
ABSTRACT
The main protease (3-chymotrypsin-like protease, 3CLpro) of SARS-CoV-2 has become a focus of anti-coronavirus research. Despite efforts, drug development targeting 3CLpro has been hampered by limitations in the currently available activity assays. Additionally, the emergence of 3CLpro mutations in circulating SARS-CoV-2 variants has raised concerns about potential resistance. Both emphasize the need for a more reliable, sensitive, and facile 3CLpro assay. Here, we report an orthogonal dual reporter-based gain-of-signal assay for measuring 3CLpro activity in living cells. It builds on the finding that 3CLpro induces cytotoxicity and reporter expression suppression, which can be rescued by its inhibitor or mutation. This assay circumvents most limitations in previously reported assays, especially false positives caused by nonspecific compounds and signal interference from test compounds. It is also convenient and robust for high throughput screening of compounds and comparing the drug susceptibilities of mutants. Using this assay, we screened 1789 compounds, including natural products and protease inhibitors, with 45 compounds that have been reported to inhibit SARS-CoV-2 3CLpro among them. Except for the approved drug PF-07321332, only five of these inhibit 3CLpro in our assays: GC376; PF-00835231; S-217622; Boceprevir; and Z-FA-FMK. The susceptibilities of seven 3CLpro mutants prevalent in circulating variants to PF-07321332, S-217622, and GC376 were also assessed. Three mutants were identified as being less susceptible to PF-07321322 (P132H) and S-217622 (G15S, T21I). This assay should greatly facilitate the development of novel 3CLpro-targeted drugs and the monitoring of the susceptibility of emerging SARS-CoV-2 variants to 3CLpro inhibitors.
Introduction
The coronavirus disease 2019 (COVID-19) pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has become a global catastrophe, particularly with the emergence and spread of the Omicron variant, which has more mutations in spike glycoprotein, resulting in stronger viral binding affinity, higher transmissibility, and antibody escape [Citation1]. Despite efforts, COVID-19 has spread around the world, resulting in more than 671 million confirmed cases and over 6.7 million deaths (https://www.worldometers.info/coronavirus/). The continued emergence of SARS-CoV-2 variants has led to decreased protection from vaccination [Citation2] and raised concerns about potential resistance to currently available anti-COVID-19 drugs [Citation3,Citation4].
The coronavirus main protease, 3-chymotrypsin-like protease (3CLpro), also known as nsp5, cleaves the viral replicase polyproteins at no less than 11 conserved sites to release all functional polypeptides required for viral replication and transcription [Citation5]. Due to its pivotal role in viral life cycle, remarkable sequence conservation (identity: 47%∼96% within same genus and 38%∼48% in different genera) with a high structural conservation of substrate-binding sites in all known human coronaviruses, and lack of homolog in human cells, 3CLpro has been considered a desirable target for broad-spectrum anti-coronavirus drug development [Citation5–7]. The potential of targeting 3CLpro for COVID-19 therapeutics has been validated in clinical settings by Paxlovid (ritonavir-boosted PF-07321332), which has been shown to reduce hospitalization despite evidence that it benefits only patients with non-severe COVID-19 [Citation8]. With the progress of COVID-19 pandemic, 3CLpro has become the focus of numerous drug discovery efforts worldwide. A growing number of studies on the identification, design, and discovery of 3CLpro inhibitors have been reported, and many compounds have been identified as 3CLpro inhibitors [Citation9–23]. However, quite a lot of these claimed inhibitors have been demonstrated to be nonspecific to 3CLpro [Citation15,Citation16,Citation20–23]. Some limitations in currently used 3CLpro assays should be held accountable for such false-positive results, which will significantly impede the development of effective 3CLpro inhibitors because attempts at activity optimization of leads that turn out to be dead ends are most costly in terms of time and resources. In addition, the emergence of 3CLpro mutations in circulating SARS-CoV-2 variants and the growing use of anti-3CLpro drugs in clinical settings have raised concern about a possible 3CLpro inhibitor susceptibility alteration in circulating variants [Citation4,Citation24,Citation25]. Both highlighted the need for a reliable 3CLpro assay for inhibitor identification and susceptibility testing.
However, SARS-CoV-2 antiviral assays that require biosafety level 3 (BSL-3) conditions are laborious, less suitable for high-throughput screening (HTS), often not accessible, and may also lead to a misunderstanding of the real mechanism of an inhibitor, which would be detrimental to further lead optimization [Citation15]. The widely used in vitro enzymatic assay based on fluorescence resonance energy transfer (FRET) frequently produces false positives with high reactive compounds or promiscuous compounds that can interact with multiple unrelated biological targets [Citation16]. Its requirement of protease expression and purification will also be challenging for the comparison of a variety of mutants (naturally occurring and/or lab-designed). To circumvent these problems, a variety of cell-based 3CLpro assays have been developed. Compared to in vitro enzymatic assays, cell-based assays do not require sophisticated processes of protein expression and purification, making the comparison of a variety of mutants more facile. It can also provide a more physiologically relevant measure of 3CLpro activity and other valuable information for small-molecule inhibitor development, such as membrane permeability, metabolic liability, and off-target effects. However, most reported cell-based assays still have some limitations. All loss-of-signal assays, including but not limited to the widely used Flip-GFP assay [Citation17] or its luciferase analogous [Citation18], probe the decrease in 3CLpro activity via reporter signal loss and thus cannot distinguish 3CLpro inhibition from cytotoxicity or other off-target effects, leading to false positives. Gain-of-signal assays, in which a decrease in 3CLpro activity results in an increase in the reporter signal, can easily distinguish inhibition of 3CLpro from cytotoxicity. But previously described gain-of-signal assays for 3CLpro, such as those based on the bioluminescence resonance energy transfer (BRET) [Citation19], the reporter protein complementation [Citation20,Citation21], protease-mediated cytotoxicity or suppression of reporters [Citation15,Citation22,Citation23], are still limited by a lack of sensitivity and/or HTS compatibility. Furthermore, all these assays using a single readout may suffer signal interference from test compounds, such as fluorescence, absorbance, quenching, or luciferase inhibition. A separate complementary assay will be needed to exclude false positives or false negatives in these assays.
To address these issues, here we described a novel gain-of-signal assay for measuring 3CLpro activity in living cells. It builds on the finding that 3CLpro causes cytotoxicity and suppresses reporter expression, which can be rescued by an inactivating mutation or treatment with a specific inhibitor. As a gain-of-signal assay, it can readily differentiate cytotoxicity or other off-target effects from true inhibition of 3CLpro. Promiscuous compounds or non-specific inhibitors targeting host-cell pathways also did not result in false positives in this assay. Compared to previously described gain-of-signal assays, it is sensitive enough to detect weak inhibition from low-potency inhibitors or a slight decrease in activity of mutated variants. Utilizing bicistronically expressed Gaussia luciferase (Gluc) and monomer red fluorescent mKate2 as an orthogonal reporter for 3CLpro activity probing, it can easily recognize false positives caused by signal interference from test compounds in a single assay. Its facileness and robustness were also illustrated in HTS of a 1785 compound library and susceptibility comparisons of seven naturally occurring mutants.
Materials and methods
Chemicals and reagents
Inhibitors and compound libraries were purchased from Topscience (Shanghai, China). Gluc substrate coelenterazine was from YEASEC (Shanghai, China). All other used chemicals are analytically pure and come from different manufacturers.
Plasmids and cells
The coding sequence of SARS-CoV-2 3CLpro (GenBank: NC_045512.2) was synthesized with codon optimization for humans by Biomed (Beijing, China). Mutants were created by site-directed mutagenesis. All constructs for mammalian expression ((A)) were created using standard molecular cloning protocols with the pCMV6-AN-mKate (OriGene, Wuxi, China) as a backbone vector.
Figure 1. SARS-CoV-2 3CLpro transfection induced cytotoxicity and suppressed reporter expression in 293T cells, and it can be rescued by treatment with the specific inhibitor. (A) Schematic of the BRET, reporters, and 3CLpro expression constructs. (B) The “WT” represents wild-type 3CLpro co-transfected with reporter as indicated, the “C145A” indicates the C145A mutant co-transfected, and the “WT+GC376” is the same as the “WT” but with a treatment of GC376 at 20 μM. The “Mock” is native cells without any treatment. (C) Western blotting results obtained from cells treated as described in (B). The arrows point to mKate2/EGFP (26 kD), 3CLpro (36 kD). Bands at about 55 kD is α-tubulin. (D) Different expression of reporters in cells treated with WT+GC376 vs. WT or C145A. Data were presented as mean ± SD (n = 3), and the significant difference was determined using the Student t-test. (E) The dual-reporter construct was used. Data with n = 2 was analysed using an ANOVA with a post-hoc Tukey's HSD test. *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001; ns: not significant.
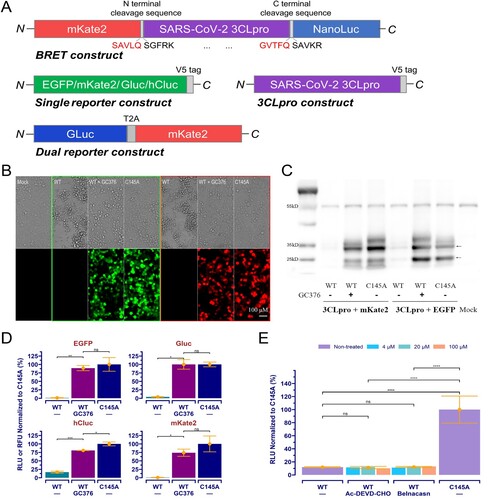
293T cells were maintained in high-glucose Dulbecco's modified Eagle medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, ExCell Bio, Shanghai, China) and 1% penicillin–streptomycin in 5% CO2 at 37°C.
Cell transfection was performed using TurboFect transfection reagent (Thermo Fisher Scientific), according to the manufacturer's protocol. The cell images were obtained using the EVOS FLoid System (Thermo Fisher Scientific).
Western blotting and reporter assay
Western blotting was performed following standard procedure. The primary antibody for V5-tag (Abcam, Shanghai, China) and peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, USA) were used for targets detection. The immunoreactive proteins were detected by SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific).
Luciferase assay was performed in the white, opaque 96-well plates using 20 μL of culture supernatant for each well, and 50 μL/well Gluc buffer (100 mM Tris-HCl at pH 8.0, 10 μM coelenterazine, 0.3% Triton X-100, and 70 mM NaI) or Cypridina Glow Assay Buffer containing 1 × Vargulin (Thermo Fisher Scientific) was added for Gluc or hCluc assay. Total chemiluminescence was acquired using the SpectraMax i3x multi-mode microplate reader (Molecular Devices). Fluorescence intensities in cells were measured using the same microplate reader.
3CLpro assay and hight throughput screening
293T cells were co-transfected with reporter and 3CLpro constructs (4:1 ratio of plasmid DNA) as described above. In each clear-bottom black 96-well plate, the untransfected cells and inactive 3CLpro mutant (C145A) co-transfected cells served as blank and positive controls, respectively. For the 3CLpro inhibition assay, compounds at the final concentrations indicated were added at 6 h post-transfection, and vehicle (0.2% DMSO) in the same volume was used as a negative control. Reporter signals were measured 54 h post-transfection, as described above. All the acquired signals were normalized to the positive controls after subtracting the mean of the blank controls. The percentage of inhibition was calculated as following formula: Inhibition % = 100% × (sample value − mean of negative controls) ÷ (mean of positive controls − mean of negative controls).
For HTS, all cells were co-transfected with the wild-type 3CLpro and the Glu-mKate2 dual reporter constructs, with 0.2% DMSO and 20 μM GC376 as negative and positive controls, respectively, and no blank controls included. Compounds were assessed once at 20 μM. The mean value of inhibition data from all samples excluding positive controls plus three times its standard deviation (SD) was taken as the threshold for hit selection. The Z’-factor [Citation26] was used to evaluate the quality and performance of HTS.
Data analysis and statistics
All data was presented as mean values with SDs, or raw data provided. The Student t-test or analysis of variance (ANOVA) with post-hoc Tukey's honestly significant difference (HSD) test was used to determine the significance of difference. The dose–response data was analysed using four-parameter logistic regression with assistance from R and package “drc” [Citation27,Citation28]. Previously reported x-ray crystal structures (PDB IDs: 6WTT [Citation10], 7VU6 [Citation13], 7VH8 [Citation29], 7TOB [Citation25]) were used for inhibitor binding sites analysis.
Results
SARS-CoV-2 3CLpro induced protease-mediated cytotoxicity and suppressed reporter expression in 293T cells
Our original design for the cellular 3CLpro assay was to express a BRET-based 3CLpro biosensor ((A)) in host cells and detect the BRET signal caused by its autocleavage. However, transfection of this construct into 293T cells resulted in a strong cytotoxicity. To confirm this observation, further cross-validation experiments accompanied by an inactive mutant (C145A) or the treatment of GC376 [Citation9] were performed using various reporters. The results showed that 3CLpro-induced cytotoxicity could be reverted by a treatment with GC376 at 20 μM ((B)), which also restored reporter expression in 3CLpro-transfected cells to a level compared to that in cells transfected with the inactive C145A mutant ((B,D)), suggesting protease activity is responsible for the cytotoxicity. Western immunoblotting ((C)) further showed the protease-mediated overall suppression of protein expression in host cells, indicating that protease-mediated cytotoxicity in host cells rather than the cleavage of reporter proteins will be an appropriate interpretation for the diminished signals of reporters.
Cytotoxicity caused by viral proteases has been documented in other coronaviruses, and the apoptosis resulting from protease-mediated cleavage in host proteins is considered the underlying mechanism [Citation30]. SARS-CoV-2 3CLpro-induced cytotoxicity has also been described [Citation15,Citation23] and characterized as apoptosis [Citation15]. We therefore examined whether inhibitors targeting the cell death pathway might exert the same effect on the reporter's expression. At concentrations up to 100 μM, both the caspase 3 inhibitor Ac-DEVD-CHO targeting apoptosis pathway [Citation31], and the caspase 1 inhibitor Belnacasan targeting pyroptosis pathway [Citation32], failed to rescue reporter expression in 3CLpro transfected cells ((E)).
It will be simple to link the reporter expression to the survival or death of cells with 3CLpro inhibited or active. However, 3CLpro-induced reporter expression suppression with no obvious cell death had also been observed and was partially related to the fact that 3CLpro suppresses the accumulation of reporter mRNA in transfected cells [Citation22]. Several studies have demonstrated that SARS-CoV-2 3CLpro cleaves numerous host proteins in 3CLpro-transfected or SARS-CoV-2-infected cells, including but not limited to those involved in apoptosis regulation, transcription and translation, Hippo signalling, and immune or inflammatory response regulation [Citation33–36]. Furthermore, TAIL substrate-targeted N-terminomics profiling of 3CLpro substrates in human cell proteomes showed that 3CLpro cleaves more than 100 host cell proteins involved in essential cellular processes [Citation36,Citation37]. Given such extensive cleavage of host proteins by 3CLpro, we consider that an inhibitor targeting one cellular pathway rather than 3CLpro will not rescue this protease-mediated cytotoxicity and/or reporter expression suppression; meanwhile, the expression of a co-transfected reporter will reflect the comprehensive effects of 3CLpro-mediated extensive host protein cleavages on living cells and provide a reliable readout for 3CLpro activity.
Validation of the reporter co-transfection-based assay for measuring 3CLpro activity and inhibitor potency in living cells
We next evaluated whether co-transfected reporter expression could provide a sensitive and quantitative measure for 3CLpro in living cells. Considering the limitations of the single reporter-based assay, as previously stated, the dual reporter construct ((A)) was used for assay development and all following studies. As proof-of-concept, a series of assays were performed in which the 3CLpro DNA was gradually replaced by equal amounts of the C145A mutant in transfection. The results showed that both the expression of Gluc and mKate2 increased exponentially with the amount of 3CLpro DNA being replaced. Linear regression analysis showed that the reporter readouts in natural logarithm are significantly correlated to the percent of 3CLpro DNA replaced. The limit of detection (LoD) calculated as three times the SD of the negative control divided by the slope of the fitted regression, also indicates satisfactory sensitivity for the Gluc and mKate2 assays ((A)). To assess if this assay is also reliable and quantitative for evaluating the potency of inhibitors, some compounds that have been reported to inhibit 3CLpro of SARS-CoV-2 were further tested. GC376 and PF-00835231 demonstrated typical dose response inhibition with a half-maximal inhibitory concentration (IC50) comparable to that reported in previous studies using cell-based 3CLpro assays or in vitro antiviral assays [Citation9,Citation10,Citation15–17,Citation19–21,Citation23] ((B,C), and Table S2). Ebselen, which inhibited SARS-CoV-2 3CLpro in enzymatic assay and SARS-CoV-2 replication in cells [Citation11], showed no inhibition at concentrations up to 100 μM ((D)). This is consistent with other studies suggesting it is not specific to 3CLpro but a promiscuous inhibitor with high reactivity to form selenosulfide bonds with multiple proteins, including SARS-CoV-2 papain-like protease [Citation15,Citation16,Citation20–23,Citation38]. Masitinib, a cellular tyrosine kinase inhibitor that showed activity against pan-coronaviruses and inhibited 3CLpro in cell-based FlipGFP/Protease-Glo assays and was co-crystallized with pure enzyme [Citation12], also tested negative ((D)). Given that loss-of-gain assays cannot discriminate 3CLpro inhibition from off-target effects on host cells and the frequent co-crystallization of other promiscuous compounds with 3CLpro [Citation16], our clear negative results, together with same findings from others [Citation20,Citation22], suggest that Masitinib's antiviral activity cannot be 3CLpro specific. According to a recent study showing that Imatinib, another tyrosine kinase inhibitor with high structural similarity to Masitinib, inhibits viral entry by directly binding Spike and causing it to change conformation [Citation39], Masitinib should exert antiviral action through the same mechanism.
Figure 2. The orthogonal dual reporter assay provides a sensitive and quantitative measurement of 3CLpro activity and inhibition by specific inhibitors in living cells. (A) Correlation analysis of reporter readouts vs 3CLpro activity. The data points plotted are from single experiment (n = 3), and the lines with grey shading (95% CIs) were predicted by fitted linear regression. (B) and (C) Dose-response curve and IC50 of the GC376 and PF-00835231 in the Gluc assay and mKate2 assays, respectively. The date point plotted is from two repeated experiments (n = 4). (D) Ebselen and masitinib exhibited no inhibition on 3CLpro in assay. The Gluc assay data from a single experiment (n = 3) is plotted.
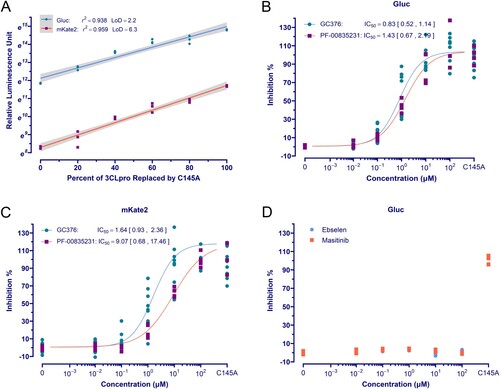
All the above demonstrated that the reporter co-transfection-based assay can be used for sensitive and quantitative detection of 3CLpro activity and inhibitor potency in living cells, while promiscuous or nonspecific compounds will not lead to false positives.
Performance, robustness, and reliability of the orthogonal dual reporter-based assay in HTS
Having established the novel gain-of-signal assay based on orthogonal dual reporter co-transfection with 3CLpro, we then explored its performance and robustness for HTS. A collection of the TargetMol Natural Product Library (1500 compounds, Table S3) and the TargetMol Protease Inhibitor Library (285 compounds, Table S4) was screened under HTS settings ((A)). Using 20 μM GC376 as a positive control, the screening exhibited excellent robustness, with a Z’-factor largely above 0.5 ((C,D)) [Citation26]. Low concentrations of GC376 (2.00 μM) and PF-00835231 (1.25 μM) randomly interpolated in screening also produced a noticeable positive signal as expected ((C,D)). Tested at 20 μM, three hits were identified that produced positive signals above the hit selection threshold in both the Gluc assay and the mKate2 assay ((C,D)). Three compounds that showed signals above the threshold only in the mKate2 assay are fluorescent compounds that interfere with mKate2 signals and were identified as false positives ((D)). Hits from primary single-point HTS were further validated by dose–response assays ((B)). S-217622 has the highest potency, with an IC50 of 0.84 μM, which is close to its IC50 against SARS-CoV-2 and other coronaviruses [Citation13], whereas Boceprevir and Z-FA-FMK have only weak to moderate potency, which is consistent with previous studies using gain-of-signal assays [Citation15,Citation19,Citation20,Citation22] (Table S2).
Figure 3. High throughput screening. (A) The schematic diagram of the HTS setting. GC376 for positive control was used at 20 μM, and 0.2% DMSO was used for negative control. (B) Dose-response curve and IC50 of hits using the Gluc assay. The data points plotted are from two repeated experiments (n = 3). (C) and (D) The results of HTS. The red dashed line is the mean value of positive controls, and the blue dashed line indicates the threshold for hit selection. The yellow triangles with annotation are GC376 (2.00 μM) and PF-00835231 (1.25 μM) randomly interpolated for performance validation.
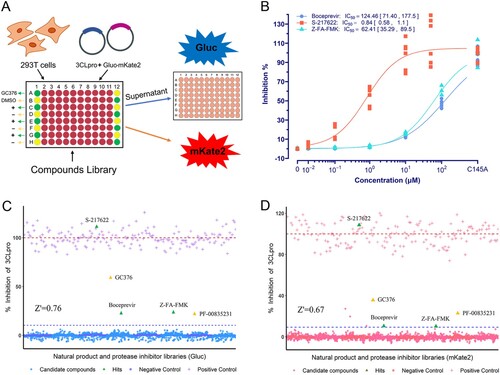
Except for three hits, all other compounds tested negative, including 37 previously claimed 3CLpro inhibitors, some of which had also shown negative results in other studies (Table S5). Included in the 285 diverse protease inhibitors screened were lots of inhibitors targeting various cellular pathways, including but not limited to those targeting cell death pathways (Table S4). Negative results from these compounds further demonstrated that inhibitors targeting the host cell pathway but not 3CLpro will not lead to false positives in this assay.
Illustration of the assay's utility in comparing the activity and susceptibilities of 3CLpro mutants
Considering the concern about potential drug resistance raised by 3CLpro mutations found in circulating SARS-CoV-2 variants [Citation4,Citation24], we next attempted to explore the possible utility of our assay for activity and susceptibility comparison in prevalent mutants. Seven mutations that are dominant in at least one lineage of circulating or previously circulated variants were investigated ().
Table 1. 3CLpro mutations investigated in this study.
Compared to the wild-type 3CLpro, the P132H mutant showed a substantial decrease in activity (30.94% ± 2.15%, p < 0.0001), and other mutants except for T21I and L205V also presented a slight but significant lower activity ((A)). For GC376; S-217622; and PF-07321332 tested, their IC50s against all seven mutants were comparable to or lower than those against the wild-type protease ((B–D)). However, three mutations led to a slope decrease in the dose–response curve of PF-07321332 (P132H) or S-217622 (G15S and T21I) ((C,D)). The variations in GC376 IC50 seem to be in connection with the changes in activity of mutants, as seen in the highest IC50 against wild-type enzyme and the lowest against the weakest P132H mutant ((B)). In theory, the IC50 of the same inhibitor required for a weak protease will be lower than that required for a strong protease in the same assay. Thus, a slight IC50 change in our assays may not make so much sense, whereas a substantial slope decrease in the dose–response curve will be more significant because it means a decrease in response at high concentration, indicating a decrease in susceptibility, as evidenced in HIV-1, for which single mutations in protease do not affect IC50 for inhibitor but decrease slope of the dose–response curve [Citation41].
Figure 4. Effect of mutations on 3CLpro activity and its susceptibility. (A) Activity assay of 3CLpro mutants investigated. The data is presented as the mean with raw data points from three repeated experiments (n = 3). The significant difference compared to WT was determined using the ANOVA with a post-hoc Tukey's HSD test. (B ∼ D) The susceptibility of 3CLpro mutants to GC376, PF-07321332, and S-217622. The data points plotted are from three repeated experiments (n = 3).
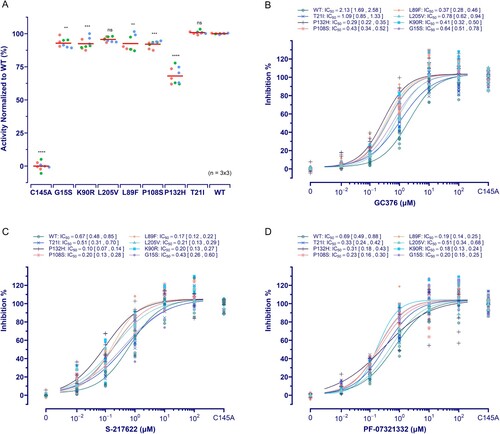
Except for P108S, which has shown lower kcat/Km values than wild-type 3CLpro in the FRET-based enzymatic assay [Citation42], previous studies using in vitro enzymatic assays have not reported significant activity or susceptibility changes of the mutants studied here [Citation4,Citation24,Citation25,Citation43]. According to the x-ray crystal structures of the inhibitor-bound SARS-CoV-2 3CLpro [Citation10,Citation13,Citation25,Citation29], other than the P108S mutation involving an allosteric site between the catalytic and dimerization domains, none of the investigated mutations participate in the SARS-CoV-2 3CLpro active site, allosteric sites, or dimerization interface ((A)). However, a recent study has demonstrated that the P132H mutation resulted in a decrease in the thermal stability of SARS-CoV-2 3CLpro, despite no compromising in catalysis or small-molecule drug inhibition in the in vitro enzymatic assay, indicating greater protein flexibility in 3CLpro, which plays an important role in broadening the substrate profile or altering ligand binding [Citation25]. P132 lies between the catalytic domain and the dimerization domain and at a turn of the flexible loop, which connects two β-sheets in the catalytic domain. The crystal structure of the SARS-CoV-2 3CLpro P132H mutant with GC376 bound showed that this mutation causes E240 to reorient and form new hydrogen bonds with H132 (water-mediated) and T198, finally resulting in a slight shift of the loop at the bottom boundary of sub-pocket S4 in substrate-binding sites ((B)) [Citation25]. Considering 3CLpro's broad substrate profile, such a shape change in the binding pocket should result in a change in 3CLpro's proteolytic activities on at least some substrates, if not all. The substantial decrease in P132H mutant activity detected in our assay well reflects this. According to the high difference in cleavage efficiencies of 3CLpro on divergent viral substrates [Citation44] and the appreciable variations of IC50s for the same inhibitor in an assay using different substrates [Citation19], we think that our assay detecting the overall effect of 3CLpro-mediated broad host substrate cleavages on cells will be more comprehensive in reflecting changes in or inhibition of 3CLpro activity than assays measuring a single substrate cleavage.
Figure 5. SARS-CoV-2 3CLpro structures with inhibitors bound. (A) 3CLpro homodimer (green and cyan) with GC376 bound (PDB ID 6WTT). The active site and two allosteric sites are circled. Mutation sites in two promoters are indicated in hot pink. Hydrogen bonds at the dimerization interface are also indicated (red dashed lines). (B) Comparison of the GC376-cocrystal structures of P132H (cyan; PDB ID 7TOB) and WT (green). GC376 and residues needed for comparison are shown as sticks. A close-up view of position 132 (top) and the substrate-binding pocket (bottom) is shown in the right panel. Water molecules are shown as red spheres. Dashed red lines indicate hydrogen bonds. S1’ and S1–S4 in red indicate sub-pockets. (C) Close-up view of PF-07321332 binding to 3CLpro (green; PDB ID 7VH8) with superimposition of P132H (cyan). The inhibitor is shown as a yellow stick. Residues participating in binding are shown as sticks, and the hydrogen bond acceptors are coloured orange. Water molecules and hydrogen bonds are shown as described in (B). (D)S-217622 binding to the 3CLpro active site (PBD ID 7VU6). Residues involving mutation are shown as purple sticks, and the π−π stacking is indicated by a dashed cyan line. Inhibitor and residues involving binding are shown as described in (C), except for the hydrogen bond between T21 and T26, which is shown in yellow. (E) Superimposition of GC376 binding to WT (green) and P132H (cyan) 3CLpro. GC376 is shown as yellow sticks or grey for its binding to WT or P132H, and residues involving inhibitor binding are shown as described above. Hydrogen bonds are indicated by red or yellow dashed lines for WT or P132H binding. (F) Superimposition of the GC376-bound P132H mutant (cyan) and WT 3CLpro (green, yellow, and grey for the three protomers in the asymmetric trimeric subunit).
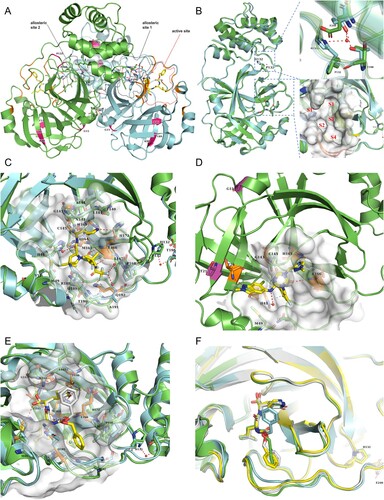
The selective change in susceptibility of P132H mutant to PF-07321332 and not to S-217622 or GC376 can be explained by the differences in their binding to S4 subsites. Upon PF-07321332 binding to the active site, its trifluoroacetyl group occupies the S4 sub-pocket and is stabilized by hydrogen bonding with Q192 and buried water molecules ((C)). This binding may be destabilized by the shift of some residues (such as P168 and Q198–Q192) in the S4 subsites. The binding of S-217622, due to no occupancy in S4 sub-pocket, will not be influenced ((D)), while GC376 can adopt various binding poses to adapt to or avoid the influence of shape change in the S4 sub-pocket ((E,F)). Similarly, the selective change in susceptibility of T21I and G15S to S-217622 versus GC376 or PF-07321332 can also explained. At S-217622's binding to 3CLpro, its 6-chloro-2-methyl-2H-indazole moiety is placed in the S1’ sub-pocket and holds hydrogen bonding with the T26 main-chain NH, while T21 sits around the S1’ sub-pocket and hydrogen bonds with the T26 side-chain OH ((D)). The substitution of T21 with nonpolar isoleucine disables this hydrogen bonding and then may destabilize the hydrogen bond between S-217622 and T26, thus weakening its binding. G15 lies in the C’ position of an α-helix, which prefers glycine [Citation45,Citation46]. The glycine to serine substitution in this position will destabilize the α-helix [Citation47] and then may affect intramolecular packing, causing a shift of downstream β-sheets containing T21 and/or T26, ultimately influencing the stabilization hydrogen bonding between S-217622 and T26. The binding of GC376 or PF-07321332 to the active site, due to no occupancy or interaction in the S1’ sub-pocket ((C,E)), will be less affected by the G15S and T21I mutations.
Taken together, these results illustrate the utility of our assay for rapidly and convincingly comparing protease activity and drug susceptibilities of 3CLpro mutants and its advantage over the in vitro enzymatic assay or other single substrate-based assays.
Discussion
Here, we described a novel gain-of-signal assay for probing 3CLpro activity in living cells, which circumvents many of the limitations of previously described approaches. In this assay, the orthogonal combination of Gluc, which has high sensitivity and extremely low compound interference [Citation48], and the bright monomeric mKate2 with low cytotoxicity will greatly facilitate recognizing the signal interference from compounds in a single assay. The naturally secreted Gluc also provides a convenient assay without a requirement of cell lysis; thus, repeated, or real-time assays on the same cells will also be feasible. As a gain-of-signal assay, it can reliably identify authentic 3CLpro inhibitors and avoid interference from promiscuous compounds or nonspecific inhibitors. Its sensitivity is high enough to detect a decrease of less than 10% in activity caused by mutations (Figures 2(A) and 4(A)), as well as to identify inhibitors with only weak potency during HTS. Furthermore, given 3CLpro's great protein flexibility [Citation10,Citation22,Citation25], broad substrate profile [Citation33–37], and divergent catalytic activity on different substrates [Citation19,Citation44], our assay, which measures the overall effect of extensive protein cleavages by 3CLpro in living cells, may provide a more comprehensive profile of 3CLpro activity in physiologically relevant conditions than most current assays that measure single-substrate cleavage. 3CLpro-induced cytotoxicity or reporter expression suppression has been used to develop assays by others as well [Citation15,Citation22,Citation23]. However, the assay using crystal violet staining [Citation23] is complicated and laborious, not well compatible with HTS, and lacks the sensitivity to identify the weak inhibitor Z-FA-FMK. Previous assays based on flow cytometry [Citation15,Citation22] are also complex, less compatible with HTS, and difficult to recognize for possible signal interference from test compounds.
Our study on 3CLpro mutants have also illustrated the facileness and reliability of this system for the systematic comparison of 3CLpro mutants. First, a substantial decrease in activity of the P132H mutant was detected ((A)). 3CLpro plays an indispensable role in viral replication by processing replicase polyproteins. Besides this, through extensive cleavages of host proteins, it contributes to the subversion or hijacking of cellular machinery and the exploiting or countering host antiviral mechanisms for efficient viral replication [Citation33–37]. Its cleaving on critical modulators of inflammatory pathways has also been linked to the enhanced cytokine production and inflammatory response observed in COVID-19 patients [Citation35]. So, a decrease in 3CLpro activity should impair viral replication and ameliorate the clinical severity of COVID-19, just as shown by the loss of viral replication fitness in a recombinant SARS-CoV-2 clone harbouring a weakening 3CLpro mutation (S144A, 1.8- fold lower kcat/Km value [Citation4]) [Citation49] and the reduced severity observed among patients infected with a SARS-CoV-2 sub-lineage carrying the P108S mutation in 3CLpro [Citation42]. The finding of activity reduction in the P132H mutant suggests a possible contribution of this mutation to the attenuated replication and pathogenicity of Omicron [Citation50] and the comparatively milder clinical course of its infection [Citation51], which will need to be further studied. Second, susceptibility changes manifested as a decrease in the slope of dose response curves were identified in mutants with different single residue substitutions. Despite no significant decrease in IC50 for three tested inhibitors found in our comparison of seven 3CLpro mutants, a clear decrease was observed in the slope of dose response curves for PF-07321332 against the P132H mutant and S-217622 against the G15S and T21I mutants ((C,D)). The structural basis for this selective influence of mutations on different inhibitors have also been clarified (Results section and ). In general, resistance mutations shift dose–response curves to the right and increase the IC50; however, it has also been documented in HIV-1 that single mutations associated with resistance to protease inhibitors do not significantly increase IC50 but reduce slope [Citation41]. Our finding of the decrease in the slope of dose–response curves for inhibitors corresponding to point mutations involving binding interactions indicates that slope decrease should be another important dimension for susceptibility analysis on 3CLpro, especially for mutants with single residue substitution. Except for G15S, which led to a 4.4-fold reduction in PF-07321332 activity (fold-change based on Ki) reported in only one report [Citation43], previous in vitro enzymatic assays have not reported significant reduced susceptibilities (defined as > 2-fold change in IC50 or Ki) [Citation4,Citation24,Citation25,Citation42,Citation43] of mutants found in this study or the decline in activity of the P132H mutant. This and the variation in results from different reports [Citation4,Citation24,Citation43] also highlight the need for comparing results from single substrate-based enzymatic assays to those from assays using more different substrates or cell-based assays.
Previous in vitro enzymatic assays have confirmed that some single substitutions around inhibitor binding sites, such as Y54, F140, S144, M165, E166, H172, A173, V186, Q189, and Q192, result in resistance [Citation4,Citation43]. Studies on SARS-CoV-2 [Citation49,Citation52] or 3CLpro-dependent chimeric vesicular stomatitis virus [Citation53] mutations selected by 3CLpro inhibitors in vitro have revealed and demonstrated that single substitutions, such as Y54C, S144A, E166A/V, L167F/V, Q192R, P252L, and T304I, confer resistance to inhibitors. However, due to their deep participation in ligand binding, most of these mutations result in a significant reduction in proteolytic activity or viral fitness, although this may be partially restored with compensatory mutations such as T21I or L50F [Citation4,Citation49,Citation52,Citation53]. With our focus on the most widespread mutations, these mutations were not investigated here due to their rarity or low prevalence in circulating or previously circulated SARS-CoV-2 variants. Only the H172Y mutant was tested for method verification. Despite showing a high background signal and less sensitivity to detect lower inhibition levels due to the large activity reduction in this mutant, our assay well revealed its resistance phenotype to PF-07321332 (Figure S1).
Given that Omicron harbouring the P132H mutation in 3CLpro has become the dominant variant globally since late 2021, the finding of a decline in the PF-07321332 dose response curve slope for the P132H mutant will be important. Although no other studies have shown an association between this mutation and PF-07321332 resistance, the rebound infection after Paxlovid treatment has been reported [Citation54–57]. The slope of the dose response curve describes how inhibition changes with drug concentrations; its decrease indicates an inhibition decline at high concentrations, namely potency loss at high inhibition levels. Typically, clinical concentrations of drugs used are well above IC50; however, sometimes an effective clinical inhibition may need a higher concentration, maybe above IC90. The M184V mutation in HIV-1, which reduces the dose response slope but has a minimal effect on IC50, has been linked to unexpected clinical failure of various protease inhibitor regimens [Citation41]. It is also worth mentioning that other substitutions (P132L/S) on P132 was found in viral isolates from several Paxlovid-treated individuals in the EPIC-HR clinical trial [Citation43]. Thus, the possible association between viral rebound after Paxlovid treatment and the P132H mutation will be worth exploring. Additionally, the recent study on mutation development in SARS-CoV-2 showed that, under PF-07321332 selection, in vitro high-level resistance can occur in a multitude of ways [Citation49]. In some pathways, precursor mutations conferring low-level resistance, such as T21I, P252L, or T304I, will first occur in viruses under low concentrations of inhibitors. As drug pressure increases, additional mutations, such as E166V or S144A, are acquired for high-level resistance. The finding of a decrease in susceptibility to PF-07321332 or S-217622 in P132H or G15S and T21I mutants indicates the potential of these mutations as founder mutations for high-level resistance. The high distal location (>8 Å) of G15, T21 and P132 from substrate-binding sites also suggests that the emergence of mutations not located near the inhibitor binding site should also be of concern. Finally, the finding that the P132H mutation affects susceptibility to PF-07321332 but not to GC376 indicates that an inhibitor that can adopt multiple poses upon target binding is less likely to be affected by “active site” residue moving. This suggests a possible strategy for designing novel 3CLpro inhibitors with a higher barrier to resistance. As much resistance can be explained by a loss or weakening of the inhibitor's binding to the target, which is always caused by side chain clashes or changes in binding pocket shape. Such a strategy may be to design an inhibitor with flexible pharmacophores or link pharmacophores using a flexible moiety that provides conformational freedom. Theoretically, if the structural flexibility of an inhibitor allows it to be easily accommodated in the various modified binding pockets of a mutant target and has no adverse effects on the important inhibitor-target interactions, the conformational flexibility and positional adaptability will make it remain active but avoid the effects of side chain clashes or changes in binding pocket shape. The former idea has been well documented in the development of nonnucleoside reverse transcriptase inhibitors [Citation58]. As a precedent for the latter design principle, the development of nucleoside analogues termed “fleximers,” which contain two or more planar moieties in the heterocyclic base and are connected by a bond that permits rotation, has also achieved some success [Citation59,Citation60].
In conclusion, we present here an orthogonal dual reporter-based gain-of-signal assay for measuring the activity of 3CLpro in living cells. The assay is convenient, sensitive, and can reliably discriminate authentic 3CLpro inhibitors from promiscuous or nonspecific inhibitors. Signal-interfering compounds can also be readily recognized within a single assay. It is also useful for detecting changes in the activity and susceptibility of 3CLpro mutants. Considering its utility and versatility, as illustrated here, and superiority over most previously described methods, this assay will be conducive to advancing our more comprehensive understanding of 3CLpro and the development of new anti-coronavirus drugs.
Supplemental Material
Download MS Excel (64.5 KB)Supplemental Material
Download MS Excel (309.7 KB)Supplemental Material
Download MS Word (137.3 KB)Acknowledgments
We would like to thank Reviewers for taking the time and effort necessary to review the manuscript. We sincerely appreciate all valuable comments and suggestions, which helped us to improve the quality of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data to understand and assess the conclusions of this study are available in this published article. The raw data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Saxena SK, Kumar S, Ansari S, et al. Characterization of the novel SARS-CoV-2 Omicron (B.1.1.529) variant of concern and its global perspective. J Med Virol. 2022;94(4):1738–1744.
- Andeweg SP, de Gier B, Eggink D, et al. Protection of COVID-19 vaccination and previous infection against Omicron BA.1, BA.2 and Delta SARS-CoV-2 infections. Nat Commun. 2022;13(1):4738.
- Stevens LJ, Pruijssers AJ, Lee HW, et al. Mutations in the SARS-CoV-2 RNA-dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci Transl Med. 2022;14(656):eabo0718.
- Hu Y, Lewandowski EM, Tan H, et al. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. bioRxiv [Preprint]. 2022:2022.06.28.497978.
- Roe MK, Junod NA, Young AR, et al. Targeting novel structural and functional features of coronavirus protease nsp5 (3CLpro, Mpro) in the age of COVID-19. J Gen Virol. 2021;102(3):001558.
- Anand K, Ziebuhr J, Wadhwani P, et al. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–1767.
- Yang H, Xie W, Xue X, et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3(10):e324.
- Therapeutics and COVID-19: living guideline, 13 January 2023. Geneva: World Health Organization; 2023 (WHO/2019-nCoV/therapeutics/2023.1).
- Owen DR, Allerton CMN, Anderson AS, et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374(6575):1586–1593.
- Ma C, Sacco MD, Hurst B, et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678–692.
- Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–293.
- Drayman N, DeMarco JK, Jones KA, et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science. 2021;373(6557):931–936.
- Unoh Y, Uehara S, Nakahara K, et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J Med Chem. 2022;65(9):6499–6512.
- Günther S, Reinke PYA, Fernández-García Y, et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science. 2021;372(6542):642–646.
- Cao W, Cho CD, Geng ZZ, et al. Evaluation of SARS-CoV-2 main protease inhibitors using a novel cell-based assay. ACS Cent Sci. 2022;8(2):192–204.
- Ma C, Tan H, Choza J, et al. Validation and invalidation of SARS-CoV-2 main protease inhibitors using the Flip-GFP and Protease-Glo luciferase assays. Acta Pharm Sin B. 2022;12(4):1636–1651.
- Froggatt HM, Heaton BE, Heaton NS. Development of a fluorescence-based, high-throughput SARS-CoV-2 3CLpro reporter assay. J Virol. 2020;94(22):e01265–20.
- O’Brien A, Chen DY, Hackbart M, et al. Detecting SARS-CoV-2 3CLpro expression and activity using a polyclonal antiserum and a luciferase-based biosensor. Virology. 2021;556:73–78.
- Hou N, Peng C, Zhang L, et al. BRET-based self-cleaving biosensors for SARS-CoV-2 3CLpro inhibitor discovery. Microbiol Spectr. 2022;10(4):e0255921.
- Rawson JMO, Duchon A, Nikolaitchik OA, et al. Development of a cell-based luciferase complementation assay for identification of SARS-CoV-2 3CLpro inhibitors. Viruses. 2021;13(2):173.
- Dey-Rao R, Smith GR, Timilsina U, et al. A fluorescence-based, gain-of-signal, live cell system to evaluate SARS-CoV-2 main protease inhibition. Antiviral Res. 2021;195:105183.
- Moghadasi SA, Esler MA, Otsuka Y, et al. Gain-of-signal assays for probing inhibition of SARS-CoV-2 Mpro/3CLpro in living cells. mBio. 2022;13(3):e0078422.
- Resnick SJ, Iketani S, Hong SJ, et al. Inhibitors of coronavirus 3CL proteases protect cells from protease-mediated cytotoxicity. J Virol. 2021;95(14):e0237420.
- Ullrich S, Ekanayake KB, Otting G, et al. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg Med Chem Lett. 2022;62:128629.
- Sacco MD, Hu Y, Gongora MV, et al. The P132H mutation in the main protease of Omicron SARS-CoV-2 decreases thermal stability without compromising catalysis or small-molecule drug inhibition. Cell Res. 2022;32(5):498–500.
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73.
- R Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2022. Available from: https://www.R-project.org/
- Ritz C, Baty F, Streibig JC, et al. Dose-response analysis using R. PLoS One. 2015;10(12):e0146021.
- Zhao Y, Fang C, Zhang Q, et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell. 2022;13(9):689–693.
- Lin CW, Lin KH, Hsieh TH, et al. Severe acute respiratory syndrome coronavirus 3C-like protease-induced apoptosis. FEMS Immunol Med Microbiol. 2006;46(3):375–380.
- Yao K, Wang K, Xu W, et al. Caspase-3 and its inhibitor Ac-DEVD-CHO in rat lens epithelial cell apoptosis induced by hydrogen in vitro. Chin Med J (Engl). 2003;116(7):1034–1038.
- Teng JF, Mei QB, Zhou XG, et al. Polyphyllin VI induces caspase-1-mediated pyroptosis via the induction of ROS/NF-κB/NLRP3/GSDMD signal axis in non-small cell lung cancer. Cancers (Basel). 2020;12(1):193.
- Zhang S, Wang J, Cheng G. Protease cleavage of RNF 20 facilitates coronavirus replication via stabilizaiton of SREBP1. Proc Natl Acad Sci U S A. 2021;118(37):e2107108118.
- Meyer B, Chiaravalli J, Gellenoncourt S, et al. Characterising proteolysis during SARS-CoV-2 infection identifies viral cleavage sites and cellular targets with therapeutic potential. Nat Commun. 2021;12(1):5553.
- Moustaqil M, Ollivier E, Chiu HP, et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerg Microbes Infect. 2021;10(1):178–195.
- Pablos I, Machado Y, de Jesus HCR, et al. Mechanistic insights into COVID-19 by global analysis of the SARS-CoV-2 3CLpro substrate degradome. Cell Rep. 2021;37(4):109892.
- Koudelka T, Boger J, Henkel A, et al. N-Terminomics for the identification of in vitro substrates and cleavage site specificity of the SARS-CoV-2 main protease. Proteomics. 2021;21(2):e2000246.
- Weglarz-Tomczak E, Tomczak JM, Talma M, et al. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci Rep. 2021;11(1):3640.
- Strobelt R, Adler J, Paran N, et al. Imatinib inhibits SARS-CoV-2 infection by an off-target-mechanism. Sci Rep. 2022;12(1):5758.
- Gangavarapu K, Latif AA, Mullen JL, et al. Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nat Methods. 2023. doi:10.1038/s41592-023-01769-3
- Sampah ME, Shen L, Jilek BL, et al. Dose-response curve slope is a missing dimension in the analysis of HIV-1 drug resistance. Proc Natl Acad Sci U S A. 2011;108(18):7613–7618.
- Abe K, Kabe Y, Uchiyama S, et al. Pro108Ser mutation of SARS-CoV-2 3CLpro reduces the enzyme activity and ameliorates the clinical severity of COVID-19. Sci Rep. 2022;12(1):1299.
- FDA, Fact sheet for healthcare providers: emergency use authorization for Paxlovid (2023); Available from: https://www.fda.gov/media/155050/download
- MacDonald EA, Frey G, Namchuk MN, et al. Recognition of divergent viral substrates by the SARS-CoV-2 main protease. ACS Infect Dis. 2021;7(9):2591–2595.
- Aurora R, Srinivasan R, Rose GD. Rules for alpha-helix termination by glycine. Science. 1994;264(5162):1126–1130.
- Aurora R, Rose GD. Helix capping. Protein Sci. 1998;7(1):21–38.
- Thomas ST, Loladze VV, Makhatadze GI. Hydration of the peptide backbone largely defines the thermodynamic propensity scale of residues at the C’ position of the C-capping box of alpha-helices. Proc Natl Acad Sci U S A. 2001;98(19):10670–10675.
- Ho PI, Yue K, Pandey P, et al. Reporter enzyme inhibitor study to aid assembly of orthogonal reporter gene assays. ACS Chem Biol. 2013;8(5):1009–1017.
- Iketani S, Mohri H, Culbertson B, et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature. 2023;613(7944):558–564.
- Shuai H, Chan JF, Hu B, et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature. 2022;603(7902):693–699.
- Wolter N, Jassat W, Walaza S, et al. Early assessment of the clinical severity of the SARS-CoV-2 Omicron variant in South Africa: a data linkage study. Lancet. 2022;399(10323):437–446.
- Jochmans D, Liu C, Donckers K, et al. The substitutions L50F, E166A, and L167F in SARS-CoV-2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir. mBio. 2023;14(1):e0281522.
- Heilmann E, Costacurta F, Moghadasi SA, et al. SARS-CoV-2 3CLpro mutations selected in a VSV-based system confer resistance to nirmatrelvir, ensitrelvir, and GC376. Sci Transl Med. 2023;15(678):eabq7360.
- Charness ME, Gupta K, Stack G, et al. Rebound of SARS-CoV-2 infection after nirmatrelvir-ritonavir treatment. N Engl J Med. 2022;387(11):1045–1047.
- Boucau J, Uddin R, Marino C, et al. Characterization of virologic rebound following nirmatrelvir-ritonavir treatment for coronavirus disease 2019 (COVID-19). Clin Infect Dis. 2023;76(3):e526–e529.
- Wang L, Volkow ND, Davis PB, et al. COVID-19 rebound after Paxlovid treatment during Omicron BA.5 vs BA.2.12.1 subvariant predominance period. medRxiv [Preprint]. 2022:2022.08.04.22278450.
- Antonelli G, Focosi D, Turriziani O, et al. Virological and clinical rebounds of COVID-19 soon after nirmatrelvir/ritonavir discontinuation. Clin Microbiol Infect. 2022;28(12):1657–1658.
- Zhan P, Liu X, Li Z, et al. Design strategies of novel NNRTIs to overcome drug resistance. Curr Med Chem. 2009;16(29):3903–3917.
- Seley KL, Zhang L, Hagos A. Fleximers.” Design and synthesis of two novel split nucleosides. Org Lett. 2001;3(20):3209–3210.
- Yates MK, Seley-Radtke KL. The evolution of antiviral nucleoside analogues: a review for chemists and non-chemists. Part II: complex modifications to the nucleoside scaffold. Antiviral Res. 2019;162:5–21.