 spike protein: potential role in vaccine and therapeutic development. Virus Res. 2020 Oct 15;288:198141.
- Chan JF, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221–236.
- Romano M, Ruggiero A, Squeglia F, et al. A structural view of SARS-CoV-2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells. 2020 May 20;9(5):1267.
- Bouvet M, Debarnot C, Imbert I, et al. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010 Apr 22;6(4):e1000863.
- Egloff MP, Benarroch D, Selisko B, et al. An RNA cap (nucleoside-2'-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21(11):2757–2768.
- Ray D, Shah A, Tilgner M, et al. West Nile virus 5'-cap structure is formed by sequential guanine N-7 and ribose 2'-O methylations by nonstructural protein 5. J Virol. 2006;80(17):8362–8370.
- Dong H, Ren S, Zhang B, et al. West Nile virus methyltransferase catalyzes two methylations of the viral RNA cap through a substrate-repositioning mechanism. J Virol. 2008 May;82(9):4295–4307.
- Dong H, Ray D, Ren S, et al. Distinct RNA elements confer specificity to flavivirus RNA cap methylation events. J Virol. 2007 May;81(9):4412–4421.
- Zhou Y, Ray D, Zhao Y, et al. Structure and function of flavivirus NS5 methyltransferase. J Virol. 2007 Apr;81(8):3891–3903.
- Schwer B, Hausmann S, Schneider S, et al. Poxvirus mRNA cap methyltransferase. bypass of the requirement for the stimulatory subunit by mutations in the catalytic subunit and evidence for intersubunit allostery. J Biol Chem. 2006 Jul 14;281(28):18953–18960.
- Bujnicki JM, Rychlewski L. Reassignment of specificities of two cap methyltransferase domains in the reovirus lambda 2 protein. Genome Biol. 2001;2(9). RESEARCH0038.
- Heilmann E, Kimpel J, Geley S, et al. The methyltransferase region of vesicular stomatitis virus L polymerase is a target site for functional intramolecular insertion. Viruses. 2019 Oct 26;11(11):989.
- Furuichi Y, Shatkin AJ. Viral and cellular mRNA capping: past and prospects. Adv Virus Res. 2000;55:135–184.
- Park GJ, Osinski A, Hernandez G, et al. The mechanism of RNA capping by SARS-CoV-2. Nature. 2022 Sep;609(7928):793–800.
- Chen Y, Cai H, Pan J, et al. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc Natl Acad Sci U S A. 2009 Mar 3;106(9):3484–3489.
- Chen Y, Guo D. Molecular mechanisms of coronavirus RNA capping and methylation. Virol Sin. 2016 Feb;31(1):3–11.
- Yan L, Yang Y, Li M, et al. Coupling of N7-methyltransferase and 3'-5’ exoribonuclease with SARS-CoV-2 polymerase reveals mechanisms for capping and proofreading. Cell. 2021 Jun 24;184(13):3474–3485e11.
- Decroly E, Imbert I, Coutard B, et al. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2'O)-methyltransferase activity [Research Support, Non-U.S. Gov't]. Journal of Virology. 2008 Aug;82(16):8071–8084.
- Bouvet M, Lugari A, Posthuma CC, et al. Coronavirus Nsp10, a critical co-factor for activation of multiple replicative enzymes. J Biol Chem. 2014 Sep 12;289(37):25783–25796.
- Minskaia E, Hertzig T, Gorbalenya AE, et al. Discovery of an RNA virus 3'->5’ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc Natl Acad Sci U S A. 2006 Mar 28;103(13):5108–5113.
- von Grotthuss M, Wyrwicz LS, Rychlewski L. mRNA cap-1 methyltransferase in the SARS genome. Cell. 2003 Jun 13;113(6):701–702.
- Bouvet M, Imbert I, Subissi L, et al. RNA 3'-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc Natl Acad Sci U S A. 2012 Jun 12;109(24):9372–9377.
- Saramago M, Barria C, Costa VG, et al. New targets for drug design: importance of nsp14/nsp10 complex formation for the 3'-5’ exoribonucleolytic activity on SARS-CoV-2. FEBS J. 2021 Sep;288(17):5130–5147.
- Decroly E, Debarnot C, Ferron F, et al. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2'-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog. 2011 May;7(5):e1002059.
- Lugari A, Betzi S, Decroly E, et al. Molecular mapping of the RNA Cap 2'-O-methyltransferase activation interface between severe acute respiratory syndrome coronavirus nsp10 and nsp16. J Biol Chem. 2010 Oct 22;285(43):33230–33241.
- Ogando NS, El Kazzi P, Zevenhoven-Dobbe JC, et al. Structure-function analysis of the nsp14 N7-guanine methyltransferase reveals an essential role in Betacoronavirus replication. Proc Natl Acad Sci U S A. 2021 Dec 7;118(49):e2108709118.
- Chen Y, Tao J, Sun Y, et al. Structure-function analysis of severe acute respiratory syndrome coronavirus RNA cap guanine-N7-methyltransferase. J Virol. 2013 Jun;87(11):6296–6305.
- Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009 Sep;8(9):724–732.
- Liu L, Dong H, Chen H, et al. Flavivirus RNA cap methyltransferase: structure, function, and inhibition. Front Biol. 2010 Aug 1;5(4):286–303.
- Aouadi W, Eydoux C, Coutard B, et al. Toward the identification of viral cap-methyltransferase inhibitors by fluorescence screening assay. Antiviral Res. 2017 Aug;144:330–339.
- Otava T, Sala M, Li F, et al. The structure-based design of SARS-CoV-2 nsp14 methyltransferase ligands yields nanomolar inhibitors. ACS Infect Dis. 2021 Aug 13;7(8):2214–2220.
- Pearson LA, Green CJ, Lin D, et al. Development of a high-throughput screening assay to identify inhibitors of the SARS-CoV-2 guanine-N7-methyltransferase using RapidFire MASS SPECTROMETRY. SLAS Discov. 2021 Jul;26(6):749–756.
- Devkota K, Schapira M, Perveen S, et al. Probing the SAM binding site of SARS-CoV-2 Nsp14 in vitro using SAM competitive inhibitors guides developing selective bisubstrate inhibitors. SLAS Discov. 2021 Oct;26(9):1200–1211.
- Basu S, Mak T, Ulferts R, et al. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of Nsp14 RNA cap methyltransferase. Biochem J. 2021 Jul 16;478(13):2481–2497.
- Ahmed-Belkacem R, Sutto-Ortiz P, Guiraud M, et al. Synthesis of adenine dinucleosides SAM analogs as specific inhibitors of SARS-CoV nsp14 RNA cap guanine-N7-methyltransferase. Eur J Med Chem. 2020 Sep 1;201:112557.
- Kasprzyk R, Spiewla TJ, Smietanski M, et al. Identification and evaluation of potential SARS-CoV-2 antiviral agents targeting mRNA cap guanine N7-methyltransferase. Antiviral Res. 2021 Sep;193:105142.
- Brecher M, Chen H, Li Z, et al. Identification and characterization of novel broad-spectrum inhibitors of the flavivirus methyltransferase. ACS Infect Dis. 2015;1(8):340–349.
- Zeng C, Wu A, Wang Y, et al. Identification and characterization of a ribose 2'-O-methyltransferase encoded by the ronivirus branch of nidovirales. J Virol. 2016 Aug 1;90(15):6675–6685.
- Sutto-Ortiz P, Tcherniuk S, Ysebaert N, et al. The methyltransferase domain of the respiratory syncytial virus L protein catalyzes cap N7 and 2'-O-methylation. PLoS Pathog. 2021 May;17(5):e1009562.
- Luan Y, Blazer LL, Hu H, et al. Design of a fluorescent ligand targeting the S-adenosylmethionine binding site of the histone methyltransferase MLL1. Org Biomol Chem. 2016 Jan 14;14(2):631–638.
- Moeller NH, Shi K, Demir O, et al. Structure and dynamics of SARS-CoV-2 proofreading exoribonuclease ExoN. Proc Natl Acad Sci U S A. 2022 Mar 1;119(9):e2106379119.
- Lin S, Chen H, Chen Z, et al. Crystal structure of SARS-CoV-2 nsp10 bound to nsp14-ExoN domain reveals an exoribonuclease with both structural and functional integrity. Nucleic Acids Res. 2021 May 21;49(9):5382–5392.
- Liu C, Shi W, Becker ST, et al. Structural basis of mismatch recognition by a SARS-CoV-2 proofreading enzyme. Science. 2021 Sep 3;373(6559):1142–1146.
- Calvet G, Aguiar RS, Melo AS, et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis. 2016 Feb 17;16:653–660.
- Martines RB, Bhatnagar J, Keating MK, et al. Notes from the field: evidence of Zika virus infection in brain and placental tissues from two congenitally infected newborns and two fetal losses – Brazil, 2015. MMWR Morb Mortal Wkly Rep. 2016;65(6):159–160.
- Chen H, Liu L, Jones SA, et al. Selective inhibition of the West Nile virus methyltransferase by nucleoside analogs. Antiviral Res. 2013 Mar;97(3):232–239.
- Chen H, Zhou B, Brecher M, et al. S-adenosyl-homocysteine is a weakly bound inhibitor for a flaviviral methyltransferase. PLoS One. 2013;8(10):e76900.
- Roingeard P, Beaumont E. Hepatitis C vaccine: 10 good reasons for continuing. Hepatology. 2020 May;71(5):1845–1850.
- Montaner JS, Lima VD, Harrigan PR, et al. Expansion of HAART coverage is associated with sustained decreases in HIV/AIDS morbidity, mortality and HIV transmission: the “HIV treatment as prevention” experience in a Canadian setting. PLoS One. 2014;9(2):e87872.
- Moreno S, Perno CF, Mallon PW, et al. Two-drug vs. three-drug combinations for HIV-1: Do we have enough data to make the switch? HIV Med. 2019 Apr;20(Suppl 4):2–12.
- Ma MJ, Qiu SF, Cui XM, et al. Persistent SARS-CoV-2 infection in asymptomatic young adults. Signal Transduct Target Ther. 2022 Mar 9;7(1):77.
- Avanzato VA, Matson MJ, Seifert SN, et al. Case study: prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised individual with cancer. Cell. 2020 Dec 23;183(7):1901–1912. e9.
- Baang JH, Smith C, Mirabelli C, et al. Prolonged severe acute respiratory syndrome coronavirus 2 replication in an immunocompromised patient. J Infect Dis. 2021 Jan 4;223(1):23–27.
- Choi B, Choudhary MC, Regan J, et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N Engl J Med. 2020 Dec 3;383(23):2291–2293.
- Kemp SA, Collier DA, Datir RP, et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature. 2021 Apr;592(7853):277–282.
- Moran E, Cook T, Goodman AL, et al. Persistent SARS-CoV-2 infection: the urgent need for access to treatment and trials. Lancet Infect Dis. 2021 Oct;21(10):1345–1347.
- Jacobs JJL. Persistent SARS-2 infections contribute to long COVID-19. Med Hypotheses. 2021 Apr;149:110538.
- Truong TT, Ryutov A, Pandey U, et al. Increased viral variants in children and young adults with impaired humoral immunity and persistent SARS-CoV-2 infection: A consecutive case series. EBioMedicine. 2021 May;67:103355.
- Taha Y, Wardle H, Evans AB, et al. Persistent SARS-CoV-2 infection in patients with secondary antibody deficiency: successful clearance following combination casirivimab and imdevimab (REGN-COV2) monoclonal antibody therapy. Ann Clin Microbiol Antimicrob. 2021 Dec 30;20(1):85.
- Gaspar-Rodriguez A, Padilla-Gonzalez A, Rivera-Toledo E. Coronavirus persistence in human respiratory tract and cell culture: An overview. Braz J Infect Dis. . 2021 Sep-Oct;25(5):101632.
- Li Z, Tharappel AM, Xu J, et al. Small-molecule inhibitors for the Prp8 intein as antifungal agents. Proc Natl Acad Sci U S A. 2021 Jan 12;118(2):e2008815118.
- Nguyenla X, Wehri E, Van Dis E, et al. Discovery of SARS-CoV-2 antiviral synergy between remdesivir and approved drugs in human lung cells. Sci Rep. 2022 Nov 2;12(1):18506.
- Yadav B, Wennerberg K, Aittokallio T, et al. Searching for drug synergy in complex dose-response landscapes using an interaction potency model. Comput Struct Biotechnol J. 2015;13:504–513.
- Vuong W, Fischer C, Khan MB, et al. Improved SARS-CoV-2 M(pro) inhibitors based on feline antiviral drug GC376: structural enhancements, increased solubility, and micellar studies. Eur J Med Chem. 2021 Oct 15;222:113584.
- Vuong W, Khan MB, Fischer C, et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun. 2020 Aug 27;11(1):4282.
- Bai B, Belovodskiy A, Hena M, et al. Peptidomimetic alpha-acyloxymethylketone warheads with Six-membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability. J Med Chem. 2022 Feb 24;65(4):2905–2925.
- Arutyunova E, Khan MB, Fischer C, et al. N-Terminal finger stabilizes the S1 pocket for the reversible feline drug GC376 in the SARS-CoV-2 M(pro) dimer. J Mol Biol. 2021 Jun 25;433(13):167003.
- Bai B, Belovodskiy A, Hena M, et al. Peptidomimetic alpha-acyloxymethylketone warheads with six-membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability. J Med Chem. 2022 Feb 24;65(4):2905–2925.
- Lu J, Chen SA, Khan MB, et al. Crystallization of feline coronavirus M(pro) With GC376 reveals mechanism of inhibition. Front Chem. 2022;10:852210.
- Decroly E, Ferron F, Lescar J, et al. Conventional and unconventional mechanisms for capping viral mRNA. Nat Rev Microbiol. 2011 Dec 5;10(1):51–65.
- Gnyszka A, Jastrzebski Z, Flis S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013 Aug;33(8):2989–2996.
- Mullard A. FDA approves an inhibitor of a novel ‘epigenetic writer’. Nat Rev Drug Discov. 2020 Mar;19(3):156.
- Ganesan A, Arimondo PB, Rots MG, et al. The timeline of epigenetic drug discovery: from reality to dreams. Clin Epigenetics. 2019 Dec 2;11(1):174.
- Dong H, Liu L, Zou G, et al. Structural and functional analyses of a conserved hydrophobic pocket of flavivirus methyltransferase [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, Non-P.H.S.]. The Journal of Biological Chemistry. 2010 Oct 15;285(42):32586–32595.
- Lim SP, Sonntag LS, Noble C, et al. Small molecule inhibitors that selectively block dengue virus methyltransferase [research support, Non-U.S. gov't]. The Journal of Biological Chemistry. 2011 Feb 25;286(8):6233–6240.
- Pan R, Kindler E, Cao L, et al. N7-Methylation of the coronavirus RNA Cap Is required for maximal virulence by preventing innate immune recognition. mBio. 2022 Feb 22;13(1):e0366221
- Bobrovs R, Kanepe I, Narvaiss N, et al. Discovery of SARS-CoV-2 Nsp14 and Nsp16 methyltransferase inhibitors by high-throughput virtual screening. Pharmaceuticals (Basel). 2021 Nov 30;14(12):1243.
- Selvaraj C, Dinesh DC, Panwar U, et al. Structure-based virtual screening and molecular dynamics simulation of SARS-CoV-2 guanine-N7 methyltransferase (nsp14) for identifying antiviral inhibitors against COVID-19. J Biomol Struct Dyn. 2021 Aug;39(13):4582–4593.
- Liu C, Zhu X, Lu Y, et al. Potential treatment with Chinese and western medicine targeting NSP14 of SARS-CoV-2. J Pharm Anal. 2021 Jun;11(3):272–277.
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73.
- Brecher M, Chen H, Liu B, et al. Novel broad spectrum inhibitors targeting the flavivirus methyltransferase. PLoS One. 2015;10(6):e0130062.
- Li Z, Brecher M, Zhang J, et al. Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res. 2017;27:1046–1064.
- Li Z, Xu J, Lang Y, et al. JMX0207, a niclosamide derivative with improved pharmacokinetics, suppresses Zika virus infection both In vitro and In vivo. ACS Infect Dis. 2020 Oct 9;6(10):2616–2628.