
Figures & data
Table 1. Summary of human prion disorders, their prevalence and main histo-molecular features.
Figure 1. Schematic representation of the putative mechanisms of PrP aggregate formation and clearance and mechanism of action of different anti-prion compounds. After synthesis, the nascent PrPC enters the lumen of the ER, where the N-terminal signal peptide is removed. Then, the protein moves to the Golgi apparatus to undergo post-translational modifications. Once fully folded, PrPC moves along the secretory pathway to the outer leaflet of the plasma membrane, where it anchors via the C-terminal GPI moiety to lipid rafts. Both cell surface and endosomal pathways are putative sites of PrPC misfolding and oligomerization. Endocytosed PrPC and misfolded PrP are rapidly recycled into the plasma membrane or removed through macroautophagy following the extracellular release of exosomes. Internalized aggregates promote the ER stress resulting in the activation of UPR and ERAD response, which, in turn, induce the degradation of misfolded PrP by the UPS system. Red solid lines indicate potential therapeutic intervention points, including ASOs targeting PrP mRNA, prion clearance (antibody therapy), prion replication (PPS and Quinacrine), and oligomers stabilization (Doxycycline). Blue arrows and dotted lines represent new potential sites of intervention (UPR and ERAD response enhancement; exosome blockade). Black solid arrows indicate molecular steps (endocytosis of PrPC; conversion of PrPC to PrPSc; macroautophagy pathways). Red solid arrows indicate the cascade triggered by the accumulation of PrP aggregates.
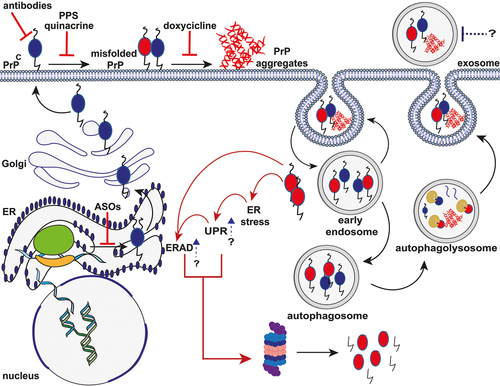
Table 2. Summary of anti-prion molecules tested by clinical studies on human prion diseases.