
ABSTRACT
Cholesterol is an important component of lipids in animal membranes. All living cells can synthesize cholesterol, but the amount of synthesis is not sufficient, and therefore cholesterol synthesized in the liver is delivered to extrahepatic tissues as a form of LDL. The liver is a primary organ to not only synthesize but also catabolize cholesterol into bile acids, which ends up to excrete with the feces. The synthetic and catabolic pathways are precisely regulated under the negative-feedback control system under the transcriptional regulation driven by several transcription factors such as the sterol regulatory element-binding proteins (SREBPs), the liver x receptor, and the farnesoid x receptor. This review summarizes various findings including our recent discoveries in the molecular mechanism of activation of SREBP that is involved in the regulation of hepatic cholesterol biosynthesis, and a novel function of the metabolic end product of cholesterol, bile acids, in skeletal muscles.
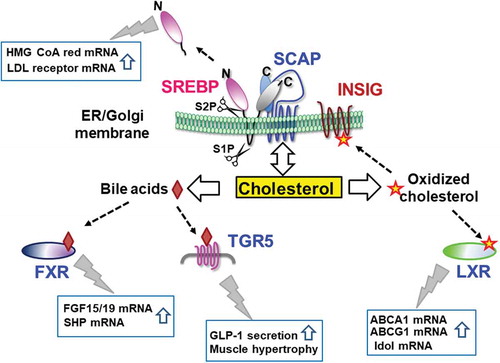
Cholesterol and its metabolites regulate the activity of SREBP, LXR, and FXR, thereby controlling their target gene expression. TGR5 activation triggers the downstream signal transduction.
KEYWORDS:
Cholesterol is synthesized from acetyl-coenzyme A (CoA) through more than 30 enzymatic reactions, making this pathway a time- and energy-consuming process. The primary organ that synthesizes cholesterol is the liver, which produces approximately 1 g of cholesterol per day. Extrahepatic tissues can also synthesize cholesterol, but not sufficiently, thus taking up cholesterol in the form of low-density lipoprotein (LDL) from the blood. The enzyme 3-hydroxy-methylglutaryl CoA (HMG CoA) reductase catalyzes the HMG CoA conversion to mevalonate and plays an important role as a rate-limiting enzyme in the cholesterol synthesis pathway. Statin, originally developed in Japan as a therapeutic drug for hypercholesterolemia, inhibits HMG CoA reductase and is taken worldwide by approximately 30 million people [Citation1]. Intracellular cholesterol levels are tightly regulated under negative feedback control, in which cholesterol or its metabolites reduce cholesterol synthesis-related gene expression and the LDL receptor gene. Excess cholesterol simultaneously degrades HMG CoA reductase protein to blunt cholesterol synthesis [Citation2]. Several intermediates in the cholesterol biosynthesis pathway also play critical physiological roles, reflecting the importance of this pathway. The cholesterol precursor molecule, lanosterol, and its metabolites trigger rapid HMG CoA reductase turnover [Citation3]. The farnesyl group controls intracellular vesicular transport and signal transduction by providing membrane-bound properties to several proteins.
Cholesterol can not be burned or decomposed in our body, so it must be converted into bile acids in the liver. When stored in the gallbladder, bile acids are secreted into the upper small intestine and serve as solubilizers to facilitate the absorption of lipids and fat-soluble vitamins. Greater than 90% of bile acids are reabsorbed in the ileum through a bile acid transporter. They are then transported back to the liver, and some of them leak into the systemic blood flow. Recent discovery of the ubiquitous G protein-coupled receptor (GPCR) bile acid-binding receptor TGR5 has begun to elucidate the largely unknown physiological function of circulating bile acids [Citation4,Citation5].
Discovery of sterol regulatory element-binding proteins
Cells maintain a steady-state level of membrane cholesterol by balancing its biosynthesis, uptake, and efflux to maintain membrane fluidity and metabolic homeostasis. Cholesterol synthesis and uptake pathways are primarily regulated at the transcriptional level through classic end product feedback inhibition of cholesterol biosynthesis enzyme gene expression and via LDLR, which mediates LDL endocytosis. The expression of cholesterol synthesis and uptake genes is enhanced under cholesterol-starved conditions, but is suppressed under cholesterol-excessive conditions [Citation6]. The promoter regions of these genes contain a shared DNA element called the sterol regulatory element (SRE), which is responsible for sterol-dependent regulation of expression. Using double-strand DNA fragments containing the SRE sequence (5′-ATCACCCCAC-3′), specific proteins were purified from nuclear extracts of cells cultured under sterol-depleted conditions. These were named the SRE-binding proteins (SREBPs), SREBP-1 and SREBP-2 [Citation7–Citation10]. Despite nuclear proteins gained from the soluble nuclear fraction, SREBPs are unexpectedly synthesized as membrane proteins inserted into the endoplasmic reticulum (ER) in a hairpin orientation with their N-termini and C-termini extending into the cytoplasm [Citation11]. After SREBPs exit the ER and enter the Golgi apparatus, they are processed by two types of cleavage enzymes, site 1 and site 2 proteases (S1P and S2P), thereby generating a soluble N-terminal-cleaved transcription factor of the basic helix-loop-helix leucine zipper family [Citation12]. This proteolytic processing enables active SREBP forms to enter the nucleus as soluble proteins, bind to SRE sequences, and stimulate gene expression of their targets. A series of studies revealed that SREBP-1 primarily regulates fatty acid synthesis-related gene expression and SREBP-2 controls cholesterol metabolism-related gene expression [Citation13,Citation14].
SREBP cleavage-activating protein
SREBPs form heterodimer complexes with the eight transmembrane domain ER membrane protein, SREBP cleavage-activating protein (SCAP). The SCAP C-terminal region contains a WD40 repeat domain known to be involved in various protein-protein interactions. The SCAP C-terminus that projects into the cytoplasm interacts with the C-terminus of SREBP-1 or SREBP-2. Structural analysis of yeast SCAP, which has a WD40 region similar in length to that found in mammals, suggests that the mammalian WD40 repeat domain might be an eight-bladed β-propeller that binds with SREBP-1 and SREBP-2 [Citation15]. This complex formation is thought to be very important for controlling SREBP’s activity as a transcription factor because SCAP, not SREBP, is deeply involved in transporting the SREBP-SCAP complex from the ER to the Golgi. Apart from being an escort protein, SCAP stabilizes the SREBP-SCAP complex. Mutant CHO cells with SCAP deficiencies have SREBPs that are unstable and rapidly broken down, leading to a marked decline in SREBP target gene expression [Citation16]. Interestingly, the SCAP C-terminal WD40 repeat domain interacts with the C-terminus of SREBP-1 and SREBP-2, which only share approximately 49% amino acid homology. This fact has generated the hypothesis that unknown proteins other than SREBPs might interact with the WD40 repeat domain, thereby affecting intracellular SREBP destiny. Using a SCAP C-terminal region that lacked its transmembrane region as a bait, we purified SCAP-binding proteins and identified heat shock protein 90 (HSP90) using mass spectrometry [Citation17]. HSP90 is known as a chaperone protein that facilitates the function and stability of its almost 200 associated proteins. As a recently identified SREBP regulator, HSP90 binds to and stabilizes SREBPs and SCAP in the ER and the Golgi. HSP90 inhibition leads to ubiquitin- and proteasome-dependent complex degradation. A series of in vitro and in vivo experiments revealed that HSP90 inhibition reduced SREBP target gene expression and lipid biosynthesis due to a decline in active SREBPs ().
Figure 1. HSP90 interacts with and stabilizes the SREBP-SCAP complex, thereby increasing lipid synthesis. HSP90 interacts with the C-terminal regions of SREBP and SCAP in the ER and Golgi. This protection increases triglyceride and cholesterol synthesis. SREBP and SCAP are rapidly degraded in the ubiquitin-proteasome pathway in the presence of the HSP90 inhibitor 17-AAG or siHSP90 RNA.

SCAP plays an indispensable role as an escort protein that transports SREBPs from the ER to the Golgi, thereby enabling SREBPs to enter ER transport vesicles that contain COPII vesicle coat proteins, such as SEC23/24 and SIR1 [Citation18,Citation19]. The loop6 domain extends into the cytoplasm between SCAP’s transmembrane domain 6 and transmembrane domain 7 and contains the COPII binding site known as the Met-Glu-Leu-Ala-Asp-Leu (MELADL) motif. This binding enables the SREBP-SCAP complex to be embedded on COPII vesicles [Citation20]. In addition to the eight transmembrane helices, SCAP possesses two large ER luminal loops. These are loop1, between transmembrane domains 1 and 2, and loop7, between transmembrane domains 7 and 8. Loop1 binds to loop7, thereby enabling SCAP to bind COPII proteins, such as SEC23 and SEC24 [Citation21]. However, loop1 binds to cholesterol when cholesterol levels in the ER membrane rise, thereby disrupting direct binding between the two loops. This triggers a conformational change that conceals the MELADL motif and prevents the SREBP-SCAP complex from exiting the ER.
Our previous finding that, in the absence of HSP90, SCAP is preferentially degraded in advance of SREBPs through a ubiquitin-proteasome pathway, led us to research its specific ubiquitin ligase. Over multiple experiments, we found that an E3 ubiquitin ligase, ring finger protein (RNF) 5, participates in SCAP ubiquitination without leading to its degradation. Another SCAP E3 ligase, RNF145, triggers SCAP ubiquitination on lysine residues within a cytoplasmic loop essential for COPII binding [Citation22]. This potentially inhibits its transport to the Golgi and subsequent SREBP-2 processing. RNF5 adds a polyubiquitin chain to a Lys residue at position 305, resulting in efficient SREBP-SCAP complex transport from the ER to the Golgi [Citation23]. Interestingly, RNF5 forms a polyubiquitin chain via the 29th Lys residue of the ubiquitin molecule, whereas the polyubiquitin chain via the 46th Lys residue generally results in substrate degradation. Mechanistically, ubiquitination modification of SCAP by RNF5 enhances the loop1 and loop7 interaction that occurs at the ER luminal side, thereby facilitating the exit of the SREBP-SCAP complex from the ER. The SCAP ubiquitination at position 305 might be a novel target for discovering therapeutic drugs or functional food ingredients that regulate SREBP activation ().
Figure 2. The E3 ubiquitin ligase RNF5 modifies SCAP via the lysine 29 polyubiquitinated chain. SCAP has eight transmembrane helices and two large luminal loops, designated loop1 and loop7. Their mutual contact allows SCAP to bind COPII proteins due to the interaction between the MELADL motif and SEC24 for transport in coated vesicles. When ER cholesterol rises, it binds to loop1. This masks the motif and hinders the SREBP-SCAP complex from exiting the ER. RNF5-induced K29 polyubiquitination of SCAP strengthens the loop1-loop7 interaction, thereby assisting ER-to-Golgi complex translocation.

Insulin-induced gene proteins
Two additional ER retention membrane proteins, insulin-induced gene 1 protein (INSIG1) and INSIG2, interact with SCAP under conditions with excess cholesterol [Citation24–Citation27]. These proteins keep the SREBP-SCAP complex in the ER membrane, thus blocking active SREBP forms from entering the nucleus. It is assumed that SREBP-2 processing is under the control of this cholesterol-dependent ER-to-Golgi transport system. SREBP-1 might be activated via a different mechanism [Citation28], given that fatty acid metabolism controlled by SREBP-1 is regulated differently from cholesterol metabolism. The precise mechanism of SREBP-1 processing, however, remains ambiguous.
INSIG1 was initially identified as a gene with highly inducible expression by insulin [Citation29,Citation30]. INSIG1 and INSIG2 are ER retention membrane proteins with six transmembrane domains that share 69% of their amino acid identity. INSIG proteins reportedly bind to oxidized cholesterols, including 22-hydroxycholesterol, 24-hydroxycholesterol, 25-hydroxycholesterol, and 27-hydroxycholesterol, in a pocket within the transmembrane spanning regions. They also interact with SCAP via transmembrane domains 3 and 4 [Citation31]. Although INSIG1 recruits two types of E3 ligases, GP78 and TRC8, INSIG2 only forms a complex with TRC8 [Citation32]. One intriguing INSIG function is that these proteins not only hinder the SREBP-SCAP complex from exiting the ER, but also simultaneously trigger HMG CoA reductase protein degradation, thereby reducing cholesterol biosynthesis. Elevation of oxidized cholesterol levels in the ER membrane stabilizes INSIGs via binding, resulting in these bifunctional effects. Conversely, INSIGs are ubiquitinated by their associated E3 ligases and rapidly degraded through the proteasome pathway under low oxidized cholesterol conditions [Citation33]. This removes the block on the SREBP-SCAP complex exiting the ER and increases SREBP target gene expression.
A recent report demonstrated that INSIG protein phosphorylation reduced oxidized cholesterol binding to these proteins, thereby disrupting the interaction between SCAP and INSIG proteins [Citation34]. It is conceivable that cancer cells increase lipogenesis for their proliferation by activating SREBPs via this process. Activated AKT phosphorylates cytosolic phosphoenolpyruvate carboxykinase 1 (PCK1), a rate-limiting enzyme in gluconeogenesis. This induces translocation of PCK1 to the ER, where PCK1 uses GTP as a phosphate donor to phosphorylate INSIG1/2. The detailed mechanism by which INSIG phosphorylation results in reduced oxidized cholesterol binding remains unclear. Another report showed that INSIG1 phosphorylation by AMP-dependent protein kinase (AMPK) reduces INSIG1’s interaction with GP78, thereby repressing its ubiquitination and degradation [Citation35]. AMPK activation might augment INSIG stability and reduce lipogenic gene expression.
In GP78-/- mice, cholesterol biosynthesis is reduced via decreased SREBP transcriptional activity due to stabilized INSIGs, or reduced GP78-mediated ubiquitination, in the face of increased HMG CoA reductase protein. This finding indicates that INSIG proteins can prevent the ER-to-Golgi translocation of the SREBP-SCAP complex, thereby depressing SREBP transcriptional activity in the absence of GP78-mediated HMG CoA reductase degradation [Citation36]. Nevertheless, how the physiological roles of INSIG1 and INSIG2 are divided in the dual regulation of SCAP and HMG CoA reductase remains unclear. However, clear functional distinctions exist between INSIG isoforms. For instance, INSIG1 is highly induced by insulin, whereas an INSIG2 splicing isoform expressed primarily in the liver, INSIG2a, is reduced by insulin [Citation37]. A recent paper demonstrated that the cAMP response element-binding protein (CREB) bZIP transcription factor, one of the insulin-induced genes, directly inhibits INSIG2a transcription through association with activating transcription factor 4 (ATF4) [Citation38]. Meanwhile, INSIG1/2-double-knockout mice overproduce sterols and their intermediates, which trigger midline facial abnormalities and death soon after birth [Citation39]. This is likely due to disturbed function of the hedgehog protein that is modified with cholesterol to control body shape development.
Bile acids and their receptors
Recent scientific findings support the hypothesis that bile acids, which are cholesterol catabolites produced only in the liver, are bioactive signaling molecules that function as ligands for FXR or the GPCR, TGR5 [Citation40]. Cholesterol can be synthesized from acetyl-CoA, but cannot decompose into smaller molecules in our body. It must be converted into bile acid to be catabolized and excreted in the feces. Bile acid, lipid, and carbohydrate homeostasis are all controlled by bile acids binding to two types of receptors. By activating FXR, bile acids induce robust transcription of the peptide hormone FGF15 and FGF19 in the ileum of mice and humans, respectively. Beneficial effects of FGF15/19 are potentially due to reduced hepatic gluconeogenesis, increased hepatic glycogen synthesis, reduced body weight and adiposity, increased peripheral glucose disposal, and increased muscle mass [Citation41]. An intriguing finding regarding TGR5’s role in energy metabolism and glucose homeostasis suggests that targeting this receptor to increase thermogenesis and glucagon-like peptide 1 (GLP-1) secretion may combat obesity and insulin resistance [Citation42–Citation44].
Bile acids synthesized in the liver are converted to bile salts by conjugation with glycine or taurine. Bile salts are secreted into the bile ducts, then stored in the gallbladder until they are secreted into the duodenum. After each meal, the gallbladder contracts to expel bile into the intestinal tract, where bile acids serve as solubilizers that facilitate lipid and fat-soluble vitamin absorption. More than 90% of bile salts are reabsorbed in the ileum through a bile acid transporter, ileal bile acid transporter, then transported back to the liver. It has been estimated that bile acids are recycled more than 10 times before they are finally eliminated. Some bile acids leak into the systemic blood flow as they return to the liver, resulting in a circulating level of approximately 10 μM. The blood bile acid concentration transiently elevates after every meal. Thus, this rise is considered to be a type of feeding signal. TGR5 has been identified as a sensor protein for this feeding signal. Once bile acid binds to TGR5, intracellular cAMP concentration elevates, protein kinase A is activated in response, CREB is activated, and downstream signals are transmitted. In the L cells of the small and large intestine, TGR5 stimulates GLP-1 secretion, one of incretin, which improves insulin sensitivity.
In parallel with the events that occur in the intestinal tract, TGR5 stimulates type II iodothyronine deiodinase gene expression in the human skeletal muscle and rodent brown adipose tissue. This enzyme converts the inactive thyroid hormone T4 form to the active T3 form in the cell [Citation45]. TGR5 activation also results in increased peroxisome proliferator-activated receptor γ coactivator-1α gene expression and enhanced mitochondria activity. Both of these processes promote thermogenesis. Taken together, these results indicate that bile acid improves insulin resistance by incretin actions via the bile acid-TGR5 axis. Furthermore, this may promote anti-obesity effects by elevating thermogenesis.
Novel functions of skeletal muscle TGR5
Skeletal muscle expresses multiple GPCR types, including the β2-adrenergic receptor and corticotropin-releasing factor receptor 2. Together with their ligands, these receptors induce cAMP accumulation in myofibers, thereby eliciting muscle hypertrophy [Citation46]. Notably, clenbuterol, a synthetic ligand of the β2-adrenergic receptor, is designated as a banned doping drug because it induces hypertrophy when administered to animals and humans [Citation47]. Because TGR5 is also expressed in skeletal muscle and activates cAMP signaling, the biological function of muscle TGR5 was investigated using a transgenic TGR5 mouse line (Tg mice) [Citation48]. These mice are altered to express human TGR5 in skeletal muscle because mice express relatively low TGR5 levels in muscle compared to humans. Constant human TGR5 expression increased muscle mass in the gastrocnemius and quadriceps by 10–15% and appeared to significantly strengthen muscle power. When the mice were fed a high-fat diet to induce impaired glucose tolerance, an oral glucose tolerance test revealed that the Tg mice showed ameliorated postprandial hyperglycemia and improved glucose metabolism. Conversely, muscle mass in TGR5-deficient mice was significantly decreased, which was accompanied by reduced muscle strength. These results suggest that TGR5 activation by blood bile acids results in muscle hypertrophy and improves glucose metabolism.
Physiological roles of bile acid-TGR5 interaction in skeletal muscle
Cell biological analyses using differentiated C2C12 myotubes revealed that ER stress elevates TGR5 gene expression. Previous work has shown that an exercise challenge test induces ER stress in mouse skeletal muscle [Citation49]. TGR5 mRNA levels significantly increased in the gastrocnemius of wild type mice following 1 h of treadmill training. The promoter region of the mouse TGR5 gene contains the ER stress response motif that is controlled by ATF6 and the transcription factor NF-Y [Citation50]. A mutation analysis of the promoter region revealed the importance of this motif for exercise-induced TGR5 transcription. Furthermore, this induction was not present in ATF6-deficient mice when compared to wild type mice. Bile acids are secreted into the small intestine as a response to feeding, resulting in high blood levels after eating. Therefore, we suggest that blood bile acid is one feeding signal whose elevation is likely to act as a signal that induces skeletal muscle hypertrophy. Likewise, bile acid-TGR5 signaling is stimulated immediately following exercise, in line with elevated TGR5 expression, thus leading to muscle hypertrophy ().
Figure 3. The proposed role of Tgr5 in skeletal muscle. ATF6 increases TGR5 expression in muscle cells, which is mimicked by exercise in vivo. TGR5 activation promotes muscle hypertrophy and muscle cell differentiation.

Perspectives
Bile acid metabolism by gut microorganisms is an important determinant of bile acid composition, which may substantially affect host metabolism. A common modification is removing the amino acid moiety of conjugated primary bile acids to create unconjugated forms that can be passively absorbed, primarily in the colon without bile acid uptake transporters. Therefore, agents that alter the microbiome can affect the composition of bile acid pools. Indeed, a recent report showed that metformin, a therapeutic drug for diabetes, altered the gut microbiome in a manner that generated host health benefits [Citation51]. The consequences of bacteria-modified bile acids on host metabolism are proactively under worldwide investigation. Novel methodological advances are likely to present new opportunities to further elucidate the effects of the microbiome-bile acid axis. Simultaneously, innovative approaches that further clarify cholesterol metabolism regulation are expected to lead to the development of therapeutic agents that are superior to statins ().
Figure 4. Cholesterol and its metabolites regulate the activity of transcription factors and their receptors. The ER membrane protein SREBP is proteolytically processed by two cleavage enzymes, site 1 and site 2 proteases, in the Golgi to generate its active soluble form. The active form translocates to the nucleus, thereby increasing target gene expression. The SREBP-associated protein SCAP controls exit of the SREBP-SCAP complex from the ER by cholesterol binding. Cholesterol metabolites, such as oxidized cholesterols and bile acids, bind and activate LXR and FXR, respectively, thereby stimulating their target gene expression. TGR5 activation by bile acids triggers GLP-1 secretion from the intestine and leads to skeletal muscle hypertrophy.

Acknowledgments
The author would like to thank Enago for the English language review.
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
References
- Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161(1):161–172.
- Johnson BM, DeBose-Boyd RA. Underlying mechanisms for sterol-induced ubiquitination and ER-associated degradation of HMG CoA reductase. Semin Cell Dev Biol. 2018;81:121–128.
- Coates HW, Brown AJ. A wolf in sheep’s clothing: unmasking the lanosterol-induced degradation of HMG-CoA reductase. J Lipid Res. 2019;60(10):1643–1645.
- Maruyama T, Miyamoto Y, Nakamura T, et al. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun. 2002;298(5):714–719.
- Kawamata Y, Fujii R, Hosoya M, et al. Shintani Y and others. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278(11):9435–9440.
- Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annu Rev Genet. 2007;41:401–427.
- Yokoyama C, Wang X, Briggs MR, et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. 1993;75(1):187–197.
- Tontonoz P, Kim JB, Graves RA, et al. ADD1: a novel helix-loop-helix transcription factor associated with adipocyte determination and differentiation. Mol Cell Biol. 1993;13(8):4753–4759.
- Sato R, Yang J, Wang X, et al. Assignment of the membrane attachment, DNA binding, and transcriptional activation domains of sterol regulatory element-binding protein-1 (SREBP-1). J Biol Chem. 1994;269(25):17267–17273.
- Hua X, Yokoyama C, Wu J, et al. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci U S A. 1993;90(24):11603–11607.
- Wang X, Sato R, Brown MS, et al. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. 1994;77(1):53–62.
- Sakai J, Rawson RB, Espenshade PJ, et al. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol Cell. 1998;2(4):505–514.
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131.
- Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. 2017;13(12):710–730.
- Gong X, Li J, Shao W, et al. Structure of the WD40 domain of SCAP from fission yeast reveals the molecular basis for SREBP recognition. Cell Res. 2015;25(4):401–411.
- Rawson RB, DeBose-Boyd R, Goldstein JL, et al. Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J Biol Chem. 1999;274(40):28549–28556.
- Kuan YC, Hashidume T, Shibata T, et al. Protein 90 modulates lipid homeostasis by regulating the stability and function of sterol regulatory element-binding protein (SREBP) and SREBP cleavage-activating protein. J Biol Chem. 2017;292(7):3016–3028.
- Sun LP, Li L, Goldstein JL, et al. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem. 2005;280(28):26483–26490.
- Espenshade PJ, Li WP, Yabe D. Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proc Natl Acad Sci U S A. 2002;99(18):11694–11699.
- Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer’s bottle to Scap’s MELADL. J Lipid Res. 2009;50(Suppl):S15–27.
- Zhang Y, Lee KM, Kinch LN, et al. Direct demonstration that loop1 of scap binds to loop7: A CRUCIAL EVENT IN CHOLESTEROL HOMEOSTASIS. J Biol Chem. 2016;291(24):12888–12896.
- Zhang L, Rajbhandari P, Priest C, et al. Inhibition of cholesterol biosynthesis through RNF145-dependent ubiquitination of SCAP. Elife. 2017;6. DOI:10.7554/eLife.28766
- Kuan YC, Takahashi Y, Maruyama T, et al. Ring finger protein 5 activates sterol regulatory element-binding protein 2 (SREBP2) to promote cholesterol biosynthesis via inducing polyubiquitination of SREBP chaperone SCAP. J Biol Chem. 2020;295(12):3918–3928.
- Yang T, Espenshade PJ, Wright ME, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110(4):489–500.
- Engelking LJ, Kuriyama H, Hammer RE, et al. Overexpression of Insig-1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin-stimulated lipogenesis. J Clin Invest. 2004;113(8):1168–1175.
- Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci U S A. 2002;99(20):12753–12758.
- Sun LP, Seemann J, Goldstein JL, et al. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci U S A. 2007;104(16):6519–6526.
- Nakakuki M, Kawano H, Notsu T, et al. A novel processing system of sterol regulatory element-binding protein-1c regulated by polyunsaturated fatty acid. J Biochem. 2014;155(5):301–313.
- Mohn KL, Laz TM, Hsu JC, et al. The immediate-early growth response in regenerating liver and insulin-stimulated H-35 cells: comparison with serum-stimulated 3T3 cells and identification of 41 novel immediate-early genes. Mol Cell Biol. 1991;11(1):381–390.
- Diamond RH, Du K, Lee VM, et al. Novel delayed-early and highly insulin-induced growth response genes. Identification of HRS, a potential regulator of alternative pre-mRNA splicing. J Biol Chem. 1993;268(20):15185–15192.
- Radhakrishnan A, Ikeda Y, Kwon HJ, et al. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A. 2007;104(16):6511–6518.
- Jo Y, Lee PC, Sguigna PV, et al. Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. Proc Natl Acad Sci U S A. 2011;108(51):20503–20508.
- Gong Y, Lee JN, Lee PC, et al. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006;3(1):15–24.
- Xu D, Wang Z, Xia Y, et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature. 2020;580(7804):530–535.
- Han Y, Hu Z, Cui A, et al. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nat Commun. 2019;10(1):623.
- Liu TF, Tang JJ, Li PS, et al. Ablation of gp78 in liver improves hyperlipidemia and insulin resistance by inhibiting SREBP to decrease lipid biosynthesis. Cell Metab. 2012;16(2):213–225.
- Yabe D, Komuro R, Liang G, et al. Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc Natl Acad Sci U S A. 2003;100(6):3155–3160.
- Zhang F, Hu Z, Li G, et al. Hepatic CREBZF couples insulin to lipogenesis by inhibiting insig activity and contributes to hepatic steatosis in diet-induced insulin-resistant mice. Hepatology. 2018;68(4):1361–1375.
- Engelking LJ, Evers BM, Richardson JA, et al. Severe facial clefting in Insig-deficient mouse embryos caused by sterol accumulation and reversed by lovastatin. J Clin Invest. 2006;116(9):2356–2365.
- Ahmad TR, Haeusler RA. Bile acids in glucose metabolism and insulin signaling – mechanisms and research needs. Nat Rev Endocrinol. 2019;15(12):701–712.
- Benoit B, Meugnier E, Castelli M, et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat Med. 2017;23(8):990–996.
- Watanabe M, Houten SM, Mataki C, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439(7075):484–489.
- Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun. 2005;329(1):386–390.
- Thomas C, Gioiello A, Noriega L, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10(3):167–177.
- Zietak M, Kozak LP. Bile acids induce uncoupling protein 1-dependent thermogenesis and stimulate energy expenditure at thermoneutrality in mice. Am J Physiol Endocrinol Metab. 2016;310(5):E346–54.
- Berdeaux R, Stewart R. cAMP signaling in skeletal muscle adaptation: hypertrophy, metabolism, and regeneration. Am J Physiol Endocrinol Metab. 2011;303(1):E1–17.
- Woodall BP, Woodall MC, Luongo TS, et al. Skeletal muscle-specific G protein-coupled receptor kinase 2 ablation alters isolated skeletal muscle mechanics and enhances clenbuterol-stimulated hypertrophy. J Biol Chem. 2016;291(42):21913–21924.
- Sasaki T, Kuboyama A, Mita M, et al. The exercise-inducible bile acid receptor Tgr5 improves skeletal muscle function in mice. J Biol Chem. 2018;293(26):10322–10332.
- Wu J, Ruas JL, Estall JL, et al. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1α/ATF6α complex. Cell Metab. 2011;13(2):160–169.
- Yoshida H, Okada T, Haze K, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20(18):6755–6767.
- Sun L, Xie C, Wang G, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med. 2018;24(12):1919–1929.