Figures & data
Table 1. Key VFs of K. pneumoniae involved in the immune evasion.
Figure 1. Cell signalling associated with K. pneumoniae induced autophagy. (A) Autophagy is required for TRPM2-AMPK-p38- and Mincle-mediated NETs formation in a ROS-dependent and an independent manner, respectively. In the ROS-dependent pathway, TRPM2 can sense ROS through AMPK-p38-autophagy to promote NETs formation during K. pneumoniae infection; In a ROS-independent manner, Mincle-mediated activation of autophagy, occurring downstream of mTOR, is required for NETs formation in response to pneumonic infection, and this autophagy activation precedes ROS effect on NETs formation; (B) The activated TLR2 receptor through Lyn promotes the fusion of autophagosome and lysosome, which enhances autophagy; (C) K. pneumoniae enters macrophage through endocytosis, which needs PI3K activation. The activated PI3K phosphorylate AKT regulates the recruitment of Rab14 to the phagosome, and manipulates phagosome maturation; (D) Atg7 is responsible for the elongation of autophagosome membrane. During K. pneumoniae infection, Atg7 can interact with phosphorylated IκBα to inhibit IκBα ubiquitination and reduce the activation of NF-κB signalling pathway and inflammation activation.
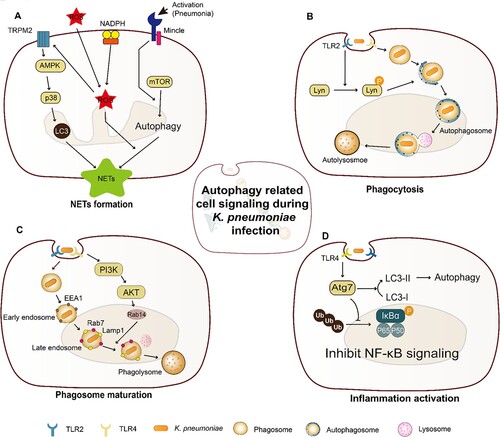
Table 2. Strategies and mechanism involved in counteracting autophagy and cell death trigged by K. pneumoniae.
Figure 2. Cell death triggered by K. pneumoniae in neutrophils. K. pneumoniae modulates the anti-apoptotic effects by downregulating the ratio of Bax to Bcl-2 and upregulating the expression of Mcl-1, resulting in the delay of Caspase 3/7 activation and the inhibition of apoptosis. K. pneumoniae also can increase the activity of flippase to reduce PS exposure, which is the signal of “eat me” for efferocytosis, followed by decreasing the efferocytic uptake of neutrophils by macrophage. In parallel, K. pneumoniae inhibits caspase 8, which promotes the formation of necroptosome complex RIPK1/RIPK3/MLKL to induce necroptosis. Necroptosis emits K. pneumoniae to the external environment, and decreases efferocytosis. (Created with BioRender. com).
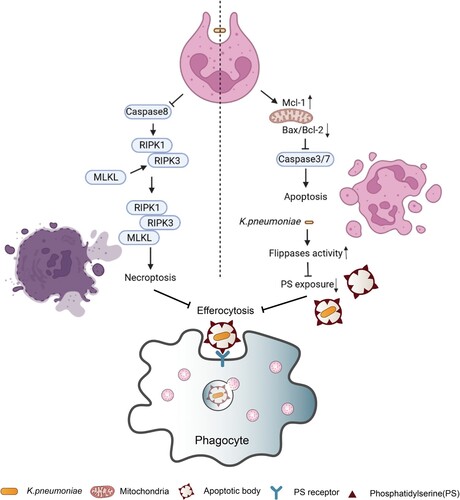
Figure 3. Pyrotosis triggered by K. pneumoniae infection. TLR4 receptor can recognize K. pneumoniae LPS and K1-CPS, and activate NLRP3 inflammasome and caspase-11. Inflammasome formation allows caspase-1 activation, resulting in mature IL-1β and IL-18 release by cleavage of pro-IL-1β and pro-IL-18, respectively. Subsequently, pore formation and release of intracellular soluble components, such as LDH and HMGB, is culminated by the induction of pyroptosis/pyronecrosis. IL-1β together with other mediators recruit new cells to the infection site, promoting efferocytosis of pyroptotic-infected cells and bacterial clearance. But this progress can be inhibited by IL-10. Simultaneously, K. pneumoniae can activate NLRC4 to promote caspase1 activation, and further increase the release of IL-1β and IL-18. NLRC4 is also associated with IL-17 releasing, which was independent with caspase1. The released IL-1β through the interaction with its receptor IL-1R promotes the secretion of ICAM-1 and VCAM-1, and further promotes neutrophil recruitment.
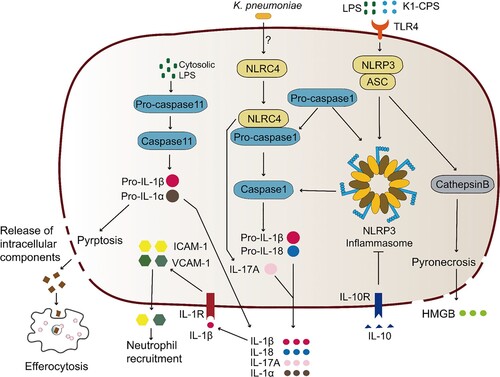
Table 3. The function of cytokines coping with K. pneumoniae infection.
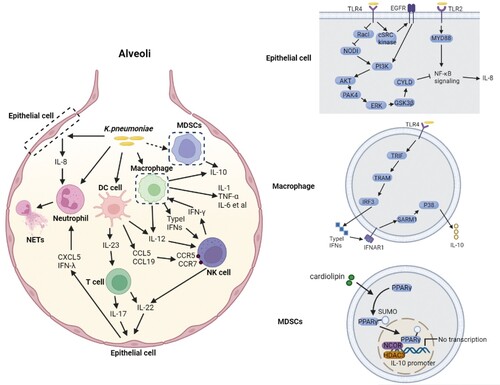
Figure 4. Complex network of cytokines coping with K. pneumoniae infection. K. pneumoniae can directly interact with macrophages, neutrophils, DC cells and epithelial cells in alveoli (left panel). The macrophage is the major immune cell in alveoli, and can produce type I IFNs upon infection with K. pneumoniae, resulting in NK cell migration and IFN-γ production. IFN-γ further activates the macrophage anti-microbial armament; both macrophages and DC cells can produce IL-12, which is required for IFN-γ production. IFN-γ production can be additionally promoted by K. pneumoniae matured DC cells through the interaction of CCL5-CCR5 receptor and CCL19-CCR7 receptor on NK cells. K. pneumoniae-infected DC cells secrete IL-23 which further induces IL-17 production by T cells to promote bacterial clearance. IL-17 promotes CXCL5 production through IL-17R on the epithelial cells to induce the recruitment of neutrophils. The recruitment of neutrophils can also be promoted by IL-8 and IFN-λ secreted by K. pneumoniae infected epithelial cells. Neutrophils are involved in the elimination of K. pneumoniae through NETs formation. IL-22 interacts with IFN-λ to help maintain the integrity of epithelial barrier to inhibit neutrophil transmigration and microbial invasion. The MDSCs produced IL-10 also has a crucial role in K. pneumoniae clearance. The synergy of these cytokines constructs the complex network of host defense against K. pneumoniae. The detailed signalling pathways of IL-8 and IL-10 production upon K. pneumoniae infection are shown on the right panel. (Created with BioRender. com).