
Abstract
Algae-derived protein has immense potential to provide high-quality protein foods for the expanding human population. To meet its potential, a broad range of scientific tools are required to identify optimal algal strains from the hundreds of thousands available and identify ideal growing conditions for strains that produce high-quality protein with functional benefits. A research pipeline that includes proteomics can provide a deeper interpretation of microalgal composition and biochemistry in the pursuit of these goals. To date, proteomic investigations have largely focused on pathways that involve lipid production in selected microalgae species. Herein, we report the current state of microalgal proteome measurement and discuss promising approaches for the development of protein-containing food products derived from algae.
Graphical Abstract

Background/introduction
Microalgae are a diverse group of microorganisms that contain essential nutrients suitable for the human diet. These include proteins, dietary fiber, polyunsaturated fatty acids, antioxidants, and bioactive compounds [Citation1]. Some microalgae species devote a large proportion of their biomass to protein (e.g., 66%), which make them a potentially ideal complementary protein source for human nutrition [Citation2–4]. In addition, microalgae growth and production have significant environmental advantages compared to animal and plant-based production practices. The rapid growth rate of microalgae ensures their high biomass production, concomitant with increased protein yield [Citation5]. Furthermore, microalgae do not need fertile land to produce protein in comparison to plant-based and animal-based products, as non-arable land can be used for their production [Citation6].
Although microalgae have been commercially cultured for more than 50 years [Citation7], some varieties, such as Nostoc, Arthrospira (Spirulina), and Aphanizomenon, have been consumed by people for thousands of years [Citation8]. Proteins derived from algae are of particular interest to meet the growing demand for dietary protein, which underpins an expectation of substantial market growth for future commercial microalgae production. Currently, microalgae-based alternative protein products are produced by several biotechnology companies, which focus on only a few varieties, such as: Chlorella vulgaris, Arthrospira sp., Nannochloropsis sp., and Dunaliella sp. Yet, there remain hundreds of thousands of microalgae species [Citation9], wherein little is known about their protein content, nutritional quality, and production characteristics.
Crude protein content varies markedly for different microalgae strains, ranging from 6 to 66% [Citation3,Citation4] and can be enhanced by modifying growing conditions. In addition, genetic engineering can be used to develop microalgae strains with specific traits that are favorable for industrial scale production, such as: rapid growth, tolerance to abiotic stresses [Citation10,Citation11], and/or specific nutritional attributes, such as higher levels of specific essential amino acids (EAAs). To this end, an in-depth understanding of the molecular mechanisms that underpin microalgae biochemistry is required, which can be accomplished by applying omic approaches, such as: genomics, transcriptomics, proteomics, and metabolomics. Critically, proteomics offers key information that can inform nutritional quality not offered by other omic measurements.
The definition of proteomics now extends beyond the identification of all proteins in a particular cell or cell compartment; it includes the analysis of post-genomic events in cells, including: characterization of protein isoforms, post-translational modifications (PTMs) of proteins, protein–protein interactions, protein structures, and protein complexes [Citation12]. Proteomics can aid genomic studies as it enables us to achieve a comprehensive understanding of metabolic pathways, and, in turn, the behavior of microalgae in response to its surrounding environmental conditions. Conversely, genomics is able to complement proteomics through information acquired by complete genome sequencing [Citation13]. However, genomic data are only available for a limited number of the thousands of known microalgae species and functional understanding of these genes is also under-represented [Citation10]. To address this, there is an important need to utilize omic technologies to broaden our understanding of a diverse range of microalgae species/strains, especially those that show promise for applications in food production.
Proteomics can provide valuable information regarding the optimal growth conditions for algal protein productivity, protein quality for human nutrition, and food health and safety. Researchers aim to track the protein complement across growth conditions to identify optimal parameters. In terms of quality and safety, proteomics also enables the identification of peptides that are liberated during passage through the human digestive tract. These measurements provide key data to support assessments regarding functional benefits or health concerns, e.g., allergens.
With a critical mass of information regarding algal proteomics and market interest in complementary protein sources, this review seeks to: (a) evaluate a range of methods that can be used to study the algal proteome; (b) summarize current knowledge regarding the algal proteome; and, (c) identify important research approaches utilizing proteomic methods that will help identify algal species that are best suited to the production of high-quality protein ingredients for the human diet.
Protein extraction methods
A majority of microalgae species and strains have a rigid cell wall that limits the accessibility of protein molecules, thus species-specific extraction methods are often required. A key consideration is the variation in cell wall structure, which can be comprised of different polysaccharides, such as: cellulose, hemicellulose, pectin, and xylan; monosaccharides like mannose and uronic acid (acidic sugar); and polymers, e.g., sporopollenin and algaenan [Citation14–16]. These rigid cell walls act as a physical barrier that can prevent digestive enzymes from accessing cell contents, thus inhibiting digestibility and nutrient availability. Accordingly, effective cell disruption methods are essential for food applications and enable the assessment of protein complement and the discovery of the putative peptides when these foods are digested as part of the diet. In addition, more intensive extraction methods featuring detergents or organic solvents are often used to provide further insight into the mechanisms by which algal protein yield and quality can be optimized.
Food-grade protein extraction methods
Until now most microalgal proteomic studies report the use of extraction techniques that are not food safe. For food applications, protein extraction methods must meet safety regulations, which limits the use of a broad range of chemicals that are highly effective in disrupting the tough cell wall of many microalgae species/strains, thereby necessitating tailored approaches. For instance, species such as Haematococcus pluvialis has a very rigid cell wall and would require more intensive cell disruption than a species like Porphyridium cruentum which has a very weak cell wall [Citation17]. In defining the protein extraction method, consideration must also be given to protein solubility and energy inputs to achieve a cost-effective process [Citation18]. A variety of disruption methods have been used to break the microalgae cell wall so that there is greater area for interaction between the soluble proteins and solvent. These disruption methods include: physical, mechanical, chemical, and biological procedures. For example, the combinations of multiple methods have been used for Chlorella pyrenoidosa and Nannochloropsis gaditana, including enzyme digestion, homogenization, and ultrasonication [Citation17,Citation19].
It has been reported that mechanical procedures, such as bead milling and high-pressure homogenization (HPH), can recover microalgal protein more efficiently than physical methods such as thermal treatment, sonication, and pulsed electric field (PEF) [Citation17,Citation20,Citation21]. Of these methods, HPH has been characterized as the most effective method for algal disruption compared to physical and chemical processes, while also representing reasonable energy costs [Citation14,Citation21]. Despite the high efficiency of such a disruption procedure to recover soluble proteins, the shear force should be chosen carefully as excessive force can degrade some proteins and negatively affect the functionality of others [Citation22]. Enzymes can serve as a viable alternative to mechanical techniques to weaken the cell walls in cases where the biomolecule is sensitive to the pressure and speed forces [Citation23]. Regardless of the effectiveness of choosing a cell disruption method and extraction method, industrial scale-up of algal protein production will require the energy cost of the technique per unit of extracted protein to be carefully assessed as highlighted by Safi et al. [Citation17]. Despite the low energy requirement of enzymatic hydrolysis, the considerable enzyme costs could pose limitations on the large-scale production of proteins within industrial settings. Using chemical procedures such as salting-out is considered as another safe method, as salts such as ammonium sulfate are subsequently removed by ultrafiltration.
The use of different methods to extract protein from microalgae has been reviewed recently [Citation18,Citation24]; however, the main comparisons were made based on total protein extracted, using estimates of total nitrogen content as a proxy protein measurement (Kjeldahl or Dumas methods) [Citation25]. Yet, measures of protein quality – including: EAA content, amino acid and protein digestibility and bioavailability and/or peptides functionality – are often overlooked. Only a small number of studies have reported measures of protein quality [Citation14,Citation17,Citation20,Citation26], highlighting a substantial knowledge gap that needs addressing and readily addressable using high quality proteome measurements.
The bottom-up proteomic approach is hitherto under-represented in algal research, particularly with regard to food-grade protein extraction. To understand fatty acids biosynthesis in Chlorella vulgaris, proteins were extracted using Milli-Q water and digested for a bottom-up proteome measurement [Citation27]. Other studies have measured the algal peptidome from food-grade extraction methods to analyze the bio-active peptides in microalgae species. In this regard, a peptidomics study of Auxenochlorella pyrenoidosa used a food-grade protein extraction, achieved by using HPH on microalgae-water slurry [Citation28]. In another peptidomics study on Arthrospira maxima, the combination of freeze–thaw cycles, ultrasonication and homogenization was used to extract algal proteins [Citation29]. While limited in their abundance, these studies show the utility of bottom-up proteome measurement when applied to food safe protein extractions.
Deep eutectic solvents (DESs) are another efficient and green method to extract proteins for the food industry. DES are generally composed of a quaternary ammonium salt as a hydrogen bond acceptor (such a choline chloride) and a hydrogen bond donor (such as glycerol) with a melting point lower than its constitutive components [Citation30]. Although this method is widely used for plant seed protein extraction [Citation31], it has not been considered for microalgal protein extraction. As such, DES may represent an interesting opportunity for microalgae food-grade protein extraction.
The application of proteome measurement to the study of edible extracted proteins from microalgae can support the identification of optimal processing methods that not only increase protein quantity but also identify proteins/peptides with nutritional and functional benefits for human health and/or identify potential allergens [Citation32,Citation33]. Consequently, while currently limited in its application, proteomics has great potential to offer a valuable tool – along with other established screening tests – to assess the quality and safety of algal protein.
Non-food-grade protein extraction methods
Non-food-grade protein extraction methods use a broad range of extraction buffers and solvents, such as: osmolytes (e.g., glycerol), detergents (e.g., sodium dodecyl sulfate, SDS), reducing agents (e.g., 2-mercaptoethanol; dithiothreitol, DTT), and denaturing reagents (e.g., urea). The selection of extraction buffer is crucial in proteomics as it effects the physicochemical and functional properties of proteins for subsequent measurement [Citation32,Citation34]. In order to obtain the most effective protein extraction, some mechanical procedures, such as ultrasonication, are used in conjunction with extraction buffers [Citation35]. The composition of several different buffers has been used to extract protein from microalgae. The two most used methods involve either precipitation and resolubilization in lysis buffer after removing unwanted components; or solubilization in lysis buffer before precipitation and subsequent resolubilization in lysis buffer or solubilized in buffered detergent following by filtering to remove small molecules. Supplementary Table S1 presents a summary of extraction methods used in microalgal proteomic research along with other information related to the respective proteomic approach. This information has been collated from published results since 2017 in order to cover contemporary efforts to measure microalgal proteomes.
Protein digestion and peptide clean-up
Bottom-up proteomics relies on protein identification through the analysis of peptides resulting from enzyme digests. The protein mixture in the sample is initially digested using proteolytic enzymes to obtain peptides to enable their analysis using mass spectrometry. Trypsin is the most commonly reported protease for protein digestion in mass spectrometry-based proteomics. However, trypsin is limited in that it does not cover the entire proteome as its access to cleavage sites can be constrained by tightly folded proteins, or hindered by PTMs, e.g., acetylated K or presence of glycans [Citation36,Citation37]. As such, alternative proteases may enable a more comprehensive assessment of the microalgal proteome. Such alternative proteolytic enzymes may include: Lys-C, Glu-C, Lys-N, Asp-N, and chymotrypsin; and nonspecific proteases, e.g., proteinase K, and elastase [Citation38,Citation39].
The choice of the proteolytic enzyme in algal proteomics relies on the abundance of the amino acids targeted by each enzyme. Arginine has been reported as a highly abundant amino acid in the protein sequences of microalgae [Citation40]; and consequently, may affect the mass spectrometric identification of tryptic peptides. The high content of acidic amino acids, i.e., aspartic and glutamic acid in cyanobacteria [Citation40], such as Spirulina, may suggest considering the use of Asp-N or Glu-C along with trypsin for protein digestion. Most algal proteomic studies have performed protein digestion using trypsin, nevertheless, a few studies have used the trypsin/Lys-C combination in Dunaliella sp [Citation41], and sequential-digestion with Lys-C and trypsin in Chlamydomonas reinhardtii [Citation42,Citation43].
In microalgal proteomic studies, the most commonly used protein digestion method is performed in-solution using a single microtube after reduction and alkylation of proteins Supplementary (Table S1). After liberating peptides using this approach, peptide clean-up or peptide desalting is required. Peptide clean-up is accomplished by removing salts and buffers using different types of reversed phase resins, such as: C18, C8 or styrene-divinylbenzene resin (SDB), in stop-and-go-extraction tip (StageTip) or micro spin column formats [Citation44]. Other standard approaches to sample preparation such as filter-aided sample preparation (FASP) have been less favored to date but are popular among the wider proteomics community.
Several novel protein digestion methods have been developed to process protein samples more rapidly and efficiently than the conventional processing methods. Suspension trapping (S-Trap) – the filter-based method- and phase-enhanced sample-preparation (SP3) – the paramagnetic bead-based approach are two examples of these recent processing methods [Citation45,Citation46]. The processing time required for centrifugation cycles in S-Trap, also available in 96-well filter plate, is reduced to less than 15 min [Citation46].
Despite the increased adoption of in-solution and filter-based digestion methods in proteomic studies, most studies on algae continue to use in-gel digestion methods (Supplementary Table S1). Since 2017, 10% of studies that reported algal proteomics used two-dimensional gel electrophoresis (2-DE), which is considered a deprecated methodology due to its limited sensitivity to identify less abundant proteins, difficulties in the identification of non-water-soluble proteins such as membrane proteins, and poor representation of proteins of high or low molecular mass or isoelectric point [Citation13,Citation47]. More advanced methods of protein digestion such as S-trap and SP3 have been recently utilized in proteomic studies of Phaeodactylum tricornutum and Symbiodinium tridacnidorum [Citation35,Citation48]. Of relevance, a recent study described the evaluation of three methods of digestion – including on-filter digestion using FASP, SP3, and in-solution – using STAGE tips for sample clean-up in Symbiodinium tridacnidorum microalgae, wherein SP3 was highlighted as the best method with the highest robustness and digestion efficiency [Citation35].
While the main reported protein processing methods are covered in this – and the previous – section, the paucity of knowledge in this space necessitates careful consideration of the efficiency of alternative and novel sample preparation methods for microalgal proteomic research.
Mass spectrometry-based analysis of microalgal proteins
Quantitative proteomics – label-free and isotopic labeling
Quantitative proteomics can be divided into two major workflows: label-free and label-based approaches. Label-free proteomics is based on: peptide sample preparation, sample separation by liquid chromatography (LC) and analysis by MS/MS, and finally data analysis including: peptide identification, quantification, and statistical analysis. In label-based techniques, peptides are commonly labeled using reagents with isobaric chemical structures that when fragmented in the mass spectrometer release reporter ions along with sequence information [Citation49]. By extension, proteins can then be inferred with measures of relative abundance across the original samples. In addition to chemical tagging, metabolic labeling has also proven popular in the proteomics community, wherein isotopically labeled amino acids are used as a nutrient for cell and animal culture. The peptides resulting from tryptic digestion then demonstrate specific differences in their masses, which can be used for quantification, while fragmentation of these peptides provides sequence information [Citation50,Citation51]. Whereas chemical labeling approaches involve using chemical reactions to in vitro label specific chemical groups within amino acids.
Although label-free methods have been the most popular approaches in microalgal proteomics, iTRAQ has been the prevalent label-based method across a range of species, including: Nostoc sp., Chlamydomonas reinhardtii, Chlamydomonas nivalis, Scenedesmus obliquus, Chlorella vulgaris, and Dunaliella salina (Supplementary Table S1) [Citation52–56]. Through iTRAQ labeling, the proteomic response of Dunaliella salina to salt stress uncovered the key role of photosynthesis and ATP synthesis for the modulation of early salinity-responsive pathways [Citation56]. In another iTRAQ-based study of two species of Chlamydomonas, the molecular mechanisms of salt stress in triggering the fatty acid accumulation were revealed [Citation53]. Therein, a decrease in the abundance of enzymes involved in the TCA cycle in salt-stressed C. nivalis was demonstrated to result in fatty acid biosynthesis. Moreover, the iTRAQ-based method attracted interest to investigate the molecular pathways responsible for fatty acid biosynthesis with the aim of biofuel production [Citation53,Citation57,Citation58]. As an example of an iTRAQ-based method for the purpose of biofuel production is a study on the mechanism of lipid accumulation under low and high nitrogen in Scenedesmus acuminatus. The results of this study revealed that fatty acid synthesis and branched-chain amino acid metabolism were increased when the nitrogen supply is low for S. acuminatus. Some algal proteomic studies that used label-free shotgun proteomics using data-dependent acquisition (DDA) mode are discussed in section “Quantitative proteomics based on acquisition modes”.
Quantitative proteomics based on acquisition modes
Depending on the MS instrumentation used, three different acquisition modes can be considered for proteome measurement: DDA, data-independent acquisition (DIA), and targeted proteomic modes, e.g., multiple reaction monitoring (MRM) [Citation59] or parallel reaction monitoring (PRM) () [Citation60]. For nearly the last three decades, liquid chromatography–tandem mass spectrometry (LC–MS/MS) in DDA mode, now widely known as shotgun proteomics, has been used for proteomic studies [Citation61]. In DDA mode, the most intense precursor ions from a MS scan event are isolated and fragmented in a series of subsequent MS/MS scan events.
Figure 1. Workflow of three acquisition modes of quantitative proteomics. Quantitative proteomics is categorized by three acquisition modes: data-dependent acquisition (DDA), data-independent acquisition (DIA), and targeted proteomic modes (MRM and PRM). DDA mode can be categorized into two major groups of label-free and label-based approaches. In label-free methods, which are also used for DIA and targeted methods, the protein and peptide samples are prepared separately and then subjected to individual LC–MS/MS analysis. In label-based methods such as metabolic labeling and chemical labeling, depending on the method of labeling, the isotope labels are incorporated into the samples and several samples can be combined and analyzed in a single experiment.
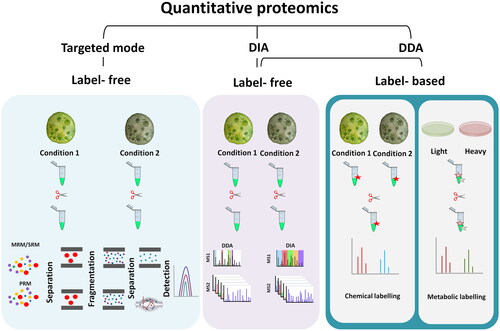
Targeted methods such as MRM and PRM are capable of reproducible and precise quantification of hundreds of proteins. However, the identities of these must be known in advance. Consequently, it is better suited to the validation phase of proteomics experiments rather than the discovery phase [Citation62].
Advances in MS instrumentation and bioinformatics have resulted in the development of the DIA-MS approach. DIA can quantify thousands of proteins in complex samples, similar to DDA [Citation59]. The first widely adopted DIA method was the sequential window acquisition of all theoretical fragment ion spectra (SWATH-MS) [Citation63]. A wide range of product ions are scanned and generated by MSMS using broad isolation windows. As DIA repeatedly scans every peptide in a sample, a complex set of MS/MS data is generated that makes data interpretation difficult if there is no reference spectral library. Therefore, to quantify DIA data, DIA spectra are compared with spectral libraries, which are annotated peptide-spectrum matches from previous DDA experiments (or predicted synthetic spectra). The most widely used approach for spectral library generation is characterizing the samples of interest for SWATH-MS in initial experiments using a DDA approach [Citation64]. However, using in silico spectral predictions resulting in DIA library-free approaches are progressing rapidly to perform library-free DIA data analysis [Citation65–67].
DDA has been used in almost all published microalgal proteomics studies, while the DIA approach has been rarely reported [Citation55,Citation68–71]. In this regard, label-free shotgun proteomics has been mostly used to study the proteomic response of algae species to environmental perturbation, such as light stress, nutrient deficiency, etc. An example of this approach concerns the study of microalgae response to light wherein different CO2 concentrations led to proteome reprogramming in Nannochloropsis oceanica [Citation72]. In another example, the DDA method was used to reveal the high abundance of ammonia and urea transporters, as well as phosphokinases and phosphate transporters, in Ostreococcus tauri in response to low-nitrogen and low-phosphorus environments [Citation73]. The membrane proteome of Ettlia oleoabundans was also studied using this method, showing the proteome response to nitrogen deprivation [Citation74]. Photosynthesis-related proteins, including Photosystem II Subunit S (PSBS) and Maintenance of Photosystem II under High Light1 (MPH1), were identified as responsive to nitrogen deprivation, suggesting a similar photoprotective mechanism to prolonged nitrogen deprivation in microalgae to that found in higher photosynthetic organisms. A range of microalgae studies were performed in recent years using the DIA approach, highlighting the increasing application of this technique in microalgae research. In one recent study, changes in the proteome of Chlorella sorokiniana following cadmium exposure were investigated [Citation68]. The suppression of photosynthesis and oxidative phosphorylation as well as the activation of photorespiration were uncovered in response to the presence of this heavy metal. This DIA study along with other algal proteomic studies that used this approach used DDA to generate DIA spectral libraries [Citation55,Citation68–70]. A DIA library-free approach has been only reported in one recent study regarding the nutrition, allergenicity, and physiochemical qualities of food-grade protein extracts in Nannochloropsis oculata [Citation75].
Post-translational modifications
PTMs are chemical modifications of a protein that involves the addition of chemical moieties to the protein or elicits a structural change which is important in cell and biological processes as they play key roles in modulating protein function, activity, stability, and/or localization [Citation76].
Among the wide range of PTMs, protein phosphorylation, acetylation, and ubiquitination are the most frequent PTMs. Phosphorylation plays a key role in the regulation of significant cellular processes, including: signal transduction pathways, replication, transcription, and response to environmental stresses. Acetylation also regulates various biological processes including protein–protein interaction, cell metabolism, and nuclear transport [Citation77]. Ubiquitination serves as a critical regulator of substrate degradation, thereby contributing to the maintenance of cellular homeostasis and the essential functions required for sustaining life activities [Citation78]. Protein methylation and glycosylation are also categorized among the top 10 major PTMs that shape biological processes [Citation77].
Several studies on microalgae PTMs have reported phosphoproteomic alterations in response to changing environmental conditions such as nutrient stress [Citation79–83]. Only a limited number of studies have investigated glycosylation pathways in Chlamydomonas reinhardtii [Citation83–85], algal protein acetylation [Citation86], and methylation [Citation87].
It has been demonstrated that phosphorylation is an effective method of enhancing the functional properties of food proteins [Citation88] and glycosylation can increase the solubility and global stability of proteins [Citation89]. Apart from PTMs that are catalyzed by enzymes, numerous nonenzymatic PTMs (nePTMs) occur, some of which result from environmental or process factors rather than biological processes, such as: glycation, carbamylation, and carbonylation. NePTM formation is prevalent in food processing and storage [Citation90,Citation91]. Therefore, NePTMs are best studied and understood using proteomics.
Data analysis
Database creation
Microalgal proteomics studies have mostly focused on C. reinhardtii as the premier reference organism given its considerable genetic characterization [Citation92]. Given the extensive number of different microalgae species, the whole genome sequence of only 105 microalgae species is publicly available which limits broad genomic and proteomic research. However, the growing availability of genomic sequences for non-model organisms is rapidly accelerating with the advancement of high-throughput sequencing technologies [Citation93].
Sequence databases are an essential component of MS-based proteomic analysis in terms of protein identification. Genome availability of the species of interest influences the efficiency of protein identification. Incomplete genome databases may lead to missing protein identifications and hinder the ability to uncover novel proteins. Using a database search also offers the opportunity to identify a higher number of peptides compared to solely employing de novo methods, since the preexisting knowledge reduces the level of evidence required for a successful identification [Citation94]. Using sequence repositories such as UniProtKB (https://www.uniprot.org), and NCBI (https://www.ncbi.nlm.nih.gov) provide access the proteomic resources for the target microalgae. In addition, algae-based databases such as Phycocosm (https://mycocosm.jgi.doe.gov/Algae/Algae.info.html), contain genomic information for more than one hundred algae species. However, when a sequence database of a non-model species is absent, cross-species identification, de novo genomic and transcriptomic analysis, and de novo protein sequencing can be used to assist with protein identification [Citation95] (). As a first step to generate a database for a non-model microalgae, retrieving sequences of closely related organisms from sequence repositories such as proteomic, genomic, and transcriptomic resources could be considered [Citation97]. In the absence of genomic data, transcriptomic data represent a useful tool for MS-based protein identifications. The use of next generation sequencing technologies in RNA-seq for transcriptome characterization can provide a high-quality sequence database relatively cheaply and rapidly, so this approach has become very important in MS-based protein identification in non-model species [Citation98]. In a membrane proteomic study of the non-model E. oleoabundans, a protein database was generated using translated transcriptome (RNA seq) of this microalga [Citation74]. In the absence of genomic and transcriptomic databases, protein identification in non-model organisms relies on cross-species identification or cross-species protein sequence similarity [Citation99]. Proteins can be identified by identification of conserved peptides of the proteins in model species, or another related species, but assembling such peptides into proteins can be problematic [Citation100].
Figure 2. Workflow of database creation for model and non-model microalgae species. Common Repository of Adventitious Proteins (cRAP) in FASTA format is added to the microalgae of interest protein FASTA to include common protein contaminants. Sequence duplicates are removed using software packages, e.g., the open source tool SeqKit [Citation96].
![Figure 2. Workflow of database creation for model and non-model microalgae species. Common Repository of Adventitious Proteins (cRAP) in FASTA format is added to the microalgae of interest protein FASTA to include common protein contaminants. Sequence duplicates are removed using software packages, e.g., the open source tool SeqKit [Citation96].](/cms/asset/bd7eabd5-7e99-483f-9afa-7cddfe422a5e/ibty_a_2283376_f0002_c.jpg)
Peptide and protein identification in non-model algae strains can also be achieved by de novo sequencing of high-quality MS/MS spectra using de novo software packages such as PEAKS, but it is still not as efficient as peptide to spectrum matching using a genome sequence [Citation101]. Overall, with a non-accessible genome sequence, using the above approaches allows proteomic analysis of any given species of interest. However, the results of protein identification in this case might not be as complete as those in model species. Using complementary databases such as Alga-PrAS (Algal Protein Annotation Suite) allow the comparative analysis of physicochemical and structural properties and PTMs in algal proteomes. This database is accessible through http://alga-pras.riken.jp.
Identification of putative allergen proteins
Allergenic proteins can be found in a broad range of protein containing foods, which can cause immune responses in susceptible individuals. These allergic reactions cause biological disorders linked to gastrointestinal and respiratory tracts or skin [Citation102]. For the most prevalent allergenic foods, such as: eggs, milk, fish, peanuts, soybeans, and wheat, extensive research effort has been undertaken to identify and quantify allergen proteins and discover marker peptides using proteomics [Citation103]. However, little is known regarding the potential allergenicity of proteins from microalgae [Citation104].
The combination of mass spectrometric analysis of algae to characterize peptides and use of in silico identification databases provides a novel/promising approach to better understand the potential allergenicity of microalgal proteins. In this regard, databases containing information for allergens are valuable tools that are publicly available at WHO/International Union of Immunological Societies (IUIS) (https://iuis.org), Allergome (https://www.allergome.org), UniProtKB (https://www.uniprot.org), Comprehensive Protein Allergen Resource (COMPARE) (https://comparedatabase.org), and Food Allergy Research and Resource Program (FARRP) (https://farrp.unl.edu). There are several programs (and accompanying web servers) such as AllerCatPro [Citation105], AllergenPro [Citation106], and Allermatch [Citation107] for helping researchers to extract the allergenicity information from various databases and predict the allergenicity of proteins of interests.
The information acquired through in silico analysis helps to identify conserved homologous proteins that may be cross-reactive with known allergens. Cross reactivity can be predicted based on their primary sequence homology, structure, and presence of B- or T-cell epitopes in the sequence [Citation104]. However, identification of IgE antibodies to proteins is not necessarily connected to clinical allergy. Hence, many other factors can trigger clinical cross-reactivity including food protein characteristics such as the stability of the allergen protein against pH, protease digestion or heat, immune response such as IgE antibody affinity, and host (patient) factors such as illness [Citation108].
There is a lack of information for microalgae allergens in the WHO/IUIS and other allergen databases as research into identifying allergenic proteins in microalgae, along with experimental data like serum IgE binding tests, is currently insufficient. Only a few studies have investigated the allergic reactions after microalgae intake [Citation109,Citation110]. Furthermore, these studies are limited to a small number of commercially available species such as Chlorella and Spirulina. A case report showed that a 11-year-old boy who consumed Chlorella tablets (2 g/day) for 3 months developed acute tubulointerstitial nephritis [Citation111]. Spirulina intake was also shown to cause systemic allergic symptoms such as shortness of breath, urticaria in two patients who also report mild oral reactions to some fresh fruit and raw vegetables [Citation110]. The identification of microalgae putative allergens has been reported recently for Chlorella and Spirulina [Citation33]. Using shotgun proteomics and bioinformatic analysis of sequence-based homology between microalgae and known allergenic proteins, Bianco et al. [Citation33] identified several putative allergens. Thioredoxins, superoxide dismutase, and C-phycocyanin beta-subunit were identified in Spirulina and calmodulin and troponin C were found in Chlorella; however, immunochemical tests are still required to validate the allergenicity of those proteins. Several putative inhalation-related, contact and food allergenic proteins were also identified using in silico methods in Nannochloropsis oculata [Citation75]. MS identification of putative allergenic proteins in other microalgae specie should be considered along with human clinical studies and quantitative measurement of immunoglobulin E (IgE) upon ingestion of the microalgae of interest.
Media composition and environmental conditions can affect metabolic pathways
Proteomic methods have been used to understand how the algal proteome is modified by a range of environmental factors. These include CO2 concentration, macronutrients, and light, which are reported to result in differing proteome response, amino acid, and total protein content of the biomass [Citation112–114]. Much of the previous proteomic focus has been on understanding conditions that favor lipid production, particularly for biofuel generation [Citation53,Citation79,Citation114,Citation115].
Carbon dioxide levels have a significant impact on protein content and algae biomass [Citation114,Citation116]. The increase in growth rate and extracellular protein composition of Chlamydomonas reinhardtii were reported in response to increased concentration of CO2 in algal culture [Citation117]. Wei et al. showed that exposing Nannochloropsis oceanica culture to high level of CO2, i.e., 50 000 ppm, resulted in higher protein content but no change in carbohydrates and lipid content. Using proteomics as well as transcriptomics revealed that at low CO2 conditions, there was lower abundance of protein synthesis-related gene products at the transcript- and protein levels, which supports the observation of lower protein content. In addition, low CO2 (100 ppm) decreased the abundance of several EAAs such as valine and isoleucine whereas non-EAAs, such as glycine, alanine, and serine, increased [Citation114]. Although high levels of CO2 can increase the biomass and protein content, carbon concentrating mechanism (CCM) activity declines in non-tolerant algae species as a result of exposure to high CO2 [Citation116]. Due to CCM shutdown, the biomass and the final protein content is adversely affected. The transcript knockdown carbonic anhydrase 2 (CA2) – one of the key components of CCM – in Nannochloropsis oceanica showed a greater photosynthesis rate and biomass production in high CO2 when compared to the wild-type strain [Citation118].
Nitrogen and phosphorus limitation act as effective pressure factors allowing the accumulation of lipids, such as triacylglycerol, in microalgae [Citation119–122]. Thus, most omics studies have been conducted from the biofuel production perspective to analyze the response of microalgae to nitrogen and phosphorus deprivation. Cai et al. demonstrated that the low C/N ratio (12:1) was beneficial to the synthesis of glutamate in heterotrophic cultivation of Chlorella vulgaris, with the biomass productivity of 0.90 g/L/day, protein content of 61.6% with EAAs at 41.8% [Citation123].
The accumulation of lipids in nitrogen and phosphorus deprived cells does not favor protein and amino acid quantity. Proteins as intracellular nitrogen storage molecules are targeted to degradation in nitrogen-deprived cells and lipids are accumulated in the cells as energy storage molecules [Citation120,Citation124]. The decrease in abundance of tRNA synthetases, translation initiation and elongation factors in proteomic studies of algae species may confirm the reduced protein biosynthesis of nitrogen-depleted cells [Citation120,Citation121]. However, less efficient carbon fixation and energy supply constraints in phosphorus-deprived cells cause a reduction in protein content [Citation122]. Ribosomal proteins and those with functional domains that are expected to be altered in reduced growth conditions decreased in abundance in P-deprived Scenedesmus sp. cells [Citation125]. Chen et al. demonstrated that the abundance of those proteins involved in photosynthesis, chlorophyll and protein biosynthesis decreased in Thalassiosira pseudonana grown in nitrogen and phosphorus-deficient cultures and shed light on the metabolic pathways and associated cellular functions in responses to stress at the proteome level.
Sunlight plays a crucial role in microalgal growth with the amount of light directly influencing biomass productivity. In raceway ponds, geographic location is especially important in defining the amount of light supplied and subsequent biomass productivity of the microalgae [Citation126]. The intensity and wavelength of light also affect protein quantity and EAA content of microalgae [Citation127,Citation128]. In Chlorella ellipsoidea protein content was raised when exposed to blue light from LEDs compared to white, green, and red LEDs [Citation128]. Protein quantity in Dunaliella salina was slightly higher in both blue and red than white light with no change in EAA content [Citation127]. In complementary work, a label-based proteomic study of Nannochloropsis oceanica showed higher carotenoid metabolism and ROS scavengers in response to red light when compared with blue light [Citation129].
Microalgae exhibit flexibility in their metabolic modes, employing photoautotrophy, heterotrophy, and mixotrophy based on factors such as light availability and carbon supply [Citation130]. Proteome and transcriptome measurements of Chlorella vulgaris were performed during the autotrophy-to-mixotrophy-to-heterotrophy transition [Citation131]. The increased growth rate that was observed during mixotrophy was suggested to be linked to enhanced stress tolerance via inositol and increased resistance to oxidative stress through thioredoxin modulation [Citation131].
In addition to studying the impact of growth medium on either protein quantity/quality or biosynthesis of a particular peptide/protein, the molecular mechanisms of microalgae tolerance to environmental factors can be explored through proteomics. The importance of the microalgae tolerance to environmental factors, such as temperature, is more valuable when cultivation is carried out in outdoor environments. Using iTRAQ-based quantitative proteome measurement, Li et al. [Citation132] demonstrated the response of Spirulina platensis to low temperature. This study confirmed the suppression of protein synthetic machinery in Spirulina exposed to low temperature of 15 °C.
High salinity is another environmental stressor that may affect lipid accumulation in a similar way to that seen following nitrogen deprivation [Citation53,Citation133]; however, protein content and proteome responses to salt stress vary depending on the algae species and their habitat. For example, total protein content increased slightly in Tetraselmis chuii in 40 ppt salt compared to control conditions. At the same time, it decreased significantly with an increased salt concentration in Chlorella vulgaris [Citation134]. The green microalgae Dunaliella salina is a model for investigating the molecular adaptation mechanisms in salt stress as it is the most tolerant photosynthetic unicellular eukaryotic organism to salinity [Citation135]. Dunaliella salina’s enormous ability to tolerate salinity and being a good source of beta-carotene make this microalgae a valuable organism for both research and industrial purposes. Although several proteomic studies investigated the salt tolerance mechanisms in Dunaliella salina [Citation56,Citation81,Citation136], there is still little known regarding the potential of this species to produce protein for human food applications.
Peptidomics: bioactive peptides in microalgae
Peptidomics is a sub-field of proteomics that qualitatively and quantitatively analyze the peptides in biological samples including bioactive peptides in food matrices [Citation137]. After protein extraction from microalgae strains, peptides may be intrinsic or generated either by using one or multiple gastrointestinal enzymes, such as trypsin, pepsin, papain, etc., or food-grade enzymes such as alcalase and flavourzyme or microbial enzymes through in situ microbial fermentation [Citation28,Citation29,Citation138,Citation139]. The subsequent quantification and identification of peptides are performed using different methods of mass spectrometric techniques, as described previously, and using relevant bioactive databases such as BIOPEP or PepBank databases [Citation140,Citation141]. Native bioactive peptides can be extracted by utilizing molecular weight cutoff (MWCO) filters [Citation142,Citation143]; however, peptidomic studies in microalgae have focused on using gastrointestinal enzymes for peptide enrichment rather than investigating the native peptides in algal protein extracts.
Potential bioactive peptides were identified in a study on Tetradesmus obliquus, four of bioactive peptides were synthesized and assessed in vitro, demonstrating a promising rate of antioxidant and angiotensin-converting enzyme (ACE) inhibitory activities [Citation139]. Using food-grade extracts from Auxenochlorella pyrenoidosa, three novel antioxidative peptides were identified [Citation28]. Although these initial findings highlight the potential functional health effects for algae derived bioactive peptides, further research is needed to establish whether algae-derived peptides are bioavailable and exert biological activity following transit of the gastrointestinal tract. In a study by Sommella et al. [Citation144], the peptides originated from in vitro digestibility method were analyzed in Spirulina. In this study, phycocyanin-derived peptides were identified in microalgal protein digests.
Future directions
In 2019, the global cultivation of microalgae was estimated at 56,456 tonnes across 10 countries, which is less than 0.2% of the total global cultivation of seaweed according to the FAO (Food and Agriculture Organization). Although there is currently substantial growth in the production and sales of microalgae-based food products [Citation145] there remains huge potential, especially when compared to other related markets, such as algal production of lipids, carotenoids, and pharmaceutical proteins, which has received extensive research interest for more than two decades [Citation146]. Multi-omics techniques have become increasingly prevalent for studying microalgal lipid synthesis, and genetic and metabolic engineering approaches have been utilized to enhance lipid production in microalgae for strain selection and enhancement. These omic studies, particularly MS-based proteomics, are essential for identifying microalgal species best suited for producing high-quality protein ingredients for human dietary purposes.
Although 2-DE is a near-deprecated technique, there remain microalgae researchers that continue to use this technique in their investigations. Given the rapid advancement in proteome science, the use of novel sample preparation and protein digestion methods should be considered before MS analysis of microalgae peptides. In addition, the explosion of genome sequencing using high-throughput technologies is already resulting in reproducible, rapid, and comprehensive protein identification. Additionally, proteogenomic approaches are aiding the detection of novel protein sequences through advanced sequencing technologies [Citation147].
Sample preparation throughput has long been a challenge in the field of proteome science, but pressure cycling technology (PCT) has recently emerged to substantially reduce sample preparation time. One example of PCT, PCT-SWATH, has been successfully used with tissue biopsy samples where it reduced sample preparation time to <6 h for 16 samples [Citation148–150]. However, the use of PCT-SWATH with plant and microalgae samples still requires evaluation and optimization. Analytical throughput can also be enhanced by high-flow chromatography applied to short-gradient proteomics, e.g., 5 min [Citation151,Citation152]. Using a 5 min LC gradient and SWATH-MS method enabled Messner et al. [Citation151] to precisely quantify the proteome of 180 human plasma samples per day. The ultra-high throughput proteomic method could be revolutionary for the advancement of the microalgal industry as it has the potential to enable screening of a wide range of microalgae species and in the genetic diversity in support of strain selection and optimization of growing conditions. Ultimately, these advancements can significantly increase biomass production, leading to the potential production of substantial amounts of dietary protein, EAAs, and bioactive peptides.
Conclusions
Microalgae has great potential as a source of dietary protein. Understanding the nutritional properties of algal proteins from a broad range of species, their functional health benefits and safety are needed to broaden their application and use. Current algal proteome investigations – specifically from a nutrition point of view – are limited in scope and many technical challenges remain. Shifting from outdated proteomic methods to modern techniques as well as developing genomic resources and optimized strains are key initiatives that are needed to accelerate the potential use of microalgae as a source of high-quality dietary protein for the population at large.
Supplemental Material
Download MS Excel (33.3 KB)Acknowledgements
S.H. would like to acknowledge scholarship support from the Commonwealth Scientific and Industrial Research Organisation (CSIRO) Early Research Career (CERC) postdoctoral fellowship.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bule MH, Ahmed I, Maqbool F, et al. Microalgae as a source of high-value bioactive compounds. Front Biosci (Schol Ed). 2018;10:197–216. doi: 10.2741/s509.
- Torres-Tiji Y, Fields FJ, Mayfield SP. Microalgae as a future food source. Biotechnol Adv. 2020;41:107536. doi: 10.1016/j.biotechadv.2020.107536.
- Wang Y, Tibbetts SM, McGinn PJ. Microalgae as sources of high-quality protein for human food and protein supplements. Foods. 2021;10:3002. doi: 10.3390/foods10123002.
- Chacón-Lee TL, González-Mariño GE. Microalgae for “healthy” foods—possibilities and challenges. Compr Rev Food Sci Food Saf. 2010;9:655–675. doi: 10.1111/j.1541-4337.2010.00132.x.
- Alishah Aratboni H, Rafiei N, Garcia-Granados R, et al. Biomass and lipid induction strategies in microalgae for biofuel production and other applications. Microb Cell Fact. 2019;18:178. doi: 10.1186/s12934-019-1228-4.
- Caporgno MP, Mathys A. Trends in microalgae incorporation into innovative food products with potential health benefits. Front Nutr. 2018;5:58. doi: 10.3389/fnut.2018.00058.
- Borowitzka MA. Commercial production of microalgae: ponds, tanks, tubes and fermenters. J Biotechnol. 1999;70:313–321. doi: 10.1016/S0168-1656(99)00083-8.
- Spolaore P, Joannis-Cassan C, Duran E, et al. Commercial applications of microalgae. J Biosci Bioeng. 2006;101:87–96. doi: 10.1263/jbb.101.87.
- Wolkers H, Barbosa MJ, Kleinegris DMM, et al. Microalgae: the green gold of the future? Large-scale sustainable cultivation of microalgae for the production of bulk commodities. Wageningen UR Food & Biobased Research; 2011. https://edepot.wur.nl/170781
- Kumar G, Shekh A, Jakhu S, et al. Bioengineering of microalgae: recent advances, perspectives, and regulatory challenges for industrial application. Front Bioeng Biotechnol. 2020;8:914. doi: 10.3389/fbioe.2020.00914.
- Fu W, Chaiboonchoe A, Khraiwesh B, et al. Algal cell factories: approaches, applications, and potentials. Mar Drugs. 2016;14:225. doi: 10.3390/md14120225.
- Tyers M, Mann M. From genomics to proteomics. Nature. 2003;422:193–197. doi: 10.1038/nature01510.
- Cho WC. Proteomics technologies and challenges. Genomics Proteomics Bioinformatics. 2007;5:77–85. doi: 10.1016/S1672-0229(07)60018-7.
- Safi C, Ursu AV, Laroche C, et al. Aqueous extraction of proteins from microalgae: effect of different cell disruption methods. Algal Res. 2014;3:61–65. doi: 10.1016/j.algal.2013.12.004.
- Bharte S, Desai K. Techniques for harvesting, cell disruption and lipid extraction of microalgae for biofuel production. Biofuels. 2021;12:285–305. doi: 10.1080/17597269.2018.1472977.
- Domozych D. Algal cell walls. In: eLS. Chichester: JohnWiley & Sons, Ltd; 2019. p. 1–11. doi: 10.1002/9780470015902.a0000315.pub4.
- Safi C, Cabas Rodriguez L, Mulder WJ, et al. Energy consumption and water-soluble protein release by cell wall disruption of Nannochloropsis gaditana. Bioresour Technol. 2017;239:204–210. doi: 10.1016/j.biortech.2017.05.012.
- Soto-Sierra L, Stoykova P, Nikolov ZL. Extraction and fractionation of microalgae-based protein products. Algal Res. 2018;36:175–192. doi: 10.1016/j.algal.2018.10.023.
- Zhang R, Chen J, Zhang X. Extraction of intracellular protein from Chlorella pyrenoidosa using a combination of ethanol soaking, enzyme digest, ultrasonication and homogenization techniques. Bioresour Technol. 2018;247:267–272. doi: 10.1016/j.biortech.2017.09.087.
- Safi C, Olivieri G, Campos RP, et al. Biorefinery of microalgal soluble proteins by sequential processing and membrane filtration. Bioresour Technol. 2017;225:151–158. doi: 10.1016/j.biortech.2016.11.068.
- Safi C, Frances C, Ursu AV, et al. Understanding the effect of cell disruption methods on the diffusion of Chlorella vulgaris proteins and pigments in the aqueous phase. Algal Res. 2015;8:61–68. doi: 10.1016/j.algal.2015.01.002.
- Postma PR, Miron TL, Olivieri G, et al. Mild disintegration of the green microalgae Chlorella vulgaris using bead milling. Bioresour Technol. 2015;184:297–304. doi: 10.1016/j.biortech.2014.09.033.
- Gomes TA, Zanette CM, Spier MR. An overview of cell disruption methods for intracellular biomolecules recovery. Prep Biochem Biotechnol. 2020;50:635–654. doi: 10.1080/10826068.2020.1728696.
- Bleakley S, Hayes M. Algal proteins: extraction, application, and challenges concerning production. Foods. 2017;6:33. doi: 10.3390/foods6050033.
- Moore JC, DeVries JW, Lipp M, et al. Total protein methods and their potential utility to reduce the risk of food protein adulteration. Compr Rev Food Sci Food Saf. 2010;9:330–357. doi: 10.1111/j.1541-4337.2010.00114.x.
- Greenly JM, Tester JW. Ultrasonic cavitation for disruption of microalgae. Bioresour Technol. 2015;184:276–279. doi: 10.1016/j.biortech.2014.11.036.
- Andrade LM, Tito CA, Mascarenhas C, et al. Chlorella vulgaris phycoremediation at low Cu+2 contents: proteomic profiling of microalgal metabolism related to fatty acids and CO2 fixation. Chemosphere. 2021;284:131272. doi: 10.1016/j.chemosphere.2021.131272.
- Zhu Z, Chen Y, Jia N, et al. Identification of three novel antioxidative peptides from Auxenochlorella pyrenoidosa protein hydrolysates based on a peptidomics strategy. Food Chem. 2022;375:131849. doi: 10.1016/j.foodchem.2021.131849.
- Montalvo GEB, Thomaz-Soccol V, Vandenberghe LPS, et al. Arthrospira maxima OF15 biomass cultivation at laboratory and pilot scale from sugarcane vinasse for potential biological new peptides production. Bioresour Technol. 2019;273:103–113. doi: 10.1016/j.biortech.2018.10.081.
- Tomé LIN, Baião V, da Silva W, et al. Deep eutectic solvents for the production and application of new materials. Appl Mater Today. 2018;10:30–50. doi: 10.1016/j.apmt.2017.11.005.
- Hewage A, Olatunde OO, Nimalaratne C, et al. Novel Extraction technologies for developing plant protein ingredients with improved functionality. Trends Food Sci Technol. 2022;129:492–511. doi: 10.1016/j.tifs.2022.10.016.
- Bose U, Broadbent JA, Juhász A, et al. Protein extraction protocols for optimal proteome measurement and arginine kinase quantitation from cricket Acheta domesticus for food safety assessment. Food Chem. 2021;348:129110. doi: 10.1016/j.foodchem.2021.129110.
- Bianco M, Ventura G, Calvano CD, et al. A new paradigm to search for allergenic proteins in novel foods by integrating proteomics analysis and in silico sequence homology prediction: focus on Spirulina and Chlorella microalgae. Talanta. 2022;240:123188. doi: 10.1016/j.talanta.2021.123188.
- Chen Y, Chen J, Chang C, et al. Physicochemical and functional properties of proteins extracted from three microalgal species. Food Hydrocolloids. 2019;96:510–517. doi: 10.1016/j.foodhyd.2019.05.025.
- Supasri KM, Kumar M, Mathew MJ, et al. Evaluation of filter, paramagnetic, and STAGETips aided workflows for proteome profiling of Symbiodiniaceae dinoflagellate. Processes. 2021;9:983. doi: 10.3390/pr9060983.
- Botting CH, Talbot P, Paytubi S, et al. Extensive lysine methylation in hyperthermophilic Crenarchaea: potential implications for protein stability and recombinant enzymes. Archaea. 2010;2010:106341. doi: 10.1155/2010/106341.
- Smith CM. Quantification of acetylation at proximal lysine residues using isotopic labeling and tandem mass spectrometry. Methods. 2005;36:395–403. doi: 10.1016/j.ymeth.2005.03.007.
- Dau T, Bartolomucci G, Rappsilber J. Proteomics using protease alternatives to trypsin benefits from sequential digestion with trypsin. Anal Chem. 2020;92:9523–9527. doi: 10.1021/acs.analchem.0c00478.
- Tsiatsiani L, Heck AJR. Proteomics beyond trypsin. FEBS J. 2015;282:2612–2626. doi: 10.1111/febs.13287.
- Kolmakova AA, Kolmakov VI. Amino acid composition of green microalgae and diatoms, cyanobacteria, and zooplankton (review). Inland Water Biol. 2019;12:452–461. doi: 10.1134/S1995082919040060.
- Elleuch J, Ben Amor F, Chaaben Z, et al. Zinc biosorption by Dunaliella sp. AL-1: mechanism and effects on cell metabolism. Sci Total Environ. 2021;773:145024. doi: 10.1016/j.scitotenv.2021.145024.
- Zhan Y, Marchand CH, Maes A, et al. Pyrenoid functions revealed by proteomics in Chlamydomonas reinhardtii. PLOS One. 2018;13:e0185039. doi: 10.1371/journal.pone.0185039.
- Pérez-Pérez ME, Mauriès A, Maes A, et al. The deep thioredoxome in Chlamydomonas reinhardtii: new insights into redox regulation. Mol Plant. 2017;10:1107–1125. doi: 10.1016/j.molp.2017.07.009.
- Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261.
- Hughes CS, Moggridge S, Müller T, et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat Protoc. 2019;14:68–85. doi: 10.1038/s41596-018-0082-x.
- HaileMariam M, Eguez RV, Singh H, et al. S-Trap, an ultrafast sample-preparation approach for shotgun proteomics. J Proteome Res. 2018;17:2917–2924. doi: 10.1021/acs.jproteome.8b00505.
- Hamzelou S, Askari H, Nobari NA. Deceptive responsive genes in gel-based proteomics. Comput Biol Chem. 2016;61:1–7. doi: 10.1016/j.compbiolchem.2015.12.001.
- Leyland B, Zarka A, Didi-Cohen S, et al. High resolution proteome of lipid droplets isolated from the pennate diatom Phaeodactylum tricornutum (Bacillariophyceae) strain pt4 provides mechanistic insights into complex intracellular coordination during nitrogen deprivation. J Phycol. 2020;56:1642–1663. doi: 10.1111/jpy.13063.
- Iliuk A, Galan J, Tao WA. Playing tag with quantitative proteomics. Anal Bioanal Chem. 2009;393:503–513. doi: 10.1007/s00216-008-2386-0.
- Lindemann C, Thomanek N, Hundt F, et al. Strategies in relative and absolute quantitative mass spectrometry based proteomics. Biol Chem. 2017;398:687–699. doi: 10.1515/hsz-2017-0104.
- DeSouza LV, Siu KWM. Mass spectrometry-based quantification. Clin Biochem. 2013;46:421–431. doi: 10.1016/j.clinbiochem.2012.10.025.
- Liu J, Zhang H, Yan L, et al. Electron transport, light energy conversion and proteomic responses of periphyton in photosynthesis under exposure to AgNPs. J Hazard Mater. 2021;401:123809. doi: 10.1016/j.jhazmat.2020.123809.
- Hounslow E, Evans CA, Pandhal J, et al. Quantitative proteomic comparison of salt stress in Chlamydomonas reinhardtii and the snow alga Chlamydomonas nivalis reveals mechanisms for salt-triggered fatty acid accumulation via reallocation of carbon resources. Biotechnol Biofuels. 2021;14:121. doi: 10.1186/s13068-021-01970-6.
- Zhang H, Zong R, He H, et al. Effects of hydrogen peroxide on Scenedesmus obliquus: cell growth, antioxidant enzyme activity and intracellular protein fingerprinting. Chemosphere. 2022;287:132185. doi: 10.1016/j.chemosphere.2021.132185.
- Zhang J, Shen L, Xiang Q, et al. Proteomics reveals surface electrical property-dependent toxic mechanisms of silver nanoparticles in Chlorella vulgaris. Environ Pollut. 2020;265:114743. doi: 10.1016/j.envpol.2020.114743.
- Wang Y, Cong Y, Wang Y, et al. Identification of early salinity stress-responsive proteins in Dunaliella salina by isobaric tags for relative and absolute quantitation (iTRAQ)-based quantitative proteomic analysis. Int J Mol Sci. 2019;20:599. doi: 10.3390/ijms20030599.
- Xue J, Balamurugan S, Li DW, et al. Glucose-6-phosphate dehydrogenase as a target for highly efficient fatty acid biosynthesis in microalgae by enhancing NADPH supply. Metab Eng. 2017;41:212–221. doi: 10.1016/j.ymben.2017.04.008.
- Shang C, Zhu S, Wang Z, et al. Proteome response of Dunaliella parva induced by nitrogen limitation. Algal Res. 2017;23:196–202. doi: 10.1016/j.algal.2017.01.016.
- Hu A, Noble WS, Wolf-Yadlin A. Technical advances in proteomics: new developments in data-independent acquisition. F1000Res. 2016;5:419. doi: 10.12688/f1000research.7042.1.
- Peterson AC, Russell JD, Bailey DJ, et al. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics. 2012;11:1475–1488. doi: 10.1074/mcp.O112.020131.
- Bantscheff M, Lemeer S, Savitski MM, et al. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem. 2012;404:939–965. doi: 10.1007/s00216-012-6203-4.
- Lange V, Picotti P, Domon B, et al. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol. 2008;4:222. doi: 10.1038/msb.2008.61.
- Gillet LC, Leitner A, Aebersold R. Mass spectrometry applied to bottom-up proteomics: entering the high-throughput era for hypothesis testing. Annu Rev Anal Chem. 2016;9:449–472. doi: 10.1146/annurev-anchem-071015-041535.
- Ludwig C, Gillet L, Rosenberger G, et al. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol Syst Biol. 2018;14:e8126. doi: 10.15252/msb.20178126.
- Sinitcyn P, Hamzeiy H, Salinas Soto F, et al. MaxDIA enables library-based and library-free data-independent acquisition proteomics. Nat Biotechnol. 2021;39:1563–1573. doi: 10.1038/s41587-021-00968-7.
- Searle BC, Swearingen KE, Barnes CA, et al. Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat Commun. 2020;11:1548. doi: 10.1038/s41467-020-15346-1.
- Demichev V, Messner CB, Vernardis SI, et al. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat Methods. 2020;17:41–44. doi: 10.1038/s41592-019-0638-x.
- León-Vaz A, Romero LC, Gotor C, et al. Effect of cadmium in the microalga Chlorella sorokiniana: a proteomic study. Ecotoxicol Environ Saf. 2021;207:111301. doi: 10.1016/j.ecoenv.2020.111301.
- Karthikaichamy A, Beardall J, Coppel R, et al. Data-independent-acquisition-based proteomic approach towards understanding the acclimation strategy of oleaginous microalga Microchloropsis gaditana CCMP526 in hypersaline conditions. ACS Omega. 2021;6:22151–22164. doi: 10.1021/acsomega.1c02786.
- Khatiwada B, Hasan MT, Sun A, et al. Proteomic response of Euglena gracilis to heavy metal exposure – identification of key proteins involved in heavy metal tolerance and accumulation. Algal Res. 2020;45:101764. doi: 10.1016/j.algal.2019.101764.
- Wang X, Meng C, Zhang H, et al. Transcriptomic and proteomic characterizations of the molecular response to blue light and salicylic acid in Haematococcus pluvialis. Mar Drugs. 2021;20:1. doi: 10.3390/md20010001.
- Wei L, You W, Gong Y, et al. Transcriptomic and proteomic choreography in response to light quality variation reveals key adaption mechanisms in marine Nannochloropsis oceanica. Sci Total Environ. 2020;720:137667. doi: 10.1016/j.scitotenv.2020.137667.
- Martin SF, Doherty MK, Salvo-Chirnside E, et al. Surviving starvation: proteomic and lipidomic profiling of nutrient deprivation in the smallest known free-living eukaryote. Metabolites. 2020;10:273. doi: 10.3390/metabo10070273.
- Garibay-Hernández A, Barkla BJ, Vera-Estrella R, et al. Membrane proteomic insights into the physiology and taxonomy of an oleaginous green microalga. Plant Physiol. 2017;173:390–416. doi: 10.1104/pp.16.01240.
- Hamzelou S, Belobrajdic D, Juhász A, et al. Nutrition, allergenicity and physicochemical qualities of food-grade protein extracts from Nannochloropsis oculata. Food Chem. 2023;424:136459. doi: 10.1016/j.foodchem.2023.136459.
- Wu X, Gong F, Cao D, et al. Advances in crop proteomics: PTMs of proteins under abiotic stress. Proteomics. 2016;16:847–865. doi: 10.1002/pmic.201500301.
- Ramazi S, Zahiri J. Posttranslational modifications in proteins: resources, tools and prediction methods. Database. 2021;2021:baab012. doi: 10.1093/database/baab012.
- Deng L, Meng T, Chen L, et al. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther. 2020;5:11. doi: 10.1038/s41392-020-0107-0.
- Roustan V, Bakhtiari S, Roustan PJ, et al. Quantitative in vivo phosphoproteomics reveals reversible signaling processes during nitrogen starvation and recovery in the biofuel model organism Chlamydomonas reinhardtii. Biotechnol Biofuels. 2017;10:280. doi: 10.1186/s13068-017-0949-z.
- Wagner V, Ullmann K, Mollwo A, et al. The phosphoproteome of a Chlamydomonas reinhardtii eyespot fraction includes key proteins of the light signaling pathway. Plant Physiol. 2008;146:772–788. doi: 10.1104/pp.107.109645.
- Wei S, Bian Y, Zhao Q, et al. Salinity-induced palmella formation mechanism in halotolerant algae Dunaliella salina revealed by quantitative proteomics and phosphoproteomics. Front Plant Sci. 2017;8:810. doi: 10.3389/fpls.2017.00810.
- Wang B, Yang J, Xu C, et al. Dynamic expression of intra- and extra-cellular proteome and the influence of epiphytic bacteria for Nostoc flagelliforme in response to rehydration. Environ Microbiol. 2020;22:1251–1264. doi: 10.1111/1462-2920.14931.
- Barolo L, Commault AS, Abbriano RM, et al. Unassembled cell wall proteins form aggregates in the extracellular space of Chlamydomonas reinhardtii strain UVM4. Appl Microbiol Biotechnol. 2022;106:4145–4156. doi: 10.1007/s00253-022-11960-9.
- Oltmanns A, Hoepfner L, Scholz M, et al. Novel insights into N-glycan fucosylation and core xylosylation in C. reinhardtii. Front Plant Sci. 2019;10:1686. doi: 10.3389/fpls.2019.01686.
- Schulze S, Oltmanns A, Machnik N, et al. N-Glycoproteomic characterization of mannosidase and xylosyltransferase mutant strains of Chlamydomonas reinhardtii. Plant Physiol. 2018;176:1952–1964. doi: 10.1104/pp.17.01450.
- Chen Z, Luo L, Chen R, et al. Acetylome profiling reveals extensive lysine acetylation of the fatty acid metabolism pathway in the diatom Phaeodactylum tricornutum. Mol Cell Proteomics. 2018;17:399–412. doi: 10.1074/mcp.RA117.000339.
- Veluchamy A, Rastogi A, Lin X, et al. An integrative analysis of post-translational histone modifications in the marine diatom Phaeodactylum tricornutum. Genome Biol. 2015;16:102. doi: 10.1186/s13059-015-0671-8.
- Li C-P, Enomoto H, Hayashi Y, et al. Recent advances in phosphorylation of food proteins: a review. LWT Food Sci Technol. 2010;43:1295–1300. doi: 10.1016/j.lwt.2010.03.016.
- Solá RJ, Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. J Pharm Sci. 2009;98:1223–1245. doi: 10.1002/jps.21504.
- Ortea I, O'Connor G, Maquet A. Review on proteomics for food authentication. J Proteomics. 2016;147:212–225. doi: 10.1016/j.jprot.2016.06.033.
- Khan S, Bhat AA. Nonenzymatic posttranslational protein modifications: mechanism and associated disease pathologies. In: Dar TA, Singh LR, editors. Protein modificomics. Amsterdam: Academic Press; 2019. p. 229–280.
- Merchant SS, Prochnik SE, Vallon O, et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science. 2007;318:245–250. doi: 10.1126/science.1143609.
- Ellegren H. Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol. 2014;29:51–63. doi: 10.1016/j.tree.2013.09.008.
- Boonen K, Hens K, Menschaert G, et al. Beyond genes: re-identifiability of proteomic data and its Implications for personalized medicine. Genes. 2019;10:682. doi: 10.3390/genes10090682.
- Champagne A, Boutry M. Proteomics of nonmodel plant species. Proteomics. 2013;13:663–673. doi: 10.1002/pmic.201200312.
- Shen W, Le S, Li Y, et al. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLOS One. 2016;11:e0163962. doi: 10.1371/journal.pone.0163962.
- Escobar-Correas S, Bose U, Broadbent JA, et al. Database construction strategies for proteome measurement of novel food ingredients. In: Koolen H, editor. Mass spectrometry for food analysis. New York (NY): Springer US; 2022. p. 133–143.
- Lopez-Casado G, Covey PA, Bedinger PA, et al. Enabling proteomic studies with RNA-Seq: the proteome of tomato pollen as a test case. Proteomics. 2012;12:761–774. doi: 10.1002/pmic.201100164.
- Samyn B, Sergeant K, Carpentier S, et al. Functional proteome analysis of the banana plant (Musa spp.) using de novo sequence analysis of derivatized peptides. J Proteome Res. 2007;6:70–80. doi: 10.1021/pr0601943.
- Carpentier SC, Panis B, Vertommen A, et al. Proteome analysis of non-model plants: a challenging but powerful approach. Mass Spectrom Rev. 2008;27:354–377. doi: 10.1002/mas.20170.
- Hughes C, Ma B, Lajoie GA. De novo sequencing methods in proteomics. Methods Mol Biol. 2010;604:105–121. doi: 10.1007/978-1-60761-444-9_8.
- Sicherer SH, Sampson HA. Food allergy. J Allergy Clin Immunol. 2010;125:S116–S125. doi: 10.1016/j.jaci.2009.08.028.
- Monaci L, De Angelis E, Montemurro N, et al. Comprehensive overview and recent advances in proteomics MS based methods for food allergens analysis. TrAC Trends Anal Chem. 2018;106:21–36. doi: 10.1016/j.trac.2018.06.016.
- Pali-Schöll I, Verhoeckx K, Mafra I, et al. Allergenic and novel food proteins: state of the art and challenges in the allergenicity assessment. Trends Food Sci Technol. 2019;84:45–48. doi: 10.1016/j.tifs.2018.03.007.
- Nguyen MN, Krutz NL, Limviphuvadh V, et al. AllerCatPro 2.0: a web server for predicting protein allergenicity potential. Nucleic Acids Res. 2022;50:W36–W43. doi: 10.1093/nar/gkac446.
- Kim C, Kwon S, Lee G, et al. A database for allergenic proteins and tools for allergenicity prediction. Bioinformation. 2009;3:344–345. doi: 10.6026/97320630003344.
- Fiers MWEJ, Kleter GA, Nijland H, et al. Allermatch™, a webtool for the prediction of potential allergenicity according to current FAO/WHO Codex Alimentarius Guidelines. BMC Bioinformatics. 2004;5:133. doi: 10.1186/1471-2105-5-133.
- Cox AL, Eigenmann PA, Sicherer SH. Clinical relevance of cross-reactivity in food allergy. J Allergy Clin Immunol Pract. 2021;9:82–99. doi: 10.1016/j.jaip.2020.09.030.
- Le TM, Knulst AC, Röckmann H. Anaphylaxis to Spirulina confirmed by skin prick test with ingredients of Spirulina tablets. Food Chem Toxicol. 2014;74:309–310. doi: 10.1016/j.fct.2014.10.024.
- Pimblett P. Spirulina allergy; a case history of two patients. World Allergy Organ J. 2020;13:100149. doi: 10.1016/j.waojou.2020.100149.
- Yim HE, Yoo KH, Seo WH, et al. Acute tubulointerstitial nephritis following ingestion of Chlorella tablets. Pediatr Nephrol. 2007;22:887–888. doi: 10.1007/s00467-006-0420-z.
- Zhan J, Rong J, Wang Q. Mixotrophic cultivation, a preferable microalgae cultivation mode for biomass/bioenergy production, and bioremediation, advances and prospect. Int J Hydrogen Energy. 2017;42:8505–8517. doi: 10.1016/j.ijhydene.2016.12.021.
- Ramsundar P, Guldhe A, Singh P, et al. Assessment of municipal wastewaters at various stages of treatment process as potential growth media for Chlorella sorokiniana under different modes of cultivation. Bioresour Technol. 2017;227:82–92. doi: 10.1016/j.biortech.2016.12.037.
- Wei L, El Hajjami M, Shen C, et al. Transcriptomic and proteomic responses to very low CO2 suggest multiple carbon concentrating mechanisms in Nannochloropsis oceanica. Biotechnol Biofuels. 2019;12:168. doi: 10.1186/s13068-019-1506-8.
- Colina F, Carbó M, Meijón M, et al. Low UV-C stress modulates Chlamydomonas reinhardtii biomass composition and oxidative stress response through proteomic and metabolomic changes involving novel signalers and effectors. Biotechnol Biofuels. 2020;13:110. doi: 10.1186/s13068-020-01750-8.
- Solovchenko A, Khozin-Goldberg I. High-CO2 tolerance in microalgae: possible mechanisms and implications for biotechnology and bioremediation. Biotechnol Lett. 2013;35:1745–1752. doi: 10.1007/s10529-013-1274-7.
- Baba M, Suzuki I, Shiraiwa Y. Proteomic analysis of high-CO2-inducible extracellular proteins in the unicellular green alga, Chlamydomonas reinhardtii. Plant Cell Physiol. 2011;52:1302–1314. doi: 10.1093/pcp/pcr078.
- Wei L, Shen C, El Hajjami M, et al. Knockdown of carbonate anhydrase elevates Nannochloropsis productivity at high CO2 level. Metab Eng. 2019;54:96–108. doi: 10.1016/j.ymben.2019.03.004.
- Hockin NL, Mock T, Mulholland F, et al. The response of diatom central carbon metabolism to nitrogen starvation is different from that of green algae and higher plants. Plant Physiol. 2012;158:299–312. doi: 10.1104/pp.111.184333.
- Park JJ, Wang H, Gargouri M, et al. The response of Chlamydomonas reinhardtii to nitrogen deprivation: a systems biology analysis. Plant J. 2015;81:611–624. doi: 10.1111/tpj.12747.
- Zhang YJ, Zhang SF, He ZP, et al. Proteomic analysis provides new insights into the adaptive response of a dinoflagellate Prorocentrum donghaiense to changing ambient nitrogen. Plant Cell Environ. 2015;38:2128–2142. doi: 10.1111/pce.12538.
- Chen X-H, Li Y-Y, Zhang H, et al. Quantitative proteomics reveals common and specific responses of a marine diatom Thalassiosira pseudonana to different macronutrient deficiencies. Front Microbiol. 2018;9:2761. doi: 10.3389/fmicb.2018.02761.
- Cai Y, Zhai L, Fang X, et al. Effects of C/N ratio on the growth and protein accumulation of heterotrophic Chlorella in broken rice hydrolysate. Biotechnol Biofuels Bioprod. 2022;15:102. doi: 10.1186/s13068-022-02204-z.
- Scarsini M, Thiriet-Rupert S, Veidl B, et al. The transition toward nitrogen deprivation in diatoms requires chloroplast stand-by and deep metabolic reshuffling. Front Plant Sci. 2021;12:760516. doi: 10.3389/fpls.2021.760516.
- Guo J, Wilken S, Jimenez V, et al. Specialized proteomic responses and an ancient photoprotection mechanism sustain marine green algal growth during phosphate limitation. Nat Microbiol. 2018;3:781–790. doi: 10.1038/s41564-018-0178-7.
- Janssen M, Wijffels RH, Barbosa MJ. Microalgae based production of single-cell protein. Curr Opin Biotechnol. 2022;75:102705. doi: 10.1016/j.copbio.2022.102705.
- Sui Y, Harvey PJ. Effect of light intensity and wavelength on biomass growth and protein and amino acid composition of Dunaliella salina. Foods. 2021;10:1018. doi: 10.3390/foods10051018.
- Baidya A, Akter T, Islam MR, et al. Effect of different wavelengths of LED light on the growth, chlorophyll, β-carotene content and proximate composition of Chlorella ellipsoidea. Heliyon. 2021;7:e08525. doi: 10.1016/j.heliyon.2021.e08525.
- Zienkiewicz A, Zienkiewicz K, Poliner E, et al. The microalga Nannochloropsis during transition from quiescence to autotrophy in response to nitrogen availability. Plant Physiol. 2020;182:819–839. doi: 10.1104/pp.19.00854.
- Sun X-M, Ren L-J, Zhao Q-Y, et al. Microalgae for the production of lipid and carotenoids: a review with focus on stress regulation and adaptation. Biotechnol Biofuels. 2018;11:272. doi: 10.1186/s13068-018-1275-9.
- Vidotti ADS, Riaño-Pachón DM, Mattiello L, et al. Analysis of autotrophic, mixotrophic and heterotrophic phenotypes in the microalgae Chlorella vulgaris using time-resolved proteomics and transcriptomics approaches. Algal Res. 2020;51:102060. doi: 10.1016/j.algal.2020.102060.
- Li Q, Chang R, Sun Y, et al. iTRAQ-based quantitative proteomic analysis of Spirulina platensis in response to low temperature stress. PLOS One. 2016;11:e0166876. doi: 10.1371/journal.pone.0166876.
- Takagi M, Karseno , Yoshida T. Effect of salt concentration on intracellular accumulation of lipids and triacylglyceride in marine microalgae Dunaliella cells. J Biosci Bioeng. 2006;101(3):223–226. doi: 10.1263/jbb.101.223.
- Haris N, Manan H, Jusoh M, et al. Effect of different salinity on the growth performance and proximate composition of isolated indigenous microalgae species. Aquacult Rep. 2022;22:100925. doi: 10.1016/j.aqrep.2021.100925.
- Wang Y, Hu B, Du S, et al. Proteomic analyses reveal the mechanism of Dunaliella salina Ds-26-16 gene enhancing salt tolerance in Escherichia coli. PLOS One. 2016;11:e0153640. doi: 10.1371/journal.pone.0153640.
- Ge Y, Ning Z, Wang Y, et al. Quantitative proteomic analysis of Dunaliella salina upon acute arsenate exposure. Chemosphere. 2016;145:112–118. doi: 10.1016/j.chemosphere.2015.11.049.
- Dallas DC, Guerrero A, Parker EA, et al. Current peptidomics: applications, purification, identification, quantification, and functional analysis. Proteomics. 2015;15:1026–1038. doi: 10.1002/pmic.201400310.
- Ko SC, Kim D, Jeon YJ. Protective effect of a novel antioxidative peptide purified from a marine Chlorella ellipsoidea protein against free radical-induced oxidative stress. Food Chem Toxicol. 2012;50:2294–2302. doi: 10.1016/j.fct.2012.04.022.
- Montone CM, Capriotti AL, Cavaliere C, et al. Peptidomic strategy for purification and identification of potential ACE-inhibitory and antioxidant peptides in Tetradesmus obliquus microalgae. Anal Bioanal Chem. 2018;410:3573–3586. doi: 10.1007/s00216-018-0925-x.
- Duchrow T, Shtatland T, Guettler D, et al. Enhancing navigation in biomedical databases by community voting and database-driven text classification. BMC Bioinformatics. 2009;10:317. doi: 10.1186/1471-2105-10-317.
- Minkiewicz P, Iwaniak A, Darewicz M. BIOPEP-UWM database of bioactive peptides: current opportunities. Int J Mol Sci. 2019;20:5978. doi: 10.3390/ijms20235978.
- Madsen CT, Refsgaard JC, Teufel FG, et al. Combining mass spectrometry and machine learning to discover bioactive peptides. Nat Commun. 2022;13:6235. doi: 10.1038/s41467-022-34031-z.
- Secher A, Kelstrup CD, Conde-Frieboes KW, et al. Analytic framework for peptidomics applied to large-scale neuropeptide identification. Nat Commun. 2016;7:11436. doi: 10.1038/ncomms11436.
- Sommella E, Salviati E, Merciai F, et al. Online comprehensive hydrophilic interaction chromatography × reversed phase liquid chromatography coupled to mass spectrometry for in depth peptidomic profile of microalgae gastro-intestinal digests. J Pharm Biomed Anal. 2019;175:112783. doi: 10.1016/j.jpba.2019.112783.
- Cai J, Lovatelli A, Aguilar-Manjarrez J, et al. Seaweeds and microalgae: an overview for unlocking their potential in global aquaculture development. Rome: FAO Fisheries and Aquaculture Circular No. 1229. FAO; 2021.
- Grama SB, Liu Z, Li J. Emerging trends in genetic engineering of microalgae for commercial applications. Mar Drugs. 2022;20:285. doi: 10.3390/md20050285.
- Sheynkman GM, Shortreed MR, Cesnik AJ, et al. Proteogenomics: integrating next-generation sequencing and mass spectrometry to characterize human proteomic variation. Annu Rev Anal Chem. 2016;9:521–545. doi: 10.1146/annurev-anchem-071015-041722.
- Cai X, Xue Z, Wu C, et al. High-throughput proteomic sample preparation using pressure cycling technology. Nat Protoc. 2022;17:2307–2325. doi: 10.1038/s41596-022-00727-1.
- Gao H, Zhang F, Liang S, et al. Accelerated lysis and proteolytic digestion of biopsy-level fresh-frozen and FFPE tissue samples using pressure cycling technology. J Proteome Res. 2020;19:1982–1990. doi: 10.1021/acs.jproteome.9b00790.
- Guo T, Kouvonen P, Koh CC, et al. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat Med. 2015;21:407–413. doi: 10.1038/nm.3807.
- Messner CB, Demichev V, Wendisch D, et al. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst. 2020;11:11–24.e4. doi: 10.1016/j.cels.2020.05.012.
- Ishikawa M, Konno R, Nakajima D, et al. Optimization of ultrafast proteomics using an LC-quadrupole-orbitrap mass spectrometer with data-independent acquisition. J Proteome Res. 2022;21:2085–2093. doi: 10.1021/acs.jproteome.2c00121.