
ABSTRACT
Specific HBsAg mutations are known to hamper HBsAg recognition by neutralizing antibodies thus challenging HBV-vaccination efficacy. Nevertheless, information on their impact and spreading over time is limited. Here, we characterize the circulation of vaccine-escape mutations from 2005 to 2019 and their correlation with virological parameters in a large cohort of patients infected with HBV genotype-D (N = 947), dominant in Europe. Overall, 17.7% of patients harbours ≥1 vaccine-escape mutation with the highest prevalence in subgenotype-D3. Notably, complex profiles (characterized by ≥2 vaccine-escape mutations) are revealed in 3.1% of patients with a prevalence rising from 0.4% in 2005–2009 to 3.0% in 2010–2014 and 5.1% in 2015–2019 (P = 0.007) (OR[95%CI]:11.04[1.42–85.58], P = 0.02, by multivariable-analysis). The presence of complex profiles correlates with lower HBsAg-levels (median[IQR]:40[0–2905]IU/mL for complex profiles vs 2078[115–6037]IU/ml and 1881[410–7622]IU/mL for single or no vaccine-escape mutation [P < 0.02]). Even more, the presence of complex profiles correlates with HBsAg-negativity despite HBV-DNA positivity (HBsAg-negativity in 34.8% with ≥2 vaccine-escape mutations vs 6.7% and 2.3% with a single or no vaccine-escape mutation, P < 0.007). These in-vivo findings are in keeping with our in-vitro results showing the ability of these mutations in hampering HBsAg secretion or HBsAg recognition by diagnostic antibodies. In conclusion, vaccine-escape mutations, single or in complex profiles, circulate in a not negligible fraction of HBV genotype-D infected patients with an increasing temporal trend, suggesting a progressive enrichment in the circulation of variants able to evade humoral responses. This should be considered for a proper clinical interpretation of HBsAg-results and for the development of novel vaccine formulations for prophylactic and therapeutic purposes.
Introduction
Infection with hepatitis B virus (HBV) still represents a major worldwide concern, accounting for a significant global disease burden and a high mortality from cirrhosis and liver cancer. In 2019, the World Health Organization (WHO) estimated that almost 300 million people are still living with HBV chronic infection, resulting in over 800,000 deaths each year, due to the progression of HBV-related liver disease [Citation1]. Currently, the annual death toll associated with HBV infection has been estimated to have exceeded that of HIV and malaria [Citation2,Citation3]. This is even more critical considering the high efficiency of HBV transmission in vivo as attested by recent calculations estimating a minimal infectious dose of only 16 virions [Citation4].
HBV is a highly evolving pathogen characterized by a high degree of genetic variability (a unique property among DNA viruses) that is caused by the lack of proof-reading activity of HBV reverse transcriptase and exacerbated by the high rate of HBV replication [Citation5]. This high degree of HBV genetic variability enables the virus to react to endogenous (i.e. immune system), and exogenous (i.e. vaccination, hepatitis B immunoglobulin, antiviral drugs) selective pressures by further modulating its genome structure. Among the different HBV-proteins, the hepatitis B surface antigen (HBsAg) consists of three different proteins encoded within the same S open reading frame (ORF S) by the alternate usage of translational start codons (i) large HBsAg (L-HBs), consisting of preS-1, preS-2 and S regions; (ii) middle HBsAg (M-HBs) consisting of pre-2 and S regions and a small-HBsAg (hereafter defined HBsAg), containing only the S region [Citation6,Citation7]. HBsAg represents the target of neutralizing antibodies thus playing a pivotal role for the development of anti-HBV protective immunity. In particular, HBsAg is translated at the endoplasmic reticulum (ER) with integration of four hydrophobic transmembrane domains into the ER membrane. HBsAg stability is then provided by the formation of intra- and inter-molecular disulfide bridges between cysteines in the lumen of the ER. This results in the formation of a long external antigenic loop defined as major hydrophilic region (MHR, aa 99–169 of HBsAg), that includes the immunodominant “a” determinant (∼aa 124–147), containing most B-cell immune-dominant epitopes eliciting neutralizing antibodies [Citation7].
So far, specific mutations within the MHR of HBsAg have been recognized to hamper the antigenicity of the viral surface glycoprotein and, in turn, its recognition by neutralizing antibodies, including those induced by vaccination [Citation8,Citation9].
These mutations (hereafter defined as vaccine-escape mutations) have been reported in individuals who have contracted HBV infection despite HBV vaccination in the setting of both horizontal or mother-to-child transmission, thus representing a potential threat for the global effectiveness of vaccination strategies [Citation10–12].
To date, the circulation of vaccine-escape mutations overtime in large populations of HBV-infected patients has been limitedly monitored, particularly in the setting of countries with a 30-years history of exposure to universal anti-HBV immunization programmes, as Italy. Likewise, paucity of information is available on their impact on markers of HBV replicative activity.
In this light, this study aims at characterizing the circulation of vaccine-escape mutations, single or in complex mutational profiles, over a time-frame of 15 years (2005–2019) and their correlation with virological parameters in one of the largest cohort of patients infected with HBV genotype-D (N = 947), the HBV dominant genotype in the European area. So far, monitoring the spread of vaccine-escape mutations is crucial to develop novel vaccine strategies thus ensuring the long-term success of global immunization programme and the achievement of WHO commitment on the elimination of viral hepatitis as a public global threat by 2030 [Citation13].
Materials and methods
Patients
This study included 947 patients with HBV genotype-D infection and detectable serum HBV-DNA, followed for clinical assessments in different Central Italy’s centres from 2005 to 2019. For each patient, the HBsAg-encoding genome region sequence (hereafter defined as HBsAg-sequence) was performed at the Virology Unit of the University Hospital Tor Vergata. Only one sequence per patient was included in the analysis.
Furthermore, a further HBs sequence was collected for 19 out of 168 patients with at least one vaccine escape mutation (median [IQR] time of 1.9 [1.5–2.8] years between the two sequences).
Information regarding patients’ demographics (age, sex, country of origin), coinfections (HIV, HDV), HBV markers (Hepatitis B e antigen [HBeAg] status; serum HBV-DNA and quantitative HBsAg), and alanine-aminotransferase levels (ALT) were recorded in an ad-hoc designed anonymous database. For ALT, the following upper normal limits (UNL) were considered: 30 U/L for women and 42 U/L for men [Citation14,Citation15].
Approval by the Ethics Committee was deemed unnecessary because, under Italian law, biomedical research is subjected to previous approval only in the hypothesis of clinical trials on medicinal products for clinical use (art. 6 and art. 9, leg. decree 211/2003). The research was conducted on data previously anonymized, according to the Italian Data Protection Code requirements (leg. decree 101/2018).
Determination of HBV markers
Serum HBV-DNA was quantified by COBAS AmpliPrep-Cobas TaqMan HBV test (Roche, Basel, CH). HBsAg quantification was carried out by Elecsys®HBsAgII assay (Roche, Basel, CH), with a lower limit of detection of 0.05 IU/ml while HBeAg qualitative detection by Elecsys®HBeAg (Roche, Basel, CH).
Population-based HBsAg sequencing
For each patient, HBsAg-sequence was obtained by Sanger sequencing of RT/S gene as previously described [Citation16]. Briefly, HBV-DNA was extracted from plasma samples using QIAmp DNA mini-kit, (Qiagen, Germantown, USA), and then the nucleotide region encoding entirely HBsAg was amplified with Amplitaq-Gold polymerase using S-HBV_F1-5′GGTCACCATATTCTTGGGAA and S-HBV_R1-5′GTGGGGGTTGCGTCAGCAAA primer pairs for first-round PCR. Polymerase chain reaction (PCR) conditions were: 93°C for 12 min, 40 cycles (94°C 50s, 57°C 50s, 72°C 90s), and 72°C for 10 min. When necessary, a second-round PCR was performed, using S-HBV_F2-5′GGTGGACTTCTCTCAATTTT and S-HBV_R2-5’TGGCGAGAAAGTGAA primer pairs. Second-round PCR conditions were: 93°C for 12 min, 40 cycles (94°C 50s, 55°C 50s, 72°C 80s), and 72°C for 10 min. Details on the nucleotide annealing regions of the primers respect to HBV-full genome (NC_003977.2) and the corresponding regions of HBsAg at amino acid levels are reported in the Supplementary Table 1 (Table S1).
PCR-products were purified and sequenced by using different sequence-specific primers and a BigDye-Terminator v.3.1 cycle sequencing kit (Applied-Biosystems, Foster City, USA) on the ABI Prism 3130XL (Applied-Biosystems, Foster City, USA).
HBsAg-sequences were analysed using SeqScape-v.2.6 software (Applied-Biosystems), then aligned using BioEdit 7.0 software (Hall, Nucleic Acid Res 1999).
Mutations were defined according to HBV genotype-D sequence (NC_003977.2). Sequences having a mixture of wild-type and mutant residues at single positions were considered to have the mutant(s) at that position.
HBV subgenotypes D were determined through phylogenetic tree constructed by Neighbor-Joining method [Citation17] on MEGA6 software [Citation18]. Branching orders reliability was assessed by bootstrap analysis of 1000 replicates.
In this study, we focused on 19 mutations systematically retrieved from literature: T116N, P120S, T126A/I/N/S, Q129H/R, T131I/N, M133I/L, C139S, K141E, P142S, D144A/E, G145A/R (). All these vaccine-escape mutations have been previously reported in literature to be involved in HBV transmission cases to vaccinated people in vivo and to be further associated with a reduced binding affinity with neutralizing antibodies and immunoglobulins in vitro ().
Table 1. List of vaccine-escape mutations defined according to in vivo and in vitro studies.
To evaluate the nucleotide diversity between longitudinal sequences, pairwise genetic distance was calculated by MEGA6 software [Citation18,Citation19].
Then, Shannon entropy score was calculated to measure the extent of amino acid variability at each amino acid position using the formula (Sn = −∑i(pi lnpi)/lnN), where pi was the frequency of each distinct amino acid and N was the total number of sequences analysed. In particular, the following subset of sequences from patients with at least one vaccine escape mutations were analysed: 168 for HBsAg N-terminus (1-98aa), MHR (99-169aa) and C-terminus (170-226aa), 42 for PreS2, 37 for PreS1, 24 for HBcAg and 15 for HBx regions. The detailed protocols for the sequencing of the genomic regions encoding PreS1, PreS2, HBcAg and HBx are reported in the Supplementary materials and the primers used for amplifying these regions are specified in Table S1.
Next-Generation sequencing (NGS) of HBsAg region
The extent of genetic heterogeneity and the intra-patient frequency of each vaccine-escape mutation was also investigated by Illumina MiSeq (Illumina, San Diego, California, USA) for a subset of 32 patients with available samples. Plasma-derived HBV-DNA was amplified according to home-made protocols targeting the nucleotide regions encoding for S-HBsAg (aa 73-226), thus covering entirely the HBsAg major hydrophilic region. Details on the nucleotide annealing regions of the primers respect to HBV-full genome (NC_003977.2) and the corresponding regions of HBsAg at amino acid levels are reported in the Table S1.
In details, primers for NGS were designed to amplify two partially overlapping amplicons covering the HBV nucleotide region coding for S-HBsAg from aa 73 to 184 and from aa 162 to 226, respectively. This primer design allowed to obtain the entire MHR region in a single amplicon, thus permitting to verify the co-presence of two or more vaccine-escape mutations in the same viral strain.
The two fragments of 332 and 384 nucleotides were generated by nested PCR using barcoded-modified primers after a first-round amplification with the same protocol, already specified for the Sanger sequencing protocol. Nested-PCR was performed with the Fast Start HiFi PCR system (Roche Diagnostics, Mannheim, Germany) under the following conditions: 1 cycle of 94°C for 5 min, 30 cycles of 94°C for 30 s, 60°C for 30 s, 72°C for 35sec, followed by a final extension at 72°C for 7 min. The amplified products were purified using Agencourt AMPure XP PCR purification beads (Beckman Coulter, Brea, CA) and quantified with the Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies, Eugene, Oregon USA) by GloMax Multidetection System (Promega, Wisconsin USA). Equimolar (12 pM) pooled PCR products were clonally amplified on captured beads in water–oil emulsion microreactors (1.2 DNA-copies/beads) and the pooled samples were lastly sequenced by MiSeq Reagent Kit v2 (500-cycles) (Illumina, San Diego, California, USA).
Raw data were obtained in fastq format. A quality control was performed by Trimmomatic [Citation20] software in order to remove adapters, PCR primers and poor-quality reads. Fastq files were analysed by VirVarSeq software (Verbist, BM) using NC_003977.2 as reference sequence. Only variants with frequency >1% were retained for further analysis.
Cell cultures and transfection
Huh7 cells were grown in a 37 °C humidified atmosphere containing 5% CO2, using Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, Inc., Gaithersburg, MD, USA) supplemented with 10% fetal bovine heat-inactivated serum and with 100 U/mL penicillin, 100 µg/mL streptomycin and 2 mM L-glutamine.
As previously described [Citation21,Citation22], plasmids encoding the wild-type (wt) and mutated HBsAg linked to a streptavidin-tag (version II, IBA Lifesciences, Göttingen, Germany) (strep-tag) were used to transfect the Huh7 cells. The strep-tag is upstream the gene encoding HBsAg, thus resulting at the N-terminus of the corresponding protein. The following complex mutational profiles, associated in patients with HBsAg negativity despite detected HBV-DNA, were tested in vitro: T116N + C139S + G145R, P120S + T126I, T126I + G145R, T126I + Q129H + D144E, T126A + T131N, T131I + M133I. As controls, the wt HBsAg and mutated HBsAg carrying each single mutation included in the complex mutational profiles were also used.
After 72 h post transfection, cell supernatants were collected and used for the quantification of strep-tagged HBsAg. In particular, the amount of strep-tagged HBsAg released in culture supernatants was quantified by using two commercial assays targeting the HBsAg (DiaSorin LIAISON® XL HBsAg Quant [Saluggia, Italy] defined as commercial assay 1 and Abbott Alinity HBsAg quant [North Chicago, USA] defined as commercial assay 2) and by using a specifically-designed ELISA capable to recognize the Strep-tag portion linked to the HBsAg (defined hereafter as Strep-tag ELISA).
Differently from the commonly used HBsAg assays, the Strep-tag ELISA is not influenced by HBsAg modifications, giving the advantage to discriminate between a reduction of HBsAg recognition (due to an altered HBsAg antigenic properties) and a decrease in HBsAg secretion. Specifically, a decreased amount of Strep-tagged-HBsAg by all the assays (one targeting the Strep-tag and two HBsAg region) is indicative of defective HBsAg secretion in the presence of a given mutation. Conversely, a decreased amount of Strep-tagged-HBsAg only by the assays targeting directly HBsAg region, is indicative of an impaired diagnostic HBsAg antigenicity.
For each mutant, at least 2 independent transfection experiments were performed, each carried out in triplicate. More detailed methods are reported in supplementary materials.
Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics, v.23.0 (Armonk, NY). Data were expressed as median (interquartile range [IQR]) for quantitative variables and as counts and percentages for qualitative variables. Chi-Squared Test of Independence based on a 2 × 2 or 3 × 2 contingency table was used for qualitative data, while Mann–Whitney test for continuous data. Only correlations with P < 0.05 were considered statistically significant. Logistic regression analyses were performed to assess factors correlated with the presence of at least 1 immune-escape mutation or complex mutational profiles, considering the following variables: gender, age, nationality, HBV D-subgenotypes, serum HBV-DNA, ALT, time windows of samples’ collection (2005–2009, 2010–2014, 2015–2019), NUC treatment. After stepwise elimination for optimized Akaike information criterion, only variables showing a P-value <0.200 in univariate analysis were included in multivariable analysis.
For in vitro experiments, statistically differences between wt and mutated HBsAg were assessed by 2-tailed Student’s T-test.
Results
Study population
The study population included 947 viremic patients with HBV genotype-D infection. Most patients were male (64.7%) and of Italian origin (73.8%), with a median (IQR) age of 51(38–62) years (). Median (IQR) serum HBV-DNA was 3.4(2.5–5.0) logIU/ml and median (IQR) HBsAg levels were 1872(402–6504) IU/ml (). The 23.6% of patients had transaminase levels two times above the upper limit normal (median [IQR] ALT: 163(103–347) IU/ml), supporting liver inflammation ().
Table 2. Patients’ characteristics.
The 58.2% of overall population was NUC-treated at the time of HBsAg-sequencing. Furthermore, 11.2% of patients were coinfected with HIV while 5.6% with HDV ().
Notably, 5.7% of patients showed atypical HBV serological profiles, defined as HBsAg negativity despite detectable viremia (median [IQR] serum HBV-DNA: 2.2[1.6–3.2] logIU/ml) or the co-positivity of HBsAg and Anti-HBs antibodies (median [IQR] anti-HBs: 29[23–107] mIU/ml) ().
By phylogenetic analysis, the predominant HBV D sub-genotype was D3 (44.8%), followed by HBV sub-genotypes D1 (26%) and D2 (23%). HBV sub-genotype D9 was detected at a lower prevalence (4.1%) while the remaining D sub-genotypes were very rarely detected in the analysed population (prevalence ≤0.7%) ().
Circulation of vaccine escape mutations
By analysing the presence of the 19 mutations associated with HBV evasion from neutralizing antibodies () in the overall population, 17.7% (168/947) of patients harboured viral variants with at least one vaccine-escape mutation.
By multivariable analysis, factors independently correlated with the detection of at least 1 vaccine-escape mutation were: HBV sub-genotype D3 (OR [95%CI]: 1.48[1.02–2.15], P = 0.03), age (OR [95%CI]: 1.16[1.09–1.24], P < 0.0001) and having ALT 2xULN (OR [95%CI]: 1.51[1.02–2.20], P = 0.03) (). Focusing on HBV sub-genotypes, 23% of patients with D3 versus 13.6% of those with non-D3 sub-genotypes carried at least 1 vaccine escape mutation (P < 0.001) ((A)). This sub-genotype is known to be largely spread in East-Europe, underling the importance of monitoring the emergence of mutations, potentially hampering the full efficacy of anti-HBV vaccination, in the setting of non-autochthonous HBV sub-genotypes.
Figure 1. Percentage of patients with at least one vaccine escape mutation across HBV genotype D sub-genotypes and prevalence of vaccine escape mutations. The histogram in panel A reports the percentage of patients harbouring ≥1 vaccine escape mutation stratified according to the different HBV D sub-genotypes identified in the study population. As reported, HBV sub-genotype D3 is characterized by the highest percentage of patients with ≥1 vaccine escape mutation compared to the other D sub-genotypes. Statistically significant differences were assessed by Chi-Squared test based on 2 × 2 contingency table. The histogram in panel B reports the prevalence of each identified vaccine escape mutation within the population of patients harbouring ≥1 vaccine escape mutation (N = 168).
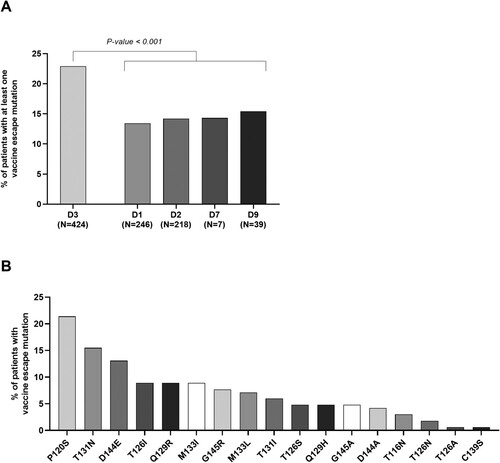
Table 3. Multivariable analysis of parameters associated with the presence of at least one vaccine escape mutation.
Notably, multivariable analysis showed an increased risk of harbouring at least one vaccine escape mutation in the time-window 2015–2019 compared to 2005–2009 (1.60[0.99–2.60], P = 0.05) (), supporting an increasing trend in the circulation of these mutations over time.
Furthermore, in the setting of 19 patients with a longitudinal sequence available after a median time of 1.9 (1.5–2.8) years, the presence of all vaccine escape mutations was confirmed also in the subsequent time point. In keeping with this result, the median (IQR) pairwise genetic distance between the two analysed sequences was 0 (0–2) substitutions/1000 nucleotides with a complete genetic identity observed in 78.9% (15/19) of patients, supporting the persistence of vaccine escape mutations over time.
In order to better elucidate if vaccine escape mutations result from a positive selective pressure, the extent of genetic variability in HBsAg MHR was compared respect to other HBV genomic regions. Interestingly, the number of mutated residues (Shannon Entropy >0) in MHR was remarkably higher than that observed in the HBx and HBcAg regions (MHR: 78.9% vs HBx: 19.5% [P < 0.0001] and vs HBcAg: 33.9% [P < 0.0001]). Similarly, a higher extent of genetic variability in MHR was also confirmed in comparison to the other HBsAg domains (MHR: 78.9% vs PreS1: 42.9% [P < 0.0001], vs PreS2: 63.6% [P = 0.05] and vs N- and C-terminus HBsAg 65.8% [P = 0.05]). Furthermore, by analysing the MHR region, the extent of genetic variability at each single position associated with vaccine escape mutations was significantly higher than that observed in the other MHR positions (Median Shannon Entropy [IQR]: 0.44 [0.14–0.63] vs 0.08 [0.03–0.24], P-value = 0.006), supporting an enrichment of genetic diversification in this immune active region.
The prevalence of the single vaccine-escape mutations observed in the overall population is reported in (B). Among patients with at least 1 vaccine-escape mutation, P120S, T131N and D144E were the most frequently observed. In particular, 21.4% (36/168) of patients harboured a viral strain with P120S mutation, 15.5% (26/168) with T131N, and 13.1% (22/168) with D144E ((B)). The largely described G145R, well known for its capability to evade humoral response, was revealed in 7.7% (13/168) of patients with vaccine-escape mutations ((B)).
Circulation of complex profiles of vaccine escape mutations
The next step of this study was focused on the circulation of complex mutational profiles, defined as the co-presence of two or more vaccine escape mutations.
Notably, complex mutational profiles were revealed in 3.1% (29/947) of patients with an increasing temporal trend over time. Indeed, the proportion of patients with complex mutational profiles raised from 0.4% (1/237) in 2005–2009 to 3.0% (12/396) in 2010–2014 and until 5.1% (16/314) in 2015–2019 (P = 0.007), suggesting an increased circulation of viral strains endowed with enhanced capability to evade humoral responses (). This datum was confirmed also by multivariable analysis showing that the risk of harbouring complex mutational profiles was 11-fold higher in the time-window 2015–2019 compared to 2005–2009 (11.04[1.42–85.58], P = 0.02) (). Multivariable analysis also showed that older patients are characterized by a higher risk to harbour complex mutational profiles ().
Figure 2. Temporal trend of circulation for complex profiles of vaccine escape mutations. The histogram reports the percentage of patients with complex profiles of vaccine-escape mutations defined as the presence of >2 vaccine escape mutations in the 3 analysed time windows: 2005–2009, 2010–2014, 2015–2019. Statistically significant difference was assessed by Chi-square for trend based on 3 × 2 contingency table.
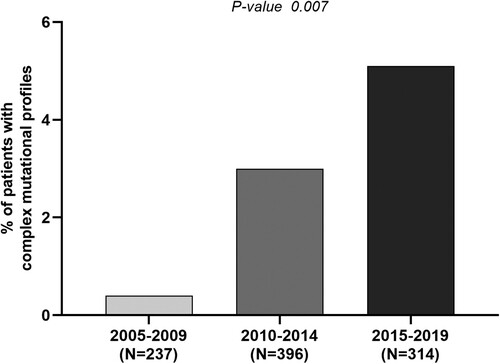
Table 4. Multivariable analysis of parameters associated with the presence of complex mutational profiles.
Impact of complex mutational profiles on virological parameters
By analysing the impact of complex mutational profiles on virological parameters, we observed a significant correlation with lower HBsAg levels: median (IQR) 40(0–2905) IU/mL for patients with complex mutational profiles vs 2078(115–6037) IU/ml for patients with only one vaccine escape mutation (P = 0.02) and vs 1881(410–7622) IU/mL for patients without them (P = 0.0004), suggesting the role of complex mutational profiles in altering HBsAg quantification ((A)).
Figure 3. Virological parameters characterizing patients with complex mutational profiles. Box plot in (A) reports the distribution of serum HBsAg observed in the group of patients with complex profiles of vaccine-escape mutations (N = 29), with a single vaccine-escape mutation (N = 139) and with no vaccine escape mutations (N = 779). Statistically significant differences were assessed by Mann–Whitney Test. The histogram (B) reports the percentage of patients with HBsAg negativity despite detectable HBV-DNA in the afore-mentioned 3 subgroups of patients. Statistically significant differences were assessed by Chi-squared Test. Box plot in (C) reports HBV-DNA levels observed in the groups of patients with complex profiles of vaccine-escape mutations (N = 18), with a single vaccine-escape mutation (N = 55) and with no vaccine escape mutations (N = 353). The analysis has been restricted to drug naïve patients. Statistically significant differences were assessed by Mann–Whitney Test.
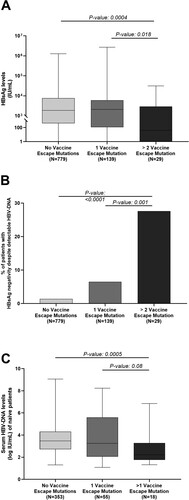
Focusing on HBsAg-negativity, the presence of complex profiles of vaccine-escape mutations also correlated with an HBsAg-negative result despite HBV-DNA positivity. Indeed, 27.6% of patients with >2 vaccine-escape mutations vs 6.5% and 1.4% of those with a single or no vaccine-escape mutations were HBsAg-negative despite active HBV replication (P = 0.001 and <0.0001) ((B)). Interestingly, HBsAg-negativity was strongly associated with the presence of T126I/A in combination with >1 additional vaccine-escape mutation (50% of patients with T126I/A-containing profiles vs 3.3% without them were HBsAg-negative, P < 0.0001). Interestingly, among HBsAg-negative patients, T126I/A frequently co-occurred with P120S and Q129H, suggesting a synergistic effect of mutations in these positions of the major hydrophilic region in altering HBsAg recognition and a potential co-evolutive selection of these mutants for their related immune escape advantage.
Furthermore, in treatment-naïve patients, we also observed a correlation between complex mutational profiles and lower serum HBV-DNA levels: median (IQR) 2.2 (1.8–3.3) logIU/ml for patients with complex mutational profiles compared to 3.2 (2.0–5.6) logIU/ml for patients with one vaccine escape mutation (P = 0.08) and to 3.5 (2.7–4.3) logIU/ml for patients without them (P = 0.0005) ((C)).
Intra-patient prevalence of vaccine escape mutations by NGS
NGS was performed on 32 patients carrying at least one vaccine-escape mutation, for which an additional serum sample was available for further sequencing analyses. No statistically significant differences in demographic and clinical characteristics were found after stratification for NGS availability (Table S2).
Firstly, the presence of all vaccine escape mutations, detected by population-based sequencing, and their co-occurrence in the same viral strain as complex mutational profiles was confirmed by NGS. Notably, NGS revealed that in the majority of patients (75%, 24/32), these vaccine-escape mutations were detected with an intra-patient prevalence higher than 90% (), supporting the full fixation of these mutations in the viral quasispecies.
Figure 4. Intra-patient prevalence vaccine escape mutations according to NGS analysis. The graph reports the intra-patient prevalence of the identified vaccine escape mutations in a subset of the patients, in which HBsAg region was analysed by NGS (N = 32). Intra-patient prevalence was expressed as percent of reads with the specific vaccine escape mutation respect to the total reads obtained for each patient. Mutations under the bottom dotted line in the graph are those with an intra-patient prevalence <20% (minority species) not detected by standard population-based sequencing. Mutation above the upper dotted line in the graph are those with an intra-patient prevalence >90% indicating their full fixation in viral quasispecies.
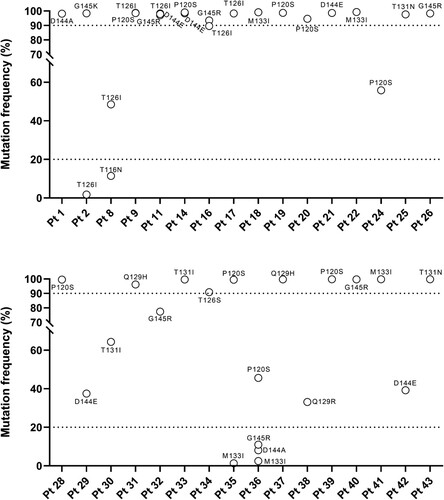
NGS also revealed the presence of four additional complex mutational profiles not detected by Sanger sequencing since their intra-patient prevalence was <20% (limit of mutational detection for Sanger sequencing) (). The evidence of additional complex mutational profiles, revealed only by NGS, suggests that immune pressure can further select additional vaccine-escape mutations during chronic infection, underlining the need to monitor finely their circulation by highly sensitive NGS assays.
Impact of vaccine escape mutations on HBsAg secretion and diagnostic antigenicity
The complex mutational profiles, identified in patients with HBsAg negativity despite detectable HBV-DNA, were tested in vitro in order to evaluate their impact on HBsAg secretion and diagnostic antigenicity, compared to wt and mutated HBsAg carrying each single mutation included in the complex mutational profiles.
By these in vitro experiments, we found that most vaccine escape mutations, either single or in complex profiles, determined > 20% decrease in HBsAg secretion compared to wt. In particular, T116N, D144E and G145R determined a 60%, 58% and 52% decreased secretion (P-values ranging from 0.003 to 0.01), while a 40% decreased HBsAg secretion was observed for the single mutations C139S and T126I as well as for the complex mutational profiles T126I + G145R and T116N + C139S + G145R (P-values = 0.001 for both). A less marked defect in HBsAg secretion was observed for P120S compared to wt (23% P-value = 0.0003) ().
Figure 5. In vitro impact of vaccine escape mutation on HBsAg secretion and diagnostic antigenicity. A plasmid encoding HBsAg linked to a streptavidin-tag version II at N-terminus was used to transfect the Huh7 cells. The amount of strep-tagged HBsAg released in culture supernatants was then quantified using a specifically-designed ELISA capable to recognize the Strep-tag linked to the HBsAg (black bars) and using two commercial assays targeting HBsAg (dark grey for commercial assay 1 and light grey for commercial assay 2). For each mutant, the amount of strep-tagged HBsAg released in supernatants of Huh7 cell cultures was expressed as percentage, considering the amount of the wild-type strep-tagged HBsAg as 100%. Results represent the mean values (+/- standard deviation) of 2 independent experiments, each led in triplicate. * indicates P values ranging from 0.05 to 0.01, ** P values from 0.01 to 0.001 and *** P values <0.001 compared to wild-type. For the underlined complex mutational profiles, HBsAg release was significantly lower than that observed for the single mutation (P120S and G145R, P-value = 0.04 for both). These statistically significant differences were observed only for commercial assay 1.
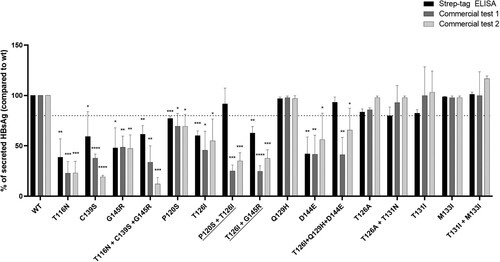
Conversely, the complex mutational profiles P120S + T126I and T126I + Q129H + D144E reduced HBsAg diagnostic antigenicity of 75% and 60% for the commercial assay 1 and 65% and 35% for the commercial assay 2, without altering HBsAg secretion ().
Notably, the complex mutational profiles P120S + T126I and T126I + G145R determined a further reduction in HBsAg quantification by the commercial assay 1 compared to the single P120S and G145R (P-value = 0.04 for both) (). These in vitro results confirmed the role of T126I, when present in complex mutational profiles, in affecting HBsAg diagnostic antigenicity observed in our clinical data.
Finally, the single mutations T126A, Q129H, T131I, M133I as well as the complex mutational profiles T126A + T131N and T131I + M133I did not affect HBsAg secretion and diagnostic antigenicity ().
Discussion
This study, led in one of the largest cohort of patients with HBV genotype D infection, shows that a conspicuous fraction of patients (18%) harboured viral variants with at least one vaccine-escape mutation. By NGS, the intra-patient prevalence of these mutations was >90% indicating their full fixation in viral quasispecies. It is plausible that these mutations are randomly generated due to the error-prone nature of HBV reverse transcriptase and then they are selected and persist over time since they can confer a selective advantage to the virus by favouring viral evasion from immunological pressure. This is supported by analysing the subset of patients with a longitudinal HBsAg-sequence available, confirming the presence of all vaccine escape mutations in the second time point analysed.
Furthermore, the positive selection of HBsAg variants related to vaccine escape is also confirmed by the higher extent of variability of MHR respect to all other HBV genetic regions and by the peculiar enrichment of genetic diversification of this immune active region.
At the same time, the selection of vaccine-escape mutations can play a role in fuelling HBV transmission also to vaccinated individuals, thus playing a critical role in preserving a reservoir of HBV infection. This can be critical particularly to those individuals with waning anti-HBs titres or with a weakened immune response [Citation23]. This concept is in line with a previous study reporting that the frequency of breakthrough HBV infections among vaccinated blood donors was significantly higher when the anti-HBs titres were <100 IU/L [Citation23].
Notably, 3.1% of patients showed complex mutational profiles (defined as the co-presence of two or more vaccine-escape mutations) whose prevalence underwent a progressive increase in a time window of 15 years (2005–2019) (result confirmed by multivariable analysis). To our knowledge, this is the first study evaluating the temporal trend of vaccine-escape mutations in an extended time-window including also recent years. Beyond the random generation of vaccine-escape mutations, it cannot be excluded that the universal vaccination may play a role in accelerating viral genetic evolution towards the accumulation of HBsAg mutations endowed with enhanced capability to evade humoral responses. This issue has been specifically addressed in a previous study led in Taiwan among HBsAg-positive children born before and after the implementation of a nationwide vaccination programme. The authors found that 8 of 103 (7.8%) in 1984, 9 of 46 (19.6%) in 1989, and 5 of 20 (25%) in 1994 harboured ≥1 mutation within HBsAg immune active regions [Citation24], suggesting an enrichment in the circulation of vaccine-escape mutations over time. Likewise, a study led in China in the post-vaccination era showed a progressive increase over time in the prevalence of the immune-escape mutations G145R/A (8.82% in the years 2005–2007, 26.32% in the years 2008–2010 and 44.44 in the years 2011–2013) [Citation25].
By multivariable analysis, an independent factor correlated with the presence of ≥1 vaccine-escape mutation was patients’ age. It is plausible that the positive correlation with age reflects a longer duration of HBV infection during which the virus undergoes a gradual process of genetic diversification to react to the endogenous selective pressure imposed by immune responses. Even more, it cannot be excluded that the status of immune-senescence, that characterizes patients with advanced age, can play a role in favouring the emergence of immune-escape [Citation26–29].
The circulation of viral strains with vaccine-escape mutations, with the potential to be transmitted to vaccinated individuals, highlights the need to develop novel vaccine formulations. In this regard, a recent study has shown that an HBV vaccine consisting of the Large-HBsAg may induce the production of antibodies against the pre-S1 domain that can efficiently neutralize HBV strains with vaccine-escape mutations (including G145R) in a non-human primate model [Citation30]. These findings pave the way for design novel vaccine formulation based on the combination of different forms of HBV surface glycoproteins [Citation30]. In this regard, a phase 3 randomized clinical trial has shown that a vaccine composed by all the different forms of HBsAg (Large, Middle and Small) is endowed by higher immunogenicity and can rapidly elicit higher anti-HBs titres than the currently available vaccine formulations based only on the Small-HbsAg [Citation31–33].
So far, therapeutic vaccination has been proposed as a novel strategy that could contribute to the achievement of HBV cure, by eliciting a robust virus-specific immunity. Therapeutic vaccination aims at mounting the production of neutralizing antibodies and the activation effector CD4 + and CD8+ T cells capable to control viral replication [Citation34]. Despite this rationale, so far the clinical efficacy of this approach has been limited [Citation34]. These results can be explained by a large variety of factors including the presence of vaccine-escape mutations that can jeopardize the full neutralization of viral particles. These aspects should be also taken into account in the studies aimed at evaluating the clinical efficacy recently proposed therapeutic approach based on the administration of monoclonal antibodies against HbsAg [Citation35].
Even more, this study shows that the co-presence of ≥2 vaccine-escape mutations correlates with low HBsAg titres and particularly with HBsAg-negative results despite active HBV replication. These in-vivo findings are in keeping with our in vitro results showing the ability of these mutations in hampering HBsAg secretion or in jeopardizing HBsAg recognition by antibodies used in diagnostic assays. In addition, some complex mutational profiles can further exacerbate the issue of altered HBsAg recognition, supporting that the phenomenon of diagnostic escape can be enhanced by the accumulation of multiple mutations in HBsAg immunodominant regions. Our results are in line with a previous study showing that pluri-mutated strains harbouring both G145R and T126I failed to be detected by different commercial assays for HBsAg screening [Citation36]. HBsAg-negativity despite viral replication can jeopardize a proper HBV diagnosis, thus fuelling the circulation of HBV infection and predisposing to liver disease progression.
So far, HBsAg-negativity is considered the surrogate marker to reflect cccDNA silencing and in turn the achievement of HBV functional cure. The contribution of vaccine-escape mutations to a false-negative HBsAg result highlights the need of integrating multiple HBV biomarkers (such as serum HBV-RNA and HBcrAg) for a proper monitoring of novel therapeutic strategies aimed at HBV functional cure.
Overall, the correlation of complex profiles of vaccine-escape mutations with low HBsAg titres and HBV-DNA levels can lead to a misinterpretation of chronic HBV infection status such as the identification of patients with HBeAg-negative infection [Citation37–39], highlighting the importance to use genotypic testing for a proper virological characterization of patients with HBV infection.
This study has focused on HBV genotype D, one of the most common circulating worldwide. Notably, a recent study showed that the neutralization activity of vaccine-induced antibodies was significantly lower for HBV genotype D than A and B supporting that the genetic backbone of different HBV genotypes can modulate HBV resistance to neutralizing antibodies [Citation30]. This is particularly relevant considering that the current HBV vaccines are based on the administration of HBsAg derived from genotype A2, circulating only in North Europe and North America. In this regard, previous studies have shown that vaccinated individuals (particularly those with low or waning anti-HBs titres) are characterized by an increased risk of acquiring HBV infection with genotypes genetically distant from A2 [Citation3,Citation23,Citation40,Citation41].
In our study, at least 1 vaccine-escape mutation was detected more frequently in patients infected with sub-genotype D3 than in the other sub-genotypes, highlighting the importance to consider also the neutralization efficacy according to different sub-genotypes.
We acknowledge that to fully verify if the increasing trend observed for vaccine-escape mutations is due to the vaccination, there is the need to analyse over a prolonged time-window a large cohort of individuals get infected with HBV despite full dosage of anti-HBV vaccination with a plasma sample available to perform HBsAg sequencing. Further ad hoc designed studies are necessary to address this issue.
In conclusions, at least 1 vaccine-escape mutation, single or in complex profiles, circulates in a not negligible fraction of HBV genotype-D infected patients with an increasing trend over time supporting a progressive enrichment in the circulation of variants able to evade humoral responses. Even more, complex profiles of vaccine-escape mutations correlate with lower HBsAg quantification and HBsAg-negativity despite ongoing viral replication. This should be considered for a proper clinical interpretation of HBsAg results and their circulation should be taken into account for the development of novel vaccine formulations for both prophylactic and therapeutic purposes aimed at achieving HBV functional cure. This is critical for ensuring the long-term success of global immunization programme and for the achievement of WHO commitment on the elimination of viral hepatitis as a public global threat by 2030.
Supplemental Material
Download Zip (48.8 KB)Acknowledgements
We thank Ilaria Maugliani for data management. We also thank Dr. Manuela Magnanti, Dr. Tara Mayte Suarez Viguria, Dr. Elisa Basile and Dr. Janet Dianetti for technical support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- WHO. Global progress report on HIV, viral sexually transmitted infections. World Health Organization; 2021 [cited 2022 Oct 21]. Available from: https://www.who.int/publications/i/item/9789240027077.
- Thomas DL. Global elimination of chronic hepatitis. N Engl J Med. 2019;380:2041–2050.
- Glebe D, Goldmann N, Lauber C, et al. HBV evolution and genetic variability: impact on prevention, treatment and development of antivirals. Antiviral Res. 2021;186:104973. doi:10.1016/J.ANTIVIRAL.2020.104973.
- Candotti D, Assennato SM, Laperche S, et al. Multiple HBV transfusion transmissions from undetected occult infections: revising the minimal infectious dose. Gut. 2019;68:313–321.
- Tong S, Revill P. Overview of hepatitis B viral replication and genetic variability. J Hepatol. 2016;64:S4–S16.
- Heermann KH, Goldmann U, Schwartz W, et al. Large surface proteins of hepatitis B virus containing the pre-s sequence. J Virol. 1984;52:396–402.
- Glebe D, Bremer C. The molecular virology of hepatitis B virus. Semin Liver Dis. 2013;33:103–112.
- Ghany MG, Ayola B, Villamil FG, et al. Hepatitis B virus S mutants in liver transplant recipients who were reinfected despite hepatitis B immune globulin prophylaxis. Hepatology. 1998;27:213–222.
- Carman WF, Karayiannis P, Waters J, et al. Vaccine-induced escape mutant of hepatitis B virus. Lancet. 1990;336:325–329.
- Lazarevic I. Clinical implications of hepatitis B virus mutations: recent advances. World J Gastroenterol. 2014;20:7653–7664.
- Coppola N, Loquercio G, Tonziello G, et al. HBV transmission from an occult carrier with five mutations in the major hydrophilic region of HBsAg to an immunosuppressed plasma recipient. J Clin Virol. 2013;58:315–317.
- Echevarría JM, Avellón A. Hepatitis B virus genetic diversity. J Med Virol. 2006;78(Suppl 1):S36–42. doi:10.1002/JMV.20605.
- World Health Organization. Combating hepatitis B and C to reach elimination by 2030: advocacy brief; 2016 [cited 2022 Oct 28]. Available from: https://apps.who.int/iris/handle/10665/206453.
- Yip TCF, Wong VWS, Wong GLH. Alanine aminotransferase level: the road to normal in 2021. Hepatol Commun. 2021;5:1807–1809.
- Valenti L, Pelusi S, Bianco C, et al. Definition of healthy ranges for alanine aminotransferase levels: a 2021 update. Hepatol Commun. 2021;5:1824–1832.
- Salpini R, Alteri C, Cento V, et al. Snapshot on drug-esistance rate and profiles in patients with chronic hepatitis B receiving nucleos(t)ide analogues in clinical practice. J Med Virol. 2013;85:996–1004.
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. doi:10.1093/oxfordjournals.molbev.a040454.
- Tamura K, Stecher G, Peterson D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729.
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–26. doi:10.1093/oxfordjournals.molbev.a040023.
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30:2114.
- Salpini R, Piermatteo L, Battisti A, et al. A hyper-glycosylation of HBV surface antigen characterizes immunosuppression-driven HBV reactivation and hinders HBsAg recognition in vitro. J Hepatol. 2018;68:S765–S766.
- Salpini R, Battisti A, Piermatteo L, et al. Key mutations in the C-terminus of the HBV surface glycoprotein correlate with lower HBsAg levels in vivo, hinder HBsAg secretion in vitro and reduce HBsAg structural stability in the setting of HBeAg-negative chronic HBV genotype-D infection. Emerg Microbes Infect. 2020;9:928–939.
- Stramer SL, Wend U, Candotti D, et al. Nucleic acid testing to detect HBV infection in blood donors. N Engl J Med. 2011;364:236–247.
- Hsu HY, Chang MH, Liaw SH, et al. Changes of hepatitis B surface antigen variants in carrier children before and after universal vaccination in Taiwan. Hepatology. 1999;30:1312–1317.
- Yan B, Lv J, Feng Y, et al. Temporal trend of hepatitis B surface mutations in the post-immunization period: 9 years of surveillance (2005-2013) in eastern China. Sci Rep. 2017;7:6669. doi:10.1038/S41598-017-07085-Z.
- Tsutsumi Y, Yamamoto Y, Shimono J, et al. Hepatitis B virus reactivation with rituximab-containing regimen. World J Hepatol. 2013;5:612.
- Martel N, Cotte L, Trabaud MA, et al. Probable corticosteroid-induced reactivation of latent hepatitis B virus infection in an HIV-positive patient involving immune escape. J Infect Dis. 2012;205:1757–1761.
- Ceccarelli L, Salpini R, Sarmati L, et al. Late hepatitis B virus reactivation after lamivudine prophylaxis interruption in an anti-HBs-positive and anti-HBc-negative patient treated with rituximab-containing therapy. J Infect. 2012;65:180–183.
- Milazzo L, Ebranati E, Cattaneo D, et al. Recurrence of another hepatitis B virus escape mutant comes back in a patient infected with HIV and low CD4 + count. J Med Virol. 2014;86:97–101.
- Washizaki A, Murayama A, Murata M, et al. Neutralization of hepatitis B virus with vaccine-escape mutations by hepatitis B vaccine with large-HBs antigen. Nat Commun. 2022;13:1–12.
- Diaz-Mitoma F, Popovic V, Spaans JN. Assessment of immunogenicity and safety across two manufacturing lots of a 3-antigen hepatitis B vaccine, Sci-B-Vac®, compared with engerix-B® in healthy Asian adults: a phase 3 randomized clinical trial. Vaccine. 2021;39:3892–3899.
- Vesikari T, Langley JM, Segall N, et al. Immunogenicity and safety of a tri-antigenic versus a mono-antigenic hepatitis B vaccine in adults (PROTECT): a randomised, double-blind, phase 3 trial. Lancet Infect Dis. 2021;21:1271–1281.
- Vesikari T, Finn A, van Damme P, et al. Immunogenicity and safety of a 3-antigen hepatitis B vaccine vs a single-antigen hepatitis B vaccine: a phase 3 randomized clinical trial. JAMA Netw Open. 2021;4:e2128652. doi:10.1001/JAMANETWORKOPEN.2021.28652.
- Knolle PA, Huang LR, Kosinska A, et al. Improving therapeutic vaccination against hepatitis B—insights from preclinical models of immune therapy against persistent hepatitis B virus infection. Vaccines (Basel). 2021;9:1333. doi:10.3390/VACCINES9111333.
- Fung S, Choi HSJ, Gehring A, et al. Getting to HBV cure: the promising paths forward. Hepatology. 2022;76:233–250.
- Sticchi L, Caligiuri P, Cacciani R, et al. Epidemiology of HBV S-gene mutants in the Liguria Region. Italy. 2013;9:568–571. doi:10.4161/hv.23236.
- Cornberg M, Wong VW-SS, Locarnini S, et al. The role of quantitative hepatitis B surface antigen revisited. J Hepatol. 2017;66:398–411.
- Höner zu Siederdissen C, Cornberg M. The role of HBsAg levels in the current management of chronic HBV infection; 2014 [cited 2018 Aug 31]. Available from: www.annalsgastro.gr.
- Lampertico P, Agarwal K, Berg T, et al. EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67:370–398.
- Luongo M, Critelli R, Grottola A, et al. Acute hepatitis B caused by a vaccine-escape HBV strain in vaccinated subject: sequence analysis and therapeutic strategy. J Clin Virol. 2015;62:89–91.
- Tacke F, Amini-Bavil-Olyaee S, Heim A, et al. Acute hepatitis B virus infection by genotype F despite successful vaccination in an immune-competent German patient. J Clin Virol. 2007;38:353–357.