
ABSTRACT
Characterized by high genetic diversity, broad host range, and resistance to adverse conditions, coupled with recent reports of neurotropic astroviruses circulating in humans, mamastroviruses pose a threat to public health. The current astrovirus classification system based on host source prevents determining whether strains with distinct tropism or virulence are emerging. By using integrated phylogeny, we propose a standardized demarcation of species and genotypes, with reproducible cut-off values that reconcile the pairwise sequence distribution, genetic distances between lineages, and the topological reconstruction of the Mamastrovirus genus. We further define the various links established by co-evolution and resolve the dynamics of transmission chains to identify host-jump events and the sources from which different mamastrovirus species circulating in humans have emerged. We observed that recombination is relatively infrequent and restricted to within genotypes. The well-known “human” astrovirus, defined here as mamastrovirus species 7, has co-speciated with humans, while there have been two additional host-jumps into humans from distinct hosts. Newly defined species 6 genotype 2, linked to severe gastroenteritis in children, resulted from a marmot to human jump taking place ∼200 years ago while species 6 genotype 7 (MastV-Sp6Gt7), linked to neurological disease in immunocompromised patients, jumped from bovines only ∼50 years ago. Through demographic reconstruction, we determined that the latter reached coalescent viral population growth only 20 years ago and is evolving at a much higher evolutionary rate than other genotypes infecting humans. This study constitutes mounting evidence of MastV-Sp6Gt7 active circulation and highlights the need for diagnostics capable of detecting it.
Introduction
Since their discovery in 1975 [Citation1], astroviruses have been identified in a myriad of hosts, including domestic and wild animals [Citation2], avian and mammalian species [Citation3], and in terrestrial and aquatic environments [Citation4]. The Astroviridae family includes two genera, Avastrovirus (infecting birds) and Mamastrovirus (infecting mammals)[Citation5]. Based on codon usage analysis, it has been proposed that the split between these two genera took place ∼310 million years ago [Citation6]. However, considering their differences in evolutionary rates [Citation7,Citation8] and each species’ conformance to distinct molecular clocks, pinpointing the date of this evolutionary split should be revisited using state-of-the-art methodologies, such as Bayesian analysis.
Despite extensive amounts of genomic data available for the astrovirus family, classification at the species level is unfortunately still based on the host source of identification of the viral strain [Citation9]. This hampers tracking of outbreaks and defining the emergence of novel strains, and thus in 2011 the International Committee on the Taxonomy of Viruses (ICTV) highlighted the need to adapt a more reliable classification method [Citation9]. Recent efforts to classify astrovirus species using amino acid genetic distances have proposed cut-off values of ∼0.671 in Mamastrovirus and ∼0.704 in Avastrovirus [Citation5]. However, these values are arbitrary and will lead to misclassifications since neither the topological distribution of the lineages nor with the pairwise sequence comparison distribution (PASC) of each genus have been reconciled.
Clinically, Mamastrovirus has been primarily associated with self-limiting gastroenteritis in humans, especially in infants, young children, and the immunocompromised [Citation10]. Astroviruses appear to be ubiquitous in other mammals, typically presenting with a subclinical course in older animals, but with clinical outcomes similar to that in humans in younger animals [Citation3]. Recent studies have revealed the implications of Mamastrovirus infections for nervous system disease in three species, including mink [Citation11,Citation12], cattle [Citation13,Citation14] and humans [Citation15–19]. These novel strains have been grouped into different clades classified as VA1/HMO-C, HMO and MLB [Citation20], yet it remains uncertain whether they represent different mamastrovirus species or genotypes possessing altered tropism. Similarly, it is unclear if these clades have epidemiological links or if they represent zoonotic events, recent emergent strains, or well-established, circulating viruses.
In the current study, we identified another novel genome with high identity to the aforementioned human astroviruses linked to neurological disorders. To provide clarity regarding the emergence and diversification of the Mamastrovirus genus, we herein propose a novel classification scheme combining PASC analysis with genetic distances (p-distance) to integrate phylogenetic distributions. Additionally, using cophylogenetic approaches, we have revealed the evolutionary links between individual mamastrovirus species and their respective hosts. Finally, by applying time-stamped Bayesian phylogenies, we uncovered the temporal dynamics of mamastrovirus strains circulating in the human population.
Results
Reconciliation of phylogenetic inferences, genetic distances and PAirwise sequence comparison defines seven distinctive species within the mamastrovirus genus
We assembled a complete astrovirus genome (AbE11938; GenBank accession OP594725) from the plasma of a febrile individual using high-throughput sequencing coupled to virus target enrichment. BLAST homology determination and preliminary phylogenetic analysis, based on Maximum Likelihood reconstruction, indicated the sequence was related to the VA1/UK clade, which contains a cluster of patients that were either immunocompromised, had neurologic disorders, or severe gastrointestinal (GI) disease, such as coeliac. AbE11938 shares amino acid identity of 99% for capsid and 97% for RNA-dependent RNA-polymerase (RdRp), with its closest reference, CMRHP6 (MH933754.1)[Citation21]. Since astroviruses typically cause mild GI symptoms, we sought to establish phylogenetic links to clinical outcome through a better understanding of the distribution of the Mamastrovirus genus.
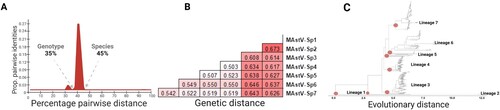
Classification of the Mamastrovirus genus into species has previously been based upon the host species of isolation, without reconciling the genetic distances with topological arrangements. A maximum-likelihood tree was constructed using all 512 unique full-length Mamastrovirus genomes available in GenBank. Eight major lineages were identified visually (Supplementary material Figure S1), however PASC analysis and the calculated pairwise distances were used to determine the appropriate taxonomic classifications. Two clear distributions were observed for all sequences, with 45% denoting the species division and 35% denoting the genotype division (A). For example, within a species sequences are at least 55% identical and within a genotype, sequences are at least 65% identical. Genetic distances were then calculated, which range from 50%−67% (e.g. > 45%) differences between each species (B). These distances agreed with cut-offs established from the PASC analysis and tree topology, resulting in only seven species (C and Supplementary material Figure S1 and Figure S2) defined in the Mamastrovirus genus (C, red dots).
Figure 1. Topological and genetic reconciliation of Mamastrovirus genus. (A) Representation of PAirwise Sequence Comparison (PASC) results obtained from the frequency distribution of pairwise distances for all 521 sequences using the SDT analysis (Supplementary material Figure S2). Cut-off values for each genetic division are indicated, denoting groupings of the same genotype level at 35% and different species values higher than 45%. (B) Genetic distances for seven main lineages obtained when grouping the strains into each redefined designation, now as distinctive species within the Mamastrovirus genus. (C) Maximum likelihood tree based on the whole genome of all 521 non-redundant genomes available at GenBank, the best-fitted model to infer the phylogenetic relationship used was the SYM + R10; the red dots indicate the seven main lineages with a genetic distance higher than 45% (Supplementary material Figure S1).
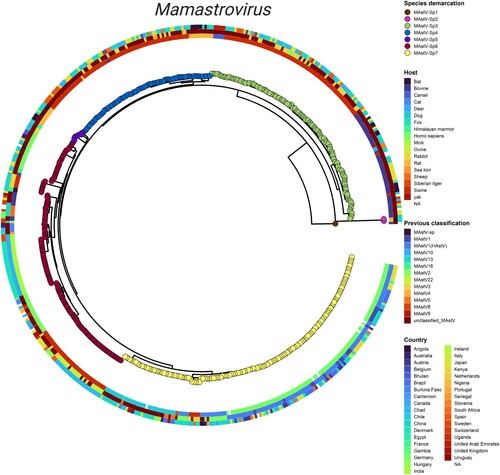
To reconcile tree topology with genetic distance, the previous classifications and additional aspects including the host and geographic distributions of the Mamastrovirus genus were incorporated into an integrated phylogeny. This representation allowed a comparison of the seven newly proposed species (central tree) with a strain’s documented host (inner circle), its previous classification (middle circle), and its country of origin (outer circle) (). Immediately evident was the lack of agreement between our species demarcations and the original classifications based on host. For example, unclassified-MastV strains are interspersed among all sequences and while groupings are apparent, there is no clear delineation from one to the next. Compared to the previous classification scheme, sequences no longer segregate by host (inner) and certainly not by country (outer). However, with the newly proposed groupings according to species (circle tips), strains now follow a pattern that correlates with a particular host(s). For example, most MastV-Sp3 (green) strains are either swine (red) or bovine (navy blue), just as all MastV-Sp7 (yellow) are primarily human, cat, dog or swine. Therefore, the Mamastrovirus genus would benefit from a reclassification into species along PASC and genetic distance lines, as it is evident that there is not a host restriction at the species level ().
Figure 2. Integrated phylogeny for the Mamastrovirus genus. Visualization of the summarized phylogenetic tree of Mamastrovirus based on whole genomes and reconciled by the PASC distribution and genetic distances. Demarcation of the species proposed in the current study (tips), host of isolation (inner ring), previous classification of species (second ring), and geographic distribution (outer ringer) are all indicated in the phylogeny. Integration of the panels was performed by using the ggtreeExtra R package.
Genotypic diversification within mamastrovirus species is driven by host-jump events
We once again turned to integrated phylogeny to identify the forces driving genetic diversity at the species level. Applying the PASC cut-offs and distance between monophyletic clades, we assigned genotypes within each species. Due to the low number of strains in GenBank, species 1, 2, and 5 were excluded from further analysis. From the genetic distances obtained (p-distance matrices, A–D) MAstV-Sp3 contains six genotypes, MAstV-Sp4 contains three genotypes, MAstV-Sp6 contains eight genotypes, and MAstV-Sp7 contains three genotypes (A – D, left). Within each species, the topological distribution and genotypic demarcations revealed the close relationship between genotype/host and rejected an association with geographic distribution (integrated phylogeny A – D). As suggested from , these trees clearly support that the expansion of genotypes within species is according to host. Therefore, we hypothesized that host-jump events are a major force favouring this diversification process. To first determine which genotypes and hosts are linked through a co-evolutionary process and uncover the dynamics of transmission chains, we used the Procrustean Approach to Co-phylogeny (PACo)[Citation22].
Figure 3. Genotypic demarcation of mamastrovirus species in relationship to their host and evolutionary reconstruction. (A-D) (left panels) Phylogenetic trees based on the whole genomes of MAstV-Sp3, MAstV-Sp4, MAstV-Sp6, and MAstV-Sp7 species, reconciled by the PASC distribution and the genetic distances (coloured table merged into the phylogenetic tree). Different genotypes (grey shading in each defined cluster and tips of the tree) for each species, host of isolation (inner ring) and geographic distribution (outer ringer) are all indicated in the phylogeny. (A-D) (right panels) Contributions of each host-virus (at the genotype level) link to the procrustean fit (centre)-jackknifed squared residuals (bars) and the upper 95% confidence intervals (error bars) resulting from applying PACo to patristic distances. Links supported among the mamastrovirus genotypes and their respective hosts are indicated by an asterisk (*). The MSQR values obtained for each viral species is represented by a red-dashed line. Resolution of the mamastrovirus phylogeny with their hosts is based on the methodology implemented in JANE. All possible codivergence, extinction, host-jumping, and lineage duplication events are described in the JANE Manual (see Supplementary Material Figures S3-S6 for clarification). A summary of the most relevant events linked to host-switch are denoted with grey dashed arrows. Host-jump events into the human population are denoted as zoonotic events. Co-speciation between MAstV-Sp7G3 (which included all the previous classified human astroviruses) and the human population is also denoted. For host species in which a genotype-host link was supported by the procrustean fit, those acting as the donor host during the jump events are located at the beginning of the arrows.
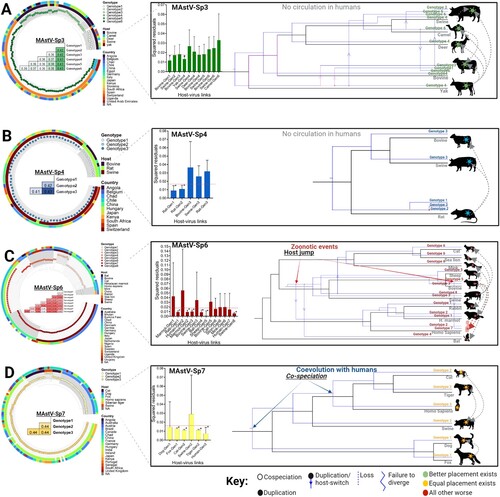
Unlike classical coevolutionary models, PACo allows for multiple host-parasite associations and directly tests the dependence of the parasite (viral genotype) phylogeny upon the host phylogeny. Principal component algorithms within PACo highlight the groupings of hosts superimposed with their associated viral genotypes, with coordinates and arrow lengths corresponding to patristic distances (Supplemental Figure S3A-S6A, upper panels). This assessment of the congruence between phylogenetic trees (virus full genomes versus host cytochrome c), or between distance matrices of taxa, is enumerated in a residual sum of squares (m2 XY) statistic reflecting patristic distances (A – D, centre histograms). By randomizing each host-virus interaction, the software models the “goodness of fit” to calculate the computationally least expensive and highest probability solution. Cutoffs (dashed red line in barplots) established independently for each species of ∼0.018 for MAstV-Sp3 and MAstV-Sp4, and ∼0.015 for MAstV-Sp6 and MAstV-Sp7, in all cases with an associated permutational p < 0.05, supported the overall congruence of viral genotype and host trees. We uncovered 14 links between Mamastrovirus and their documented host which contributed relatively little to m2 XY values (A – D asterisks). This suggests that this viral genotype and its host are in equilibrium and therefore these highly significant relationships likely represent co-evolutionary associations.
For MAstV-Sp3, genotypes 1 and 4 appear to have evolved with their bovine and yak hosts, respectively (A). Likewise, for species MAstV-Sp4, genotypes 1 and 2 are linked with rats (B). Within species MAstV-Sp6, several genotypes show a coevolutionary pattern: genotype 1 with rabbit, genotype 2 with the marmot, genotype 3 with bovine, genotype 4 with bat, genotype 5 with the sheep, and genotype 8 with swine (C). Interestingly, for the two genotypes circulating in human from this species (genotype 2 and genotype 7), the coevolutionary hypothesis with a human host was rejected, indicating these genotypes in human are both the result of a jump from another mammal (C). Unexpectedly for species MAstV-Sp7, all three genotypes have co-evolved with their respective host (or host range), rather than being incidental hosts: genotype 1 is linked with fox (canine coevolution), genotype 2 with tigers and cats (a feline coevolution), and genotype 3 with Homo sapiens, indicating MAstV-Sp7GT3 has already adapted to humans (D). As for the remaining links (without asterisks, p > 0.05, and with large error bars) where a particular genotype has been found in a given host but the coevolutionary hypothesis was rejected, these likely represent host-jumping events (A – D).
To understand the dynamics of transmission within mamastrovirus species, for example, establishing who is the donor and who is this receptor following a host-switch event, which strains fail to adapt after a host split (e.g. “failure to diverge”), and which die off, reconciliation analyses of phylogenies among the different viral genotypes (A – D right panels-blue topologies) and their respective hosts (A – D right panels-black topologies) were conducted using the JANE software package [Citation23]. As with PACo, each of these interactions is assigned a cost and all potential reconciliations, taking into account dynamic programming algorithms that find optimal solutions in polynomial time, are randomly evaluated and scored to arrive at the solution with the minimal score value (SV; see Supplementary Information Fig.S3B-S6B-lower panels). Reconciliation of the MAstV-Sp3 tree with the host tree revealed a maximum of six host switches following duplication events (dotted arrows). This highlights the fact that most of the diversification within Species 3 resulted from host-switch events from genotype 4 that gave rise to genotypes 2, 3, and 5, the latter of which diversified into genotype 6 (A). The maintenance of genotype 4 across four different host species (yak, domestic bovine, deer, and pigs) without additional diversification indicates there were four failures to diverge. Finally, there was one “loss” event for genotype 4 in swine (A), for an overall SV of 16 for Species 3. The combination of patristic distances and the reconciliation analysis for this mamastrovirus species indicated that bovine hosts (Yak and domestic bovine) acted as the donors. The remaining hosts act as receptors, for which swine principally appears to play the role of mixing-vessel (A). Only two duplication events and two failures to diverge were obtained from the MAstV-Sp4 tree (B), with a cost of SV = 4. Despite the lack of clear evidence for a host switch within the topological arrangement, we infer from the PACo analysis that a host jump occurred from rat to cows and pigs, given the lack of coevolutionary links to bovines and swine and the fact that only genotype 3 is found circulating in these hosts following the duplication event in rats (B).
Several events were suggested for MAstV-Sp6, including a co-speciation, four host-switches after duplications, five failures to diverge and two losses (C), with a total cost of SV = 18. Genotypes 2 and 7 circulating in human were the result of host-switch events after duplications. Genotype 2 appears to have emerged as a consequence of a host-switch event that further diversified in human and marmots, the latter of which also encountered an additional duplication event that failed to diversify in rabbits (e.g. loss). Since genotype 2 showed a coevolutionary link with the marmot host by patristic distance analysis, it is likely that a zoonosis caused the jump from the marmots to humans. For genotype 7 (MAstV-Sp6G7) we observed evidence of a host-switch event caused by the duplication of genotype 5 in its bovine host. Thus, bovines were the source of this zoonotic introduction into the human population.
Finally, for MAstV-Sp7, this was the only species in which two co-speciation events were identified: one branching with canines (genotype 1: dogs and fox) and a second divided between felines (genotype 2: cat and tiger) and humans (genotype 3) (D). In addition, three failures to diverge were also observed (genotype 1: dog with fox; genotype 2: cat with tiger; genotype 2: pigs with felines), all with a total cost of reconciliation of SV = 3. These results agree with patristic distances from PACo wherein each genotype was co-evolutionarily linked to their respective host (D). The only host species that did not reflect a coevolutionary relationship with any of the genotypes of MAstV-Sp7 was swine, which appears to have acquired genotype 2 from felines (D).
Intra-genomic recombination events identified within different mamastrovirus species suggests a Dobzhansky-Muller model of incompatibilities restricting inter-genotypic events.
To uncover additional forces driving the genetic diversity within the Mamastrovirus genus, and to prevent the potential bias that recombination events could introduce into a time-stamped analysis, a comprehensive analysis of recombination events among different mamastrovirus species was performed. Using the Recombination Detection Program (RDP) software package, we identified a total of 19 statistically significant recombinant sequences (p < 0.0001) with varying breakpoint positions and representation across species (Supplementary material Table S1). The distribution frequency of recombinant sequences was low among the species analysed, with five strains identified in MAstV-Sp3 (3.9%), two in MAstV-Sp4 (2.4%), four in MAstV-Sp6 (2.9%), and eight recombinant strains found in MAstV-Sp7 (4.9%) ( and Supplementary information Figure S7 and Table S1).
Figure 4. Mamastrovirus recombination is restricted to within intra-genotypic boundaries. Panels display recombination events detected by RDP5v5 software in (A) MAstV-Sp3, (B) MAstV-Sp4, (C) MAstV-Sp6, and (D) MAstV-Sp7. Events were supported by at least three detection methods and a statistical significance of p < 0.01 after Bonferroni correction (see Supplemental information Table S3), but for simplicity, only Bootscan analysis results are shown where breakpoints had a clear signal and bootstrap values of 75% or higher were obtained (left). The major-parent, minor-parent, and recombinant sequences are mapped onto the phylogenetic tree for each species (right). Major-minor parent interactions are denoted in green, major parent-recombinant strain interactions in red, and minor parent-recombinant strain interactions in blue (left panels).
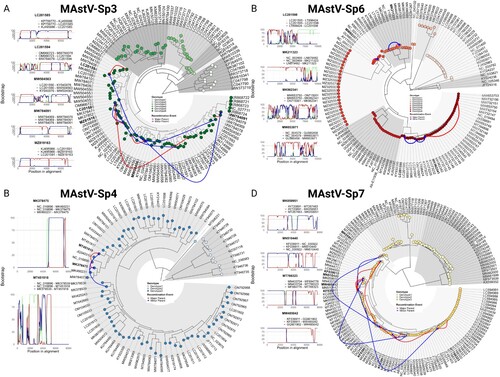
We next explored the relationships between the recombinant sequences and their respective parental strains. Species-specific phylogenetic trees were used to identify major-parent, minor-parent, and recombinant sequences. Notably, all major and minor parents of the recombinant strains were located within the same genotype for each species. For example, in MAstV-Sp3, all the recombinant strains and their respective parentals were grouped within genotype 6 (A), all of which were isolated from a swine host (A, and Supplementary material Table S1). Similarly, both recombinant strains and their respective parentals within MAstV-Sp4 were identified in genotype 3 (B), once again from a swine host (B and Supplementary material TableS1). In MAstV-Sp6 (C), one genotype 5 recombinant strain (Accession: MK211323) was isolated from a bovine host and its parental strains (Major: NC_002469) were also from a sheep and bovine host (Minor: LN879482). Another genotype 6 recombinant strain (Accession: MW853971) isolated from a Sea lion host had both parental strains isolated from a mink host. Finally, two genotype 8 recombinant strains (Accession: MK962341 and LC201598) had all the parental and the recombinant strains isolated from a swine host (also see and Supplementary information Table S1). In the case of MAstV-Sp7, all of the recombinant strains and their parentals from humans were identified in genotype 3 (D), with 4/8 strains previously identified by Babkin et al.[Citation24] (Supplementary material Figure S7) and four newly identified in the current study ( and also see and Supplementary material Table S1). It is important to note that none of the recombinant strains identified here were located within the zoonotic genotypes (G2 and G7) of MAstV-Sp6. Indeed, the fact that all recombinant strains and their respective parentals were restricted to the same genotype for each particular species () suggests that recombination events in Mamastrovirus genus follows a Dobzhansky-Muller model of incompatibilities, wherein once strains have differentiated to a certain degree they are no longer able to share genetic material [Citation25,Citation26].
Mamastrovirus infections in humans were driven by co-speciation and zoonosis events.
We next deployed time-stamped phylogeny to gauge whether the host jump events observed in MAstV-Sp6 were in the distant past or represent recent, emergent viruses of concern. This time scale was then compared to MAstV-Sp7 which we show has co-speciated with humans. The initial evaluation for the temporal signal revealed that for both species a heterochronous model was favoured (), meaning they conform to molecular clocks and enable dating. Thus, by using BEAST to calibrate time to the most recent common ancestor (tMRCA) and estimate the evolutionary rates, we observe that MAstV-Sp6G2 may have emerged nearly 200 years ago in 1826 (from a marmot host-jump) and has an evolutionary rate of 3.2 × 10−4 sites/year (A-left). Interestingly, MAstV-Sp6G7, to which our new strain AbE11938 belongs, emerged very recently in 1982 (from a bovine host-jump) and has a mutation rate of 4.3 × 10−2 sites/year, more than two logs higher (A-left). This indicates MAstV-Sp6G7 members are rapidly evolving and quickly adapting to their human host. In comparison, the “human astrovirus,” MAstV-Sp7G3, was predicted to have emerged more than two thousand years ago in approximately 73 B.C. with a 8.8 × 10−5 sites/year mutation rate, nearly a thousand times lower than MAstV-Sp6G7 (A-right). This low value mirrors that of other RNA viruses known to be in equilibrium with their host and agrees with our co-evolutionary analysis (D).
Figure 5. Evolutionary history of mamastroviruses infecting humans. (A) Time-calibrated maximum clade credibility (MCC) trees for both mamastrovirus species identified as circulating in the human population (left: MAstV-Sp6; right: MAstV-Sp7). Time-resolved phylogenies show the time for the most recent common ancestor (tMRCA) and the evolutionary rates for the genotypes circulating in humans. Host and clinical manifestations observed in the genotypes of interest are denoted. For MAstV-Sp7G3 the groups previously defined by Zhou et al. [Citation8] are denoted, the most recent demographic expansion of Group I is indicated by a red arrow. (B) (left panel) Demographic history of three human mamastrovirus genotypes inferred via Bayesian skyline plot (BSP) with coalescent tree prior and an exponential, uncorrelated clock model. The shading represents the 95% highest posterior density (HPD) of the product of generation time (τ) and effective population size (Ne). The line tracks the inferred median of Neτ. (right panel) Zoomed-in ML-phylogeny for the MAstV-Sp6 genotype 7 which includes the previously classified VA1/UK clade. Colour codings are embedded into the phylogeny to indicate tropism (inner), country of isolation (middle) and host (outer) where known.
![Figure 5. Evolutionary history of mamastroviruses infecting humans. (A) Time-calibrated maximum clade credibility (MCC) trees for both mamastrovirus species identified as circulating in the human population (left: MAstV-Sp6; right: MAstV-Sp7). Time-resolved phylogenies show the time for the most recent common ancestor (tMRCA) and the evolutionary rates for the genotypes circulating in humans. Host and clinical manifestations observed in the genotypes of interest are denoted. For MAstV-Sp7G3 the groups previously defined by Zhou et al. [Citation8] are denoted, the most recent demographic expansion of Group I is indicated by a red arrow. (B) (left panel) Demographic history of three human mamastrovirus genotypes inferred via Bayesian skyline plot (BSP) with coalescent tree prior and an exponential, uncorrelated clock model. The shading represents the 95% highest posterior density (HPD) of the product of generation time (τ) and effective population size (Ne). The line tracks the inferred median of Neτ. (right panel) Zoomed-in ML-phylogeny for the MAstV-Sp6 genotype 7 which includes the previously classified VA1/UK clade. Colour codings are embedded into the phylogeny to indicate tropism (inner), country of isolation (middle) and host (outer) where known.](/cms/asset/854bd05c-2dd6-4c10-a47f-0a78edc24bd0/temi_a_2217942_f0005_oc.jpg)
Table 1. BETS analysis comparing the fit to the data of two models (heterochronous) the constrained model (isochronous).
Bayesian skyline plots were performed to assess the time-dependent growth of the viral population size (τ) given by effective number of infections (Ne) over time for a given genotype tree of interest (B-left). Having been in circulation for more than 2 millennia, MAstV-Sp7G3 (yellow) expectedly exhibits a maintenance of its genetic diversity, visualized by the plateau in the estimated growth of the viral population over time. This stable trend changed during 1950 to 2000 when an increase in genetic diversity of this lineage was observed, presumably linked to the diversification of a new sub-lineage in group 1 (A left panel, red arrow). During the last 10 years this genotype has experienced a decrease in genetic diversity to reach the initial levels of maintenance (B, yellow) indicative of virus-host equilibrium. MAstV-Sp6G2 (orange) has also maintained an elevated level of genetic diversity for at least 100 years following its emergence, (B) however, it has experienced a drop in the last 25 years. This lineage has been sampled in two different species including humans and marmots, so this behaviour could reflect this host-jump and be an indication of lower levels of adaptation in humans. Alternatively, this decline in the genetic diversity during this period of time could also be the effect of low sampling. Compared to the other two human astrovirus lineages, MAstV-Sp6G7 (B-firebrick) has the lowest genetic diversity overall, experiencing a sharp decrease after its emergence which is emblematic of a bottleneck effect (B). This characteristic could be the consequence of its recent host-jump from the presumed donor species (bovine), for which one would expect a lower level of adaptation. While diversification appears to be on the upswing in recent years (B right), limited sampling could once again mask the true behaviour of this lineage. Nevertheless, MAstV-Sp6G7 appears to have quickly spread across the globe in the last 20 years, and it is notable that most of these strains cause either severe gastrointestinal or neurotropic disease in humans (B-right).
Discussion
The high genetic diversity present in the Mamastrovirus genus [Citation27] and their capacity to infect a broad range of hosts [Citation9] hinders the definition of outbreaks, the tracing of emerging strains, the identification of zoonoses and epidemiological links to this viral genus. In order to timely and accurately respond to these events, a reliable identification and classification scheme for viral agents causing or linked to disease is required. In response, we have proposed a standardized taxonomic organization for the demarcation of species ( and ) and genotypes (). By integrating the phylogenetic structure of the entire genus with pairwise sequence distributions and genetic distances (), we established cut-off values that can be easily adopted by the specialist in the field. This should enable rapid classification of astrovirus strains and facilitate the detection of epidemiological relationships. This will bring clarity to evolutionary trends in circulating strains (cryptic circulation or reintroduction into the population) or signal the emergence of new ones in the Mamastrovirus genus.
The emergence and rapid spread of SARS-CoV-2 [Citation28] has highlighted the challenges facing public health from emerging infectious diseases. Zoonotic viruses originating from reservoir species (frequently mammals) represent a particular concern, as the jump to humans can result in a change of tropism and severity that cause disease syndromes [Citation29]. In the current study, we identified two zoonotic events resulting from independent introductions into the human population by different animal sources: MAstV-Sp6G2, which has been circulating in marmots and MAstV-Sp6G7, which emerged from the diversification of MAstV-Sp6G5 circulating in ruminants (bovine and sheep) (). A notable takeaway from our JANE analysis is the evidence for a host jump from MAstV-Sp6G5, present in and causing neurologic disease in minks [Citation11,Citation12] and cows [Citation13], to the resulting MAstV-Sp6G7 in humans presenting with similar symptoms. Typically, viral species require both hosts to be in close proximity to one another to promote a jumping event, yet the prolonged stability of astroviruses in different environments (aquatic and terrestrial) provides a route to new hosts without them being physically in contact [Citation9,Citation27]. Therefore, the Mamastrovirus genus as a whole can be considered a zoonotic threat, but the MAstV-Sp6 species classified here is deserving of special attention ( and ). Having only emerged ∼260 years ago with the more recent arrival of genotype 7 less than 50 years ago, its higher mutational rate compared to other lineages () underscores the potential danger posed by this species.
Standing in stark contrast is the coevolution of all three genotypes of the MAstV-Sp7 species with their respective hosts (), especially MAstV-Sp7G3 (historically recognized as human astroviruses) with its human host. Our results help explain the low level of pathogenicity shown by these strains in the hosts in which they have been identified [Citation3]. The resolved time-stamped phylogeny evidenced the ancient history of these viruses (A) and the demographic reconstruction illustrated their levels of genetic stability for most of their time in circulation (B). A similar pattern for the demographic reconstruction of this species was obtained by Zhou et al. [Citation8], who focused their study on the capsid protein gene. When deconvoluted, the BSP analysis of Zhou et al.[Citation8] observed a drastic change in the demographic growth of what they defined as human astrovirus Group 1 (HAstV-1), (sub-lineage denoted with red arrow in A), during 1975 to 1995 with a peak around ∼1985 [Citation8]. We also observed this same increase in genetic diversity of MAstV-Sp7G3 (B). Hence, we agree with the notion that this sub-lineage of MAstV-Sp7G3 could become the most prevalent in the human population in the near future [Citation8].
Our analysis of Mamastrovirus recombination revealed a low incidence, but with distinct distribution patterns that reflect the different evolutionary histories and selective pressures on species ( and Supplementary Table S1). Recombination frequency varies among different viral families, seemingly higher in those with a diminished capacity to accumulate mutations in order to avoid error catastrophe, by regenerating mosaic genomes that are fit to survive a host response [Citation30]. The low frequency of recombinant strains could be the consequence of different characteristics of viruses in the Mamastrovirus genus, including that the typical course of infections is acute in most of the hosts species [Citation31] and the diversification of these viruses following a host-jump limits the homology shared among genomes. A key observation from our recombination analysis was that parental strains were located within the same genotype of each species. This is consistent with the Dobzhansky-Muller model of incompatibilities imposing restrictions on inter-species or inter-genotypic recombination [Citation25, Citation26] and agrees with our assertion that Mamastrovirus diversification is mainly driven by host-jump events. The recombination analysis further bolsters the claim from the coevolutionary analysis that swine act as mixing-vessels for the emergence of novel strains of mamastrovirus. Swines on the one hand were determined to be an “acceptor host” for different species and on the other hand this is the host where most of the recombinant strains were identified ( and and Supplementary information Table S1). Other studies have identified putative recombinants (reviewed in [Citation27]), however, we were only able to confirm those strains reported by Babkin et al.[Citation24] ( and Supplementary information Table S1 and Figure S7). Detection of recombinants can be influenced by several factors including the sequence dataset itself, but we believe the principal determinant is the methodology used (Simplot vs RDP5). Simplot software relies mainly upon a BootScan search within a sliding window, whereas RDP5 has additional methods and statistical methodologies to discard putative false positives, besides just BootScan [Citation32]. Agreeing upon a consistent methodological approach for assessing recombination in Mamastrovirus would prove beneficial.
Previous studies have raised serious concerns about the circulation of neurotropic astrovirus in humans [Citation15,Citation16,Citation18,Citation20,Citation33]. Our combined integrated-phylogeny and virus-host coevolutionary reconstruction provide evidence that these strains (MAstV-Sp6G7) are the result of a recent zoonotic event from a bovine source and that they are in the process of adaptation and diversification in the human population. An important aspect to highlight is the confusion caused by the current (hopefully previous) classification system of Mamastrovirus and the resulting inability to track these strains [Citation20]. With varying names such as VA1, VA1/HMO-C, UK1, MLB2, MLB1 BF34 [Citation20] or simply “unclassified” (B, right), this has only served to increase the difficulties in understanding the role of these emergent strains producing new clinical outcomes. We demonstrated that the strains of genotype MAstV-Sp6G7 found in patients with neurological disorders have a unique evolutionary history with different evolutionary rates ( and ), distinct from those strains in MAstV-Sp6G2 linked to pediatric diseases ( and ) or the well-known human astroviruses (MAstV-Sp7G3) ( and ). It is important to note that in all cases of neurotropism these strains have been characterized in immunocompromised patients [Citation15–19]. This evokes another lesson learned from the current COVID-19 pandemic, in which the emergence of variants of concern (VOC) or variants of interest (VOI) were linked to the circulation of SARS-CoV-2 in immunocompromised patients[Citation34]. Thus, this type of individual represents an ideal niche for the expansion of viral diversity and the acquisition of viral fitness [Citation35], characterized by an increased mutational profile [Citation36] that favours its further spread across the susceptible population, either cryptically [Citation37] or with clinical manifestations [Citation38]. An important limitation of this study is the low number of sequences from this genotype. However, we already have an indication that these strains can escape established diagnostic assays for astrovirus detection by real-time PCR or enzymatic immunoassays [Citation39], in addition to the fact that all available strains from this group have been identified through NGS. Therefore, further studies to understand (i) the status of “unknown” regarding circulation of this particular group, and (ii) the development of specific diagnostic tools for the detection of this defined genotype, will be required. Our reporting of another strain from this group provides mounting evidence that MAstV-Sp6G7 is actively circulating.
Methods
Sample collection
Plasma samples (n = 228) with disease of unknown etiology were obtained from iSpecimen, Inc. (Lexington, MA). Follow-up serology testing differed for each subject and was determined by symptoms and presumed diagnosis. Sample AbE11938, which was positive for HAstV by NGS, was evaluated for rubella and measles IgG and tested negative.
Library preparation sequencing and genome assembly
Plasma samples were extracted on an m2000sp using the TNA + Proteinase K protocol (Abbott Molecular, Des Plaines, IL, USA). Nucleic acid was eluted in 60 µL and frozen at −80°C until use. Total nucleic acid extracts were reverse transcribed using SuperScript IV kit (Invitrogen Life Technologies) followed by 2nd strand synthesis with Sequenase V2.0 T7 DNA pol (Affymetrix). cDNA was purified using Agencourt AMPpure XP beads (Beckman Coulter). Double-stranded DNA/cDNA were “tagmented” and barcoded with Nextera XT indices lacking 5’ biotin tags using 24 cycles of amplification (IDT, Coralville IA; Illumina, Carlsbad CA). Nextera libraries were purified with Agencourt AMPpure XP beads (Beckman Coulter) and quantified on a 2200 TapeStation (Agilent) and Qubit fluorometer (Life Technologies). Libraries were pooled (n = 20) and enriched for viruses using the Pan Viral probe set (n > 600,000 probes) (Twist Biosciences, San Francisco, CA) now known as the Comprehensive Viral Research Panel (CVRP). Hybridization and amplification steps were followed according to manufacturer instructions. Four captures of 88 samples total were sequenced together on an Illumina MiSeq and obtained >45 million reads.
Processing of NGS data through SURPI [Citation40] and confirmation by BLAST revealed the presence of human astrovirus reads in the AbE11938. An astrovirus consensus sequence was assembled and annotated using CLC Genomics Workbench 21.0, filtering out reads below a Q score of 30 and applying the following parameters: match = 1, mismatch = 2, insertion = 3, deletion = 3, length fraction = 0.8, and similarity fraction = 0.8. From 54,730 total reads following enrichment, 6,241 mapped to HAstV (closest reference MH933754.1) to yield full genome coverage at a depth of 99X. The complete genome sequence of HAstV isolate AbE11938 was deposited in GenBank under accession number OP594725.
Dataset collection, filtering and subsampling
Two sequence datasets (M = 521 sequences) containing all Mamastrovirus genus complete genome sequences and all the sequences of cytochrome b from hosts in which those viral members have been isolated (H = 17 sequences) were downloaded from the GenBank database (http://www.ncbi.nlm.nih.gov/) accessed on 11 October 2022. Dataset M filtration and deduplication analysis were performed as described in Perez et al. [Citation41]. To associate metadata with this dataset, additional information was extracted, including year of collection, country, previous classification, and host using the script gbmung, https://github.com/sdwfrost/gbmunge, as well as by manual curation (Supplementary material Table S1 and Table S2) The filtered sequences were aligned using the algorithm G-INS-i within MAFFT [Citation42].
Phylogenetic tree reconstruction
Phylogenetic inference of the Mamastrovirus genus was conducted using whole genomes and Maximum Likelihood (ML) probabilistic methods [Citation43]. For ML inference, the methodology recently described in Pikula et al. [Citation44] was used with some modifications. ML-phylogenetic trees derived from amino acid alignments and selection of the best-fit model were both computed with the IQ-TREE 2 programme [Citation45]. Confidence levels for branches were determined in IQ-TREE by Shimodaira test with 10,000 bootstrap replicates and trees were then visualized and edited in ggtree and ggtreeExtra [Citation46].
Taxonomical demarcation analysis using pairwise sequence comparison (PASC) and sequence demarcation tool (SDT)
As we previously employed in Perez et al. [Citation41] for the Picobirnaviridae family using complete genomes, we implemented Pairwise Sequence Comparison (PASC) [Citation47] and the Sequence Demarcation Tool (SDT) [Citation48] to determine the levels of taxonomic demarcation within the Mamastrovirus genus. Briefly, all 521 unique sequences were used as input in the SDT software, which applies a robust Needleman-Wunsch (NW)-based pairwise alignment approach with a pairwise identity calculation that ignores positions containing indels. This methodology is primarily intended to objectively assign ICTV-endorsed taxonomic classifications of genotype, species and genus based on pairwise identity demarcation thresholds [Citation49]. In parallel, pairwise nucleotide p-distances were calculated using MEGA X to determine genetic distances [Citation50]. Different matrices of nucleotide divergence between groups were generated using 500 bootstrap replicates to estimate variance. Due to the random distribution of gaps and the low level of similarity reflected in the PASC results, all cases of INDELs were treated as pairwise deletions to avoid loss of information.
Co-phylogenetic analysis
The presence of co-phylogeny, measured by the degree of co-speciation between the different genotypes identified within the different species of the Mamastrovirus genus and their respective hosts, was tested as previously described by Rios et al. [Citation51]. Briefly, the Procrustes Application to Cophylogenetic Analysis (PACo) [Citation22] tool was used to generate a graphical assessment determining the fit of the virus phylogeny onto the host phylogeny and produce a goodness-of-fit statistic. The significance of the statistic value was established by randomization of the host-parasite association data using the R-function in PACo [Citation22]. For this purpose, three input files were provided: two containing the reconstructions of the phylogenetic structure for the host and mamastrovirus species, and the third containing a binary matrix coding the host-genotype associations. The R script was run as described in the PACo Manual in Balbuena et al. [Citation22], available for free at http://www.uv.es/cophylpaco/index.html. The diversification events between the different genotypes and their respective hosts were explored using JANE software v4 [Citation23]. Cospeciation, duplication, host switch, loss and/or failure to diverge events were explored using default parameters and selecting the solution with the lowest costs.
Recombination analysis
Searches for recombinant sequences and identification of the best-supported breakpoints for all sequences in the dataset were performed as described in Pikula et al. [Citation44] with some modifications. Briefly, we used a combination of six methods implemented in Recombination Detection Program RDP5 v5: [Citation32] RDP, GENECONV, Bootscan, MaxChi, Chimera, SisScan and 3SEQ. Programmes were executed with modifications to parameter settings determined according to the guidelines in the RDP5 manual recommended for the analysis of divergent viruses. Recombinant sequences were identified when scoring below the highest acceptable p value of 0.01, and Bonferroni’s multiple comparison correction was applied. Only those sequences detected by more than three methods were considered recombinants. To eliminate false positives, we confirmed recombination results by three different approaches: (1) a markedly different outcome of the PHI test [Citation52], (2) well-supported bootstrap values >75% in BootScan analysis, and (3) confirmation splits in the trees by Shimodaira-Hasegawa test.
Time-stamped phylogenetic and demographic analysis of the mamastrovirus species and genotypes circulating in humans
The sequences from mamastrovirus species six (MAstV-Sp6) and seven (MAstV-Sp7), after removing recombinants, were initially inspected for the presence of a temporal signal using the Bayesian Evaluation of Temporal Signal test (BETS)[Citation53], as we recently described in Orf et al. [Citation54]. Briefly, BETS was conducted using BEAST v.1.10.4 [Citation55] contrasting two competing models: heterochronous vs isochronous. If the heterochronous model improves the statistical fit of the data, the use of a molecular clock to calibrate the data is warranted [Citation53]. The estimation of the log marginal likelihood of the competing models was assessed by path sampling (PS) [Citation56] and stepping-stone sampling (SS) [Citation57]. For both mamastrovirus species, an uncorrelated relaxed local clock was selected with an exponential coalescent prior with a mean of 10 and offset of 0.5. The initial ML-trees estimated by IQ-TREE2 were used as starting trees in the temporal Bayesian analyses. For both species, two Markov chain Monte Carlo (MCMC) chains were run over 200 million states using BEAST v1.10.5 with sampling every 20,000 states. The BEAGLE v.3.1.0 library was used to enhance computational speed [Citation58]. Both chains were combined after 10% of states were removed (burn-in step). We diagnosed the runs by examining trace plots and effective samples sizes (ESS >200) using Tracer v1.7 [Citation59]. In addition, for genotypes two and seven of the mamastrovirus species 6 (MAstV-Sp6G2 and MAstV-Sp6G7) and genotype 3 of species seven (MAstV-Sp7G3), a demographic reconstruction considering the effective number of infections Ne, increased by the mean viral generation time (τ) through time, was examined using the same molecular clock, but considering a Bayesian skyline plot (BSP) as prior. The dynamics of these genotypes were estimated using the same parameters for the runs as above and visualized using ggplot2 in R.
Supplemental Material
Download Zip (1.1 MB)Acknowledgments
We thank Dr. Gregory S. Orf for assistance with a parsing script in R. Author Contributions: Conceptualization, LJP and MGB; methodology, LJP and MGB; software, LJP; validation, KF, LJP; formal analysis, KF, LJP and MGB; investigation, KF, LJP and MGB; data curation, KF LJP and MGB; writing original draft preparation, LJP and MGB; writing – review and editing, LJP, MGB and GAC; visualization, LJP and MGB; supervision, GAC; project administration, MGB and GAC; funding acquisition GAC. All authors have read and agreed to the published version of the manuscript.
Disclosure statement
LJP, KF, MGB, and GAC are all employees and Abbott shareholders.
Data availability
Accessions with relevant metadata are contained in Supplemental Tables S1 and S2. Scripts for generating figures are available upon request. Raw data files for the analysis of coevolutionary analysis using PACo, JANE and the temporal analysis using BEAST are available on Github at https://github.com/LesterJP/Temporal-and-coevolutionary-analyses-reveal-the-events-driving-the-emergence-and-circulation-of-huma.
References
- Appleton H, Higgins PG. Letter: viruses and gastroenteritis in infants. Lancet (London, England). 1975 Jun 7;1(7919):1297.
- Donato C, Vijaykrishna D. The broad host range and genetic diversity of Mammalian and Avian Astroviruses. Viruses. 2017 May 10;9(5).
- Chapter 27 - Caliciviridae and Astroviridae. In: MacLachlan NJ, Dubovi EJ, editors. Fenner's veterinary virology. 5th ed. Boston: Academic Press; 2017. p. 497–510.
- Mendenhall IH, Smith GJD, Vijaykrishna D. Ecological drivers of virus evolution: astrovirus as a case study. 2015;89(14):6978–6981.
- Family - Astroviridae. In: King AMQ, Adams MJ, Carstens EB, et al., editors. Virus taxonomy. San Diego: Elsevier; 2012. p. 953–959.
- van Hemert FJ, Berkhout B, Lukashov VV. Host-related nucleotide composition and codon usage as driving forces in the recent evolution of the Astroviridae. Virology. 2007 May 10;361(2):447–54.
- Babkin IV, Tikunov AY, Zhirakovskaia EV, et al. High evolutionary rate of human astrovirus. Infection, Genetics and Evolution. 2012 Mar;12(2):435–42.
- Zhou N, Zhou L, Wang B. Molecular evolution of classic human astrovirus, as revealed by the analysis of the capsid protein gene. Viruses. 2019 Aug 1;11(8.
- Roach SN, Langlois RA. Intra- and cross-species transmission of astroviruses. Viruses. 2021;13(6):1127.
- Boujon CL, Koch MC, Seuberlich T. Chapter five - the expanding field of mammalian astroviruses: opportunities and challenges in clinical virology. In: Beer M, Höper D, editor. Advances in virus research. Vol. 99. Academic Press; 2017. p. 109–137.
- Lu RG, Li SS, Hu B, et al. The first evidence of shaking mink syndrome-astrovirus associated encephalitis in farmed minks, China. Transbound Emerg Dis. 2022 Sep 4.
- Blomstrom AL, Widen F, Hammer AS, et al. Detection of a novel astrovirus in brain tissue of mink suffering from shaking mink syndrome by use of viral metagenomics. J Clin Microbiol. 2010 Dec;48(12):4392–6.
- Li L, Diab S, McGraw S, et al. Divergent astrovirus associated with neurologic disease in cattle. Emerg Infect Dis. 2013;19(9):1385–92.
- Selimovic-Hamza S, Sanchez S, Philibert H, et al. Bovine astrovirus infection in feedlot cattle with neurological disease in western Canada. Can Vet J. 2017 Jun;58(6):601–603.
- Brown JR, Morfopoulou S, Hubb J, et al. Astrovirus VA1/HMO-C: an increasingly recognized neurotropic pathogen in immunocompromised patients. Clin Infect Dis. 2015 Mar 15;60(6):881–8.
- Naccache SN, Peggs KS, Mattes FM, et al. Diagnosis of neuroinvasive astrovirus infection in an immunocompromised adult with encephalitis by unbiased next-generation sequencing. Clin Infect Dis. 2015 Mar 15;60(6):919–23.
- Lum SH, Turner A, Guiver M, et al. An emerging opportunistic infection: fatal astrovirus (VA1/HMO-C) encephalitis in a pediatric stem cell transplant recipient. Transpl Infect Dis. 2016 Dec;18(6):960–964.
- Krol L, Turkiewicz D, Nordborg K, et al. Astrovirus VA1/HMO encephalitis after allogeneic hematopoietic cell transplantation: significant role of immune competence in virus control. Pediatr Blood Cancer. 2021 Dec;68(12):e29286.
- Bami S, Hidinger J, Madni A, et al. Human astrovirus VA1 encephalitis in pediatric patients with cancer: report of 2 cases and review of the literature. J Pediatric Infect Dis Soc. 2022 Sep 29;11(9):408–412.
- Wildi N, Seuberlich T. Neurotropic astroviruses in animals. Viruses. 2021 Jun 23;13(7.
- Yinda CK, Vanhulle E, Conceição-Neto N, et al. Gut virome analysis of cameroonians reveals high diversity of enteric viruses. Including Potential Interspecies Transmitted Viruses. mSphere. 2019 Jan 23;4(1.
- Balbuena JA, Miguez-Lozano R, Blasco-Costa I. PACo: a novel procrustes application to cophylogenetic analysis. PLoS One. 2013;8(4):e61048.
- Conow C, Fielder D, Ovadia Y, et al. Jane: a new tool for the cophylogeny reconstruction problem. Algorithms for Molecular Biology. 2010 2010/02/03;5(1):16.
- Babkin IV, Tikunov AY, Sedelnikova DA, et al. Recombination analysis based on the HAstV-2 and HAstV-4 complete genomes. Infection, Genetics and Evolution. 2014 Mar;22:94–102.
- Siobain D, Burch CL, Turner PE. Evolution of host specificity drives reproductive isolation among RNA viruses. Evolution. 2007;61(11):2614–2622.
- Niknamian S. On the neglected shifting balance theory, Bateson–Dobzhansky–Muller model & quantum evolution plus the role of mitochondrial membrane potential (MMP) impact on COVID-19. Open access J Oncol Med. 2021;4(2.
- Wohlgemuth N, Honce R, Schultz-Cherry S. Astrovirus evolution and emergence. infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases. Apr. 2019;69:30–37.
- Zhu Z, Lian X, Su X, et al. From SARS and MERS to COVID-19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respiratory Research. 2020 2020/08/27;21(1):224.
- Grubaugh ND, Ladner JT, Lemey P, et al. Tracking virus outbreaks in the twenty-first century. Nat Microbiol. 2019 Jan;4(1):10–19.
- Holmes EC. Error thresholds and the constraints to RNA virus evolution. Trends in Microbiology. 2003 Dec;11(12):543–6.
- Simon-Loriere E, Holmes EC. Why do RNA viruses recombine? Nature Reviews Microbiology. 2011 2011/08/01;9(8):617–626.
- Martin DP, Varsani A, Roumagnac P, et al. RDP5: a computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evolution. 2020;7(1.
- Deiss R, Selimovic-Hamza S, Seuberlich T, et al. Neurologic clinical signs in cattle with astrovirus-associated encephalitis. J Vet Intern Med. 2017 Jul;31(4):1209–1214.
- Viana R, Moyo S, Amoako DG, et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in Southern Africa. Nature. 2022 2022-03-24;603(7902):679–686.
- McCrone JT, Hill V, Bajaj S, et al. Context-specific emergence and growth of the SARS-CoV-2 Delta variant. Nature. 2022 Oct;610(7930):154–160.
- Perez LJ, Orf GS, Berg MG, et al. The early SARS-CoV-2 epidemic in Senegal was driven by the local emergence of B.1.416 and the introduction of B.1.1.420 from Europe. Virus Evol. 2022;8(1):veac025.
- Sun H, Dickens BL, Jit M, et al. Mapping the cryptic spread of the 2015-2016 global Zika virus epidemic. BMC Medicine. 2020 Dec 17;18(1):399.
- Ciuoderis KA, Berg MG, Perez LJ, et al. Oropouche virus as an emerging cause of acute febrile illness in Colombia. Emerg Microbes Infect. 2022 Dec;11(1):2645–2657.
- Smits SL, van Leeuwen M, van der Eijk AA, et al. Human astrovirus infection in a patient with new-onset celiac disease. J Clin Microbiol. 2010 Sep;48(9):3416–8.
- Naccache SN, Federman S, Veeraraghavan N, et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Research. 2014 Jul;24(7):1180–92.
- Perez LJ, Cloherty GA, Berg MG. Understanding the genetic diversity of Picobirnavirus: a classification update based on phylogenetic and pairwise sequence comparison approaches. Viruses. 2021 Jul 28;13(8.
- Katoh K, Kuma K-i, Toh H, et al. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Research. 2005;33(2):511–518.
- Dhar A, Minin VN. Maximum likelihood phylogenetic inference. In: Kliman RM, editor. Encyclopedia of evolutionary biology. Oxford: Academic Press; 2016. p. 499–506.
- Pikula A, Smietanka K, Perez LJ. Emergence and expansion of novel pathogenic reassortant strains of infectious bursal disease virus causing acute outbreaks of the disease in Europe. Transbound Emerg Dis. 2020 Jul;67(4):1739–1744.
- Nguyen LT, Schmidt HA, von Haeseler A, et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015 Jan;32(1):268–74.
- Xu S, Dai Z, Guo P, et al. Ggtreeextra: compact visualization of richly annotated phylogenetic data. Mol Biol Evol. 2021 Aug 23;38(9):4039–4042.
- Bao Y, Chetvernin V, Tatusova T. Improvements to pairwise sequence comparison (PASC): a genome-based web tool for virus classification. Archives of Virology. 2014 2014/12/01;159(12):3293–3304.
- Muhire BM, Varsani A, Martin DP. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLOS ONE. 2014;9(9):e108277.
- Rios L, Nunez JI, Diaz de Arce H, et al. Revisiting the genetic diversity of classical swine fever virus: A proposal for new genotyping and subgenotyping schemes of classification. Transbound Emerg Dis. 2018 Aug;65(4):963–971.
- Kumar S, Stecher G, Li M, et al. MEGA x: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution. 2018;35(6):1547–1549.
- Rios L, Coronado L, Naranjo-Feliciano D, et al. Deciphering the emergence, genetic diversity and evolution of classical swine fever virus. Sci Rep. 2017 Dec 20;7(1):17887.
- Bruen TC, Philippe H, Bryant D. A simple and robust statistical test for detecting the presence of recombination. Genetics. 2006;172(4):2665–2681.
- Duchene S, Lemey P, Stadler T, et al. Bayesian evaluation of temporal signal in measurably evolving populations. Mol Biol Evol. 2020 Nov 1;37(11):3363–3379.
- Orf GS, Perez LJ, Meyer TV, et al. Purifying selection decreases the potential for Bangui orthobunyavirus outbreaks in humans. Virus Evol. 2023;9(1):vead018.
- Suchard MA, Lemey P, Baele G, et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evolution. 2018 2018-01-01;4(1.
- Lartillot N, Philippe H. Computing Bayes factors using thermodynamic integration. Systematic Biology. 2006 2006-04-01;55(2):195–207.
- Xie W, Lewis PO, Fan Y, et al. Improving marginal likelihood estimation for Bayesian phylogenetic model selection. Systematic Biology. 2011 2011-03-01;60(2):150–160.
- Ayres DL, Darling A, Zwickl DJ, et al. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst Biol. 2012 Jan;61(1):170–3.
- Rambaut A, Drummond AJ, Xie D, et al. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst Biol. 2018 Sep 1;67(5):901–904.