
ABSTRACT
Autophagy induced in cancer cells during chemotherapy is classified into two types, which differ depending on the kind of cells or anticancer drugs. The first type of autophagy contributes to the death of cells treated with drugs. In contrast, the second type plays a crucial role in preventing anticancer drug-induced cell damages; the use of an autophagy inhibitor is considered effective in improving the efficacy of chemotherapy. Thus, it is important to determine which type of autophagy is induced during chemotherapy. Here, we showed that a novel inhibitor of RNA polymerase I, suppresses growth, induces cell cycle arrest and promotes apoptosis in leukemia cell lines. The number of apoptotic cells induced by co-treatment with CX-5461 and chloroquine, an autophagy inhibitor, increased compared with CX-5461 alone. Thus, the autophagy which may be induced by CX-5461 was the second type.
Graphical Abstract
The number of apoptotic cells induced by co-treatment with CX-5461 and CQ increased compared with CX-5461 alone.
Autophagy, a degradation process in which cellular organelles and proteins are engulfed by autophagosomes, is essential for cell homeostasis and metabolism [Citation1]. Autophagy promotes cell survival in response to cellular stressors, such as starvation, infection, and hypoxia. Moreover, several studies indicate that autophagy also promotes cancer survival during chemotherapy [Citation2,Citation3]. Autophagy is classified into two types. The first contributes to the death of cancer cells treated with anticancer agents, referred to as autophagic cell death [Citation4–Citation7]. This programmed cell death is termed type II, in contrast to apoptosis, programmed cell death type I [Citation8]. Many studies demonstrate that autophagic activity may also modulate apoptotic cell death [Citation8]. Autophagic cell death leads to a decrease in the number of surviving cancer cells, and the use of autophagy inhibitors is not effective for cancer treatment. On the contrary, second type autophagy may prevent cell death during chemotherapy [Citation2–Citation4,Citation7,Citation9]. This type of autophagy suppresses the activity of anticancer drugs, the use of autophagy inhibitors may improve the efficacy of chemotherapy. Several clinical trials have examined this hypothesis [Citation2,Citation10–Citation12]. However, the type of autophagy induced depends on cancer cells types and anticancer drugs used.
A large nucleolus is a morphological feature of cancer cells [Citation13]. The nucleolus is the main site of ribosome biogenesis. Thus, a large nucleolus can increase ribosome production; numbers of ribosomes are increased in cancer cells [Citation14]. The ribosome is the central protein synthesis organelle, composed of ribosomal RNAs and proteins. The ribosomal RNAs are transcribed from ribosomal DNA genes by Pol I. Pol I thus plays a crucial role in cell viability and proliferation. Several studies demonstrated that Pol I is up-regulated in cancer cells [Citation15,Citation16].
CX-5461, a new class of small molecule targeted anti cancer therapeutics, selectively inhibits Pol I transcription relative to Pol II transcription [Citation17]. CX-5461 exhibits anticancer activity in epithelial cancer cells and potent in vivo anti-tumor effect on murine xenograft models of human solid tumors [Citation17–Citation19].
CX-5461 is effective in treating hematologic malignancy [Citation20–Citation23] and is currently being evaluated in clinical trials for the treatment of hematopoietic malignancies [Citation24,Citation25]. However, whether CX-5461 induces autophagy in acute myeloid leukemia (AML) is not known. Hence, we examined anticancer and autophagy-inducing properties of CX-5461 on human AML cell lines. Here, we show that the number of apoptotic cells induced by co-treatment with CX-5461 and chloroquine, an autophagy inhibitor, increased compared with CX-5461 alone. This finding suggests a possible role of autophagy on leukemia cells is opposite from its role in solid tumors [Citation17,Citation18]. Thus, CX5461 may be useful for AML treatment.
Materials and methods
Cell lines and reagents
HL-60, KG-1, and THP-1 cells, human acute myeloid leukemia cell lines, were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS). CX-5461 was purchased from AdooQ BioScience (Irvine, CA, USA) and chloroquine (CQ) from Cell Signaling Technology (Danvers, MA, USA). CX-5461 was dissolved in dimethylsulfoxide and stored at −80°C. CQ was dissolved in distilled water and stored at 4°C.
Cell viability assays
Half-maximal inhibitory concentrations (IC50s) were assessed with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assays. Cell suspensions were plated into 96-well plates in the presence of the drug or solvent, incubated at 37°C for 1–2 days, and analyzed using the MTT assay [Citation9].
Cell cycle analysis
The cells were fixed with 70% methanol for 30 min, treated with FxCycle PI/RNase Staining Solution (Thermo Fisher Scientific, Waltham, MA, USA) for 30 min at room temperature. Samples were analyzed with a BD FACS CANTO II flow cytometer and Diva software (Becton Dickinson, Franklin Lakes, NJ, USA).
Immunoblotting analysis
Cell lysates were prepared in Laemmli sample buffer (Bio-Rad Laboratories Inc., Hercules, CA, USA) containing 2-mercaptoethanol, then heated for 5 min at 95°C. Proteins cell lysate were separated via SDS-PAGE and transferred to a polyvinylidene difluoride membranes. Membranes were blocked with 2% skim milk in PBS-T (phosphate-buffered saline with Tween 20) for 60 min at room temperature. Membranes were then incubated with appropriate antibodies overnight, washed three times with PBS-T and incubated with HRP-conjugated goat anti-rabbit antibodies for 30 min. Specific proteins were detected using an enhanced chemiluminescence detection system. Primary antibodies against LC3A/B, C-PARP, and β-actin were obtained from Cell Signaling Technology (Danvers, MA, USA). Primary antibody against β-tubulin was obtained from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). HRP-conjugated rabbit secondary antibody was purchased from GE Healthcare Life Sciences (Piscataway, NJ, USA).
Apoptosis assay
Apoptosis was assessed using an Annexin V Apoptosis Detection Kit (BD Pharmingen, San Diego, CA, USA). All samples were analyzed with a BD FAC SCANTO II and Diva software (Becton Dickinson Franklin Lakes, NJ, USA).
Autophagy assay
Autophagy was evaluated using a DALGreen Autophagy Detection Kit (DOJINDO, Kumamoto, Japan). Cells were stained for 30 min at 37°C and analyzed a BD FAC SCANTO II and Diva software (Becton Dickinson Franklin Lakes, NJ, USA).
Immunofluorescence staining
Cells were centrifuged in cytospin slides, fixed for 15 min in 100% methanol at −20°C, and blocked with blocking buffer containing 5% FBS and 0.3% Triton X-100 in phosphate-buffered saline (pH 8.0) for 60 min. Immunofluorescence staining was performed using a rabbit monoclonal anti-LC3A/B antibody to detect autophagosomes, and Alexa Fluor 488 conjugated and IgG (Molecular Probes, Eugene, OR, USA) were used as secondary antibodies. Nuclei were stained with Hoechst 33,342 (Dojindo, Kumamoto, Japan). Cells were observed under a BX51 fluorescence microscope (Olympus, Tokyo, Japan).
Quantitative reverse transcriptase polymerase reaction (qRT-PCR)
Total RNA was extracted from the cells using an RNeasy Kit (QIAGEN, Hilden, Germany). cDNA synthesis used a Ready-To-Go You-Prime First-Strand Beads (GE Healthcare, Little Chalfont, UK). qRT-PCR used SYBR Green (Life Technologies, Carlsbad, CA, USA), and signals during PCR were detected using a Step One Plus Realtime PCR System (Life Technologies). CX-5461 selectively inhibits rRNA synthesis, but not DNA, mRNA or protein synthesis [Citation17], and human housekeeping gene β-actin was used as an internal control for data normalization. Data were analyzed using the software and PCR system. The pre-rRNA forward primer was 5ʹ-CTGAGGGAGCTCGTCGGTGT-3ʹ, and its reverse primer was 5ʹ-GCAGAGCGCCTCCGAAGTCA-3ʹ [Citation20]. The β-actin forward primer was 5ʹ-ACGAGGCCCAGAGCAAGAG-3ʹ, and its reverse primer was 5ʹ-GACGATGCCGTGCTCGAT-3ʹ.
Statistical analyzes
Data were expressed as means ± standard deviation (SD). Between-group comparisons were performed using t-tests and Tukey-Kramer’s multiple comparisons of means tests. Statistical analyzes were performed with EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria). More precisely, EZR is a modified version of R commander, a tool designed to add statistical functions frequently used in biostatistics [Citation26].
The effect of co-treatment was measured using the combination index (CI) and analyzed with CalcuSyn software (version 2.0, Biosoft). Synergy, additivity, and antagonism are defined as CI < 1, CI = 1, and CI > 1, respectively.
Results and discussion
CX-5461 suppresses pre-rRNA translation
We examined the capability of CX5461 to inhibit Pol I transcription in AML. HL-60 cells were treated with indicated concentrations of CX-5461 for 1 h, and the inhibition of Pol I transcription was examined using pre-rRNA translation by RT-qPCR analysis. CX-5461 suppressed pre-rRNA translation in HL-60 cells (). Thus, treatment with CX-5461 suppresses ribosome biogenesis via the inhibition of Pol I transcription.
Figure 1. CX5461 suppressed the growth of acute leukemia cell lines.

CX-5461 inhibits the proliferation of AML cell lines
To investigate the suppressive effects of CX-5461 on growth, we treated AML cell lines with CX-5461 and assessed their proliferation at 24 and 48 h using the MTT assay. CX-5461 had a concentration-dependent growth-suppressive effect on these cell lines, with IC50 values of 38 nM at 24 h and 28.3 nM at 48 h in HL-60 cells (). The IC50 values of the other cell lines are presented in .
Next, we measured the effect of CX-5461 on cell cycle progression in these cell lines. Propidium iodide (PI) staining after treatment with CX-5461 revealed an increased proportion of HL-60 cells in the G2/M phase, whereas KG-1 and THP-1 cells were mostly in the S phase (). Therefore, cell-cycle arrest induced by CX-5461 displayed diversity dependent on cell type.
CX-5461 induces apoptosis in AML cell lines
Apoptosis ultimately causes cell death after inducing cell cycle arrest. Although several studies have shown that treatment with CX-5461 does not induce apoptosis in solid tumors [Citation17–Citation19], others have demonstrated that it does induce apoptosis during hematological malignancy [Citation20–Citation22]. We therefore assessed whether CX-5461 induced apoptosis in AML cell lines using Annexin V/PI dual staining. Treatment with CX-5461 increased the number of apoptotic cells in these cell lines (). Immunoblotting analysis of apoptosis in these cell lines [Citation17,Citation18] further revealed that treatment with CX-5461 increased cleaved poly-ADP-ribose polymerase, confirming that CX-5461 induced apoptosis in these cell lines ().
Figure 2. CX5461 induced apoptosis in acute myeloid leukemia cell lines.

CX-5461 has the potential to induce autophagy in AML cell lines
Several studies have indicated that autophagy is associated with cancer cell survival during chemotherapy [Citation2,Citation3]. To investigate whether CX-5461 induces autophagy in leukemia cells, autophagosomes were assessed via immunofluorescence staining. Treatment with CX-5461 caused the accumulation of autophagosomes in HL-60 cells (). Next, immunoblotting analysis revealed that treatment with CX-5461 caused the accumulation of LC3A/B-II (). Furthermore, autophagosomes were assessed using DALGreen Autophagy Detection Kit. CX-5461 also increased fluorescence intensity in a dose-dependent manner (Supplemental Figure 1). These results suggest that CX-5461 has the potential to induce autophagy in AML cell lines.
Figure 3. Treatment with CX5461 enhanced autophagy in acute myeloid leukemia cell lines.

Inhibition of autophagy enhances CX-5461-induced apoptosis
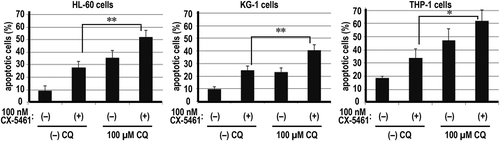
Recent studies have demonstrated two types of autophagy induced by anticancer agents. The first type induces cell death [Citation4–Citation7] defined as type II programmed cell death [Citation5–Citation7]. By contrast, the second type of autophagy plays a role in preventing cell death [Citation2–Citation4,Citation7]. Thus, we examined whether the role of autophagy in cell survival during CX-5461 treatment is the first or second type. Validation of the role of autophagy by co-treatment with anticancer drugs and autophagy inhibitors is generally accepted [Citation7]. The treatment of AML cell lines with 0–100 µM CQ caused the accumulation of LC3A/B-II (). Using Annexin V/PI dual staining, we compared the proportion of surviving cells during the inhibition of autophagy. The number of apoptotic cells induced by co-treatment with CX-5461 and CQ increased compared with CX-5461 alone (). Thus, the autophagy observed in these cell lines is the second type, which prevents cell death.
Figure 4. Co-treatment with chloroquine and CX5461 enhanced the efficacy of CX5461.

Because CI is used to determine the synergistic, additive, or antagonistic effects of a drug combination [Citation27], we analyzed CI to identify the effect of co-treatment with CQ and CX-5461. We used CalcuSyn software to calculate CI. Synergy, additivity, and antagonism are defined as CI < 1, CI = 1, and CI > 1, respectively. Cells were treated with two different concentrations of CX-5461 and CQ (). In HL-60 and THP-1 cells, a relatively low concentration of CX-5461 (50 nM) caused an additive effect, whereas a high concentration of CX-5461 (100 nM) produced a synergistic effect. In KG-1 cells, a synergistic effect was observed after using any combination of CX-5461 and CQ. These results indicated that the inhibition of autophagy enhanced the anticancer effect of CX-5461.
Therefore, we demonstrated herein that CX-5461 has the potential to induce the second type of autophagy that prevents anticancer drug-induced cell damage in leukemia cell lines. This finding indicated that under these circumstances, the role of the autophagy on leukemia cells is different from that of solid tumors [Citation17,Citation18] and suggested that the use of an autophagy inhibitor during CX5461 treatment may be useful for AML treatment.
Author contributions
Substantial contributions to conception and design: SO, KM. Data acquisition, data analysis, and interpretation: SO, KM, MK, AY, FK. Drafting the article or critically revising it for important intellectual content: SO, KM, MK, AY, FK.
Supplemental_Information.pdf
Download PDF (184.6 KB)Acknowledgments
We are grateful to Chikage Kawai and Masumi Itadani (Kawasaki Medical School, Japan) for technical assistance and to the Central Research Institute of Kawasaki Medical School for technical support.
Disclosure statement
The authors declare that they have no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Muzushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741.
- Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy. 2006;2:85–90.
- Chittaranjan S, Bortnik S, Dragowska WH, et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin Cancer Res. 2014;20:3159–3173.
- Aveic S, Pantile M, Polo P, et al. Autophagy inhibition improves the cytotoxic effects of receptor tyrosine kinase inhibitors. Cancer Cell Int. 2018;18:63.
- Sui X, Chen R, Wang Z, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838.
- Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–266.
- Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7:457–465.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222.
- Okamoto S, Tsujioka T, Suemori S, et al. Withaferin A suppresses the growth of myelodysplasia and leukemia cell lines by inhibiting cell cycle progression. Cancer Sci. 2016;107:1302–1314.
- Samaras P, Tusup M, Nguyen-Kim TDL, et al. Phase I study of a chloroquine-gemcitabine combination in patients with metastatic or unresectable pancreatic cancer. Cancer Chemother Pharmacol. 2017;80:1005–1012.
- Manic G, Obrist F, Kroemer G, et al. Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol. 2014;1:e29911.
- Shi TT, Yu XX, Yan LJ, et al. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother Pharmacol. 2017;79:287–294.
- Derenzini M, Trerè D, Pession A, et al. Nucleolar function and size in cancer cells. Am J Pathol. 1998;152:1291–1297.
- Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192.
- Rossetti S, Wierzbicki AJ, Sacchi N. Mammary epithelial morphogenesis and early breast cancer. Evidence of involvement of basal components of the RNA Polymerase I transcription machinery. Cell Cycle. 2016;15:2515–2526.
- White RJ. RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol. 2005;6:69–78.
- Drygin D, Lin A, Bliesath J, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71:1418–1430.
- Li L, Li Y, Zhao J, et al. CX-5461 induces autophagy and inhibits tumor growth via mammalian target of rapamycin-related signaling pathways in osteosarcoma. Oncol Targets Ther. 2016;9:5985–5997.
- Chen H, Duo Y, Hu B, et al. PICT-1 triggers a pro-death autophagy through inhibiting rRNA transcription and AKT/mTOR/p70S6K signaling pathway. Oncotarget. 2016;7:78747–78763.
- Hein N, Cameron DP, Hannan KM, et al. Inhibition of Pol I transcription treats murine and human AML by targeting the leukemia-initiating cell population. Blood. 2017;129:2882–2895.
- Lee HC, Wang H, Baladandayuthapani V, et al. RNA polymerase I inhibition with CX-5461 as a novel therapeutic strategy to target MYC in multiple myeloma. Br J Haematol. 2017;177:80–94.
- Bywater MJ, Poortinga G, Sanij E, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22:51–65.
- Devlin JR, Hannan KM, Hein N, et al. Combination therapy targeting ribosome biogenesis and mRNA translation synergistically extends survival in MYC-driven lymphoma. Cancer Discov. 2016;6:59–70.
- Harrison SJ, Khot A, Brajanovski N, et al. A phase 1, open-label, dose escalation, safety, PK and PD study of a first in class Pol1 inhibitor (CX-5461) in patients with advanced hematologic malignancies (HM). J Clin Oncol. 2015;33:e22212.
- Khot A, Brajanovski N, Cameron D, et al. First-in-human RNA polymerase I transcription inhibitor CX-5461 in patients with advanced hematologic cancers: results of a phase I dose-escalation study. Cancer Discov. 2019;9:1036–1049.
- Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48:452–458.
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzym Regul. 1984;22:27–55.