Figures & data
Figure 4. Molecular mechanisms of autophagy In macroautophagy, the cargo is sequestered within a unique double membrane cytosolic vesicle, an autophagosome. The autophagosome itself is formed by expansion of the phagophore. The autophagosome undergoes fusion with a late endosome or lysosome to form an autolysosome, in which the sequestered material is degraded. Microautophagy refers to the sequestration of cytosolic components directly by lysosomes through invaginations within their limiting membrane. Chaperone-mediated autophagy involves direct translocation of unfolded substrate proteins (KFERQ-like motif) across the lysosome membrane through the action of a cytosolic and lysosomal chaperone heat shock cognate protein of 70 kDa (HSC70), and the integral membrane receptor lysosome-associated membrane protein type 2A (LAMP-2A). Mitochondria can be removed by receptor mediated mitophagy using NIX, BNIP3, or FUNDC1, which interact with LC3; or via PTEN-induced putative kinase 1 (Pink1) and Parkin-mediated ubiquitination (Ub) of voltage-dependent anion channel 1 VDAC1), recognized by the adapter p62 for removal of stressed.
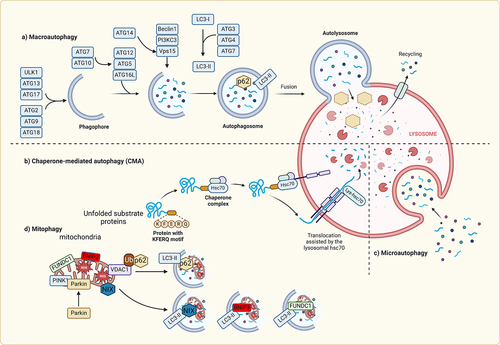
Figure 1. Signaling pathways activated by DAMPs All immunogenic nucleic acids bind cytosolic DNA sensors or RNA sensors, including retinoid acid-inducible gene I (RIG-I)-like receptors (RLRs), which are required for subsequent recognition by specific pattern recognition receptors to activate innate immune responses. DAMPs such as HMGB1, S100 proteins (S100s), and heat shock proteins (HSPs) recognize the receptor for advanced glycation end products (RAGE), TLR4, or triggering receptor expressed on myeloid cells-1 (TREM-1) and activate the MyD88-MAPK-NF-jB pathway. HMGB1 and RAGE activate the TLR9-MyD88 dependent pathway, which contributes to autoimmune pathogenesis. CD24 is a negative receptor to inhibit the DAMP-induced TLR4 pathway. ATP binding of the P2X7 receptor and uric acid, as well as asbestos and alum, increase activation of caspase-1 by the NLRP3 inflammasome to promote secretion of IL-1β and IL-18.
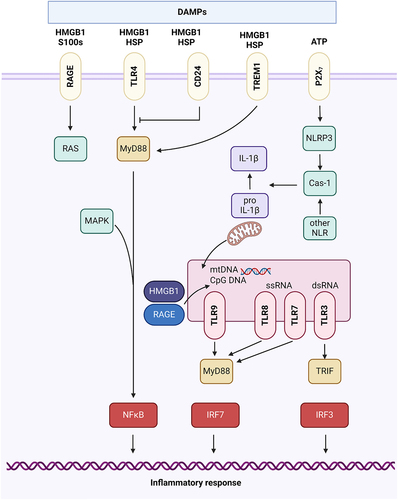
Figure 2. Innate immune activation contributes to the progression of heart failure Upon injury within the myocardium, the innate immune system is activated by various factors such as PAMPs and DAMPs. Activation of the innate immune system is mediated by signaling via NLRs, TLRs, and CLRs to mediate downstream proinflammatory effects such as production of proinflammatory cytokines and chemokines, inflammasome assembly, and promote immune cell infiltration into cardiac tissue, thus contributing to the pathogenesis of heart failure. Within the ischemic heart, DAMPs are known to activate TLR4-NFkB signaling pathway to promote production of specific cytokines leading to sterile inflammation in the heart. Casp-1, caspase-1; CLR, C-type lectin receptor; DAMP, damage-associated molecular pattern; ECM, extracellular matrix; HF, heart failure; HSP, heat-shock protein; IL, interleukin; NLR, NOD-like receptor; PAMP, pathogen-associated molecular pattern; TLR, Toll-like receptor; TNF, tumor necrosis factor.
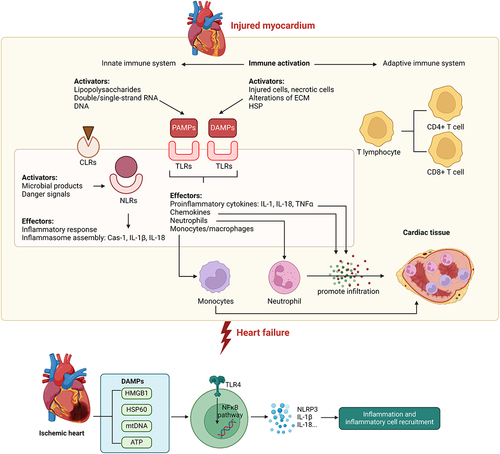
Figure 3. Progression from myocardial infarction to heart failure. Myocardial infarction leads to the death of heart muscle cells, causing the release of DAMPs and the production of pro-inflammatory substances. These substances attract immune cells, which infiltrate the damaged area and clear away cellular debris. As a response to cell death caused by the lack of blood flow and subsequent restoration, the heart initiates remodeling and fibrosis to compensate. In the resolution phase, the inflammation is reduced by anti-inflammatory cytokines, but a low-level chronic inflammation persists. The number of cardiomyocytes decreases due to myocardial necrosis, but their size increases as a result of increased mechanical strain and compensatory hypertrophy.
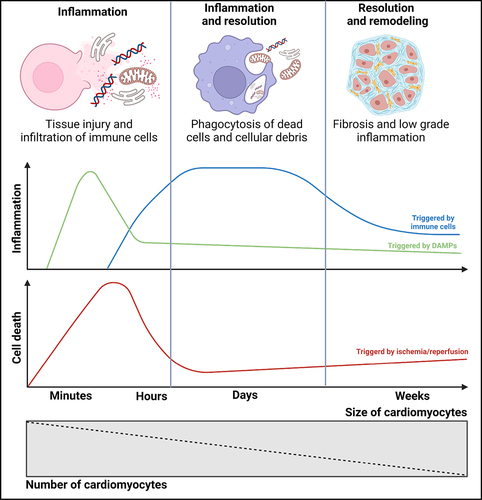
Figure 5. Targeting autophagy and inflammation in heart failure (A) Adiponectin is a cytokine that can affect both autophagy and inflammation. Adiponectin is positively correlated with autophagy upregulation. Adiponectin exhibits anti-inflammatory effects by inhibiting TLR-NF-kB signaling and promoting alternative macrophage polarization. (B) Activation of AMPK, such as via PXL770, upregulates autophagy, mitophagy, and exerts anti-inflammatory effects. These processes collectively provide protection against the development of heart failure.
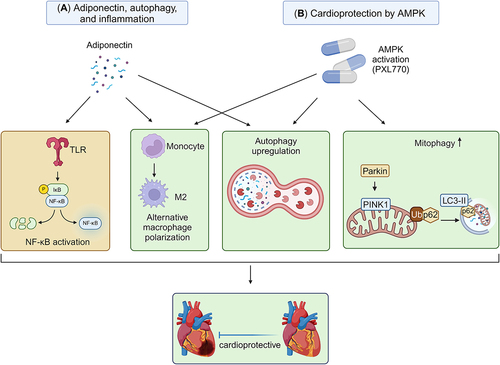
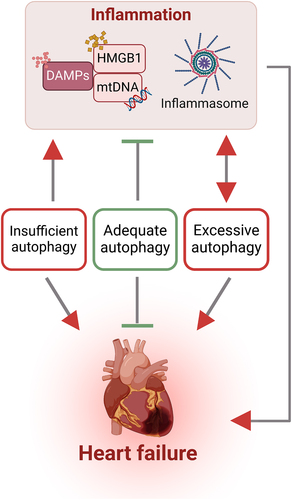
Figure 6. Interaction between autophagy and inflammation in heart failure The intricate interplay between sterile inflammation and autophagy is a central aspect of heart failure pathogenesis. This association encompasses the involvement of DAMPs (exemplified by mtDNA and HMGB1) and activation of inflammasomes which can be modulated by autophagy. Furthermore, the activation of inflammasomes results in amplification of autophagic activity to mitigate cellular damage from various stressors. Collectively, various forms of autophagy at the appropriate time and magnitude can be cardioprotective, whilst too much or too little are associated with worsening of adverse events.