
ABSTRACT
The study was aimed to investigate the effect of alpha-lipoic acid (ALA) on human umbilical vein endothelial cells (HUVECs) injury induced by hydrogen peroxide (H2O2) and to explore its possible mechanisms. We established the H2O2-induced HUVECs injury model and the ALA treatment groups in which HUVECs were co-incubated with H2O2 (250 μmol/L) and different final concentrations of ALA (100,200,400 μmol/L) for 48 h. Cell survival rate assay and LDH activity assay were carried out. The levels of related proteins were performed by Western Blot. We observed that H2O2 administration resulted in an increase in the LDH activity and a decrease in cell survival rate. The expression levels of Nox4, Bax, NF-κB p65, Caspase-9, Caspase-3, iNOS, VCAM-1 and ICAM-1 were up-regulated, while the expression level of Bcl-2 was down-regulated. All these factors were significantly improved by ALA treatment. In brief, ALA treatment ameliorates H2O2-induced HUVECs damage by inhibiting inflammation and oxidative stress.
Graphical abstract
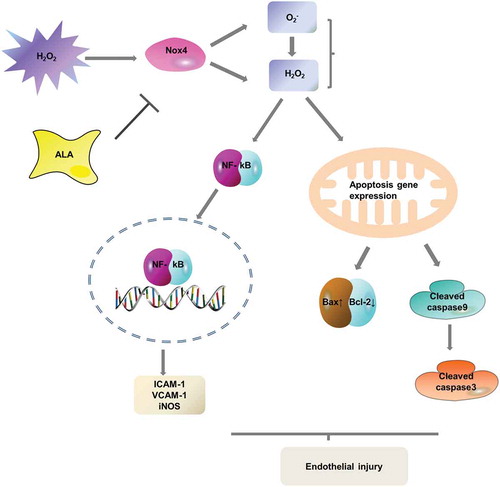
Alpha-lipoic acid attenuates H2O2-induced human umbilical vein endothelial cells injury by inhibiting apoptosis and the expression of inflammatory-related factors.
Atherosclerosis is a progressive inflammatory cardiovascular disorder and the main cause of cardiovascular morbidity and mortality worldwide. The pathogenesis of atherosclerosis is a chronic inflammatory process in which various damaging factors affect vascular endothelial cells, resulting in endothelial cell dysfunction and release of various leukocyte adhesion molecules, which adhesion at the activation site of endothelial cells [Citation1].
Endothelial dysfunction leads to abnormal blood circulation, atherosclerosis, inflammation, stroke and even cancer [Citation2,Citation3]. Endothelial cell apoptosis promotes atherosclerosis and may be further aggravated by oxidative stress. In general, oxidative stress is considered to be a major element causing cell dysfunction of vascular endothelial cells [Citation4] and the apoptosis of endothelial cells may be caused by the accumulation of reactive oxygen species (ROS) [Citation5,Citation6]. In addition, ROS act as signaling molecules that promote the activation of the inflammatory response of vascular endothelial cells, leading to vascular inflammation and endothelial dysfunction [Citation7]. Furthermore, the translocation of NF-κB from cytoplasm to nucleus triggers the expression of inflammatory-related factors such as vascular cell adhesion molecule-1 (VCAM-1) and inter-cellular adhesion molecule-1 (ICAM-1) [Citation8]. Therefore, it has been inferred that drugs that block NF-κB signal transduction and adhesion molecules’ expression in vitro may also have significant effects on the inflammatory response in vivo. It has been shown [Citation9,Citation10] that H2O2-induced oxidative stress in endothelial cells upregulates the apoptotic gene, Bax, and the caspase signaling pathway. Hence, it is essential to understand the roles of ROS in vascular endothelial cells’ physiology and pathophysiology. A more elaborate investigation of how H2O2 influences endothelial cell apoptosis and inflammation will help formulate a new strategy for treatment or prevention of cardiovascular diseases.
Previous studies have found increased expression of adhesion molecules such as ICAM-1 and VCAM-1 at lesion-prone arterial sites and atherosclerotic lesions [Citation11,Citation12]. The genetic defect of the mouse adhesion molecules are associated with reduction in atherosclerosis [Citation13]. Therefore, the regulation of the interaction between mononuclear cells and endothelial cells may be an important target for the prevention and treatment of atherosclerosis.
In general, abnormal production of proinflammatory mediators such as iNOS plays an important role in the pathogenesis of cardiovascular diseases. The overexpression of inflammatory mediators causes vascular damage which can be alleviated by inhibiting the expression of pro-inflammatory mediators [Citation14,Citation15].
Currently, antioxidants are used to reduce the production of intracellular ROS and limit its harmful effects on the body. α-Lipoic acid (ALA) has been proven to be one of the most important antioxidants that can improve vascular function [Citation16].
It shown that ALA has protective effect on early atherosclerosis in diabetic rats [Citation17]. Many studies have confirmed that ALA improves vascular function and reduces the burden of atherosclerotic plaque [Citation18,Citation19]. ALA meliorates vascular dysfunction by regulating lipid metabolism and activating AMPK in endothelial cells [Citation20]. Jones et al [Citation21] found that ALA/DHLA redox coupling enhances antioxidant defense and human endothelial cells function. The role of ALA in preventing oxidative damage in EA-hy926 cells has been demonstrated [Citation22]. Shen et al proposed that H2O2-induced human umbilical vein endothelial cells (HUVECs) can mediate apoptosis through the mitochondrial pathway [Citation23]. However, the mechanism of ALA in repairing endothelial function is still unclear. To our knowledge, little research has been done on the mechanism by which ALA inhibits apoptosis induced by H2O2 in HUVECs. To explore ALA’s mechanism of action, its effects on HUVECs induced by H2O2 were studied from two aspects: inflammation and apoptosis induced by oxidative stress.
Our study provides a possible mechanism for the anti-apoptosis effect of ALA and provides important insight into the theoretical basis for the prevention of cardiovascular diseases.
Materials and methods
Materials
HUVECs were provided by the Biochemistry and Molecular Biology Laboratory (Hebei College of Traditional Chinese Medicine). Dulbecco’s modified Eagle medium (DMEM), fetal bovine serum (FBS) and trypsin were purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). ALA, H2O2, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and dimethyl sulfoxide (DMSO) were from Sigma–Aldrich (St Louis, MO, USA). RIPA lysis buffer was from Beyotime Institute of Biotechnology (Haimen, China). The lactate dehydrogenase (LDH) assay kit was purchased from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Bicinchoninic acid (BCA) protein assay kit was from Beyotime Institute of Biotechnology. Antibodies against Bcl-2 (2876), Bax (2772), cleaved Caspase-3 (9664), cleaved Caspase-9 (9501), NF-κB p65 (4764), GAPDH (5174), β-actin (4967) and Histone H3 (9715) were purchased from Cell Signaling Technology (Danvers, MA, USA); Antibodies against VCAM-1 (sc-1504), ICAM-1 (sc-1511), iNOS (sc-650) and Nox4 (sc-30141) were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). All the secondary antibodies were purchased from Beyotime Institute of Biotechnology. All other reagents used in the experiments were of analytical grade.
Cell culture and experimental grouping
HUVECs were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco) and incubated at 37°C in 5% CO2. After cell monolayers were fused, they were digested with digestion solution containing 2.5 g/L trypsin and 0.2 g/L disodium EDTA (EDTA-Na2) and seeded in a 6-well cell culture plate at a density of 1 × 104 cells/well. After 24 h of incubation, the supernatant was discarded. The cells were then randomly divided into three groups: (1) control group, in which the cells were incubated in DMEM medium for 48 h; (2) H2O2 injury group, in which cells were exposed to H2O2 at 50 to 400 μmol/L (final concentration in the wells) for 48 h; the cell viability was analyzed to determine the appropriate H2O2 concentration for the H2O2 injury model in subsequent experiments; (3) H2O2 + ALA group, in which cells were co-incubated with H2O2 (250 μmol/L) and different concentrations of ALA (final concentrations 100,200, 400 μmol/L) for 48 h.
MTT assay
The viability of HUVECs was performed by the MTT assay as previously described [Citation24]. In brief, the control group and the experimental groups mentioned above were inoculated into 96-well plates. Each group of experiments was repeated six times. After 48 h of incubation, 20 μL of MTT solution were added into the wells for another 4 h, and then 150 μL of DMSO were added to each well. After gently shaking the plates for 10 min, the absorbance (A) of each well at 490 nm was measured with a microplate reader (Bio-Rad, Hercules, CA, USA). The formula used: cell survival rate (%) = A490 (sample wells – blank wells)/A490 (control wells – blank wells) * 100
LDH activity assay
To determine the extent of cell damage, the supernatants of the cell cultures were collected and the LDH activity was measured by an LDH kit (Nanjing Jiancheng Bioengineering Institute). The absorbance was read at a wavelength of 440 nm and enzyme activity was expressed as units per L.
LDH activity (U/L) = (ODtest – ODblank)/(ODstandard – ODblank) * standard concentration * 1000 * dilution ratio of sample
Flow cytometry analysis of apoptosis
Cell apoptosis was analyzed by flow cytometry. The treated cells and the control cells were collected separately after digestion with trypsin and centrifugation to remove the medium. The cells were washed once with phosphate-buffered saline (PBS) and fixed in 70% pre-chilled ethanol overnight at 4°C. After centrifugation, cells were washed with ice-cold PBS and incubated with 100 µg/mL RNase A for 30 min at 37°C. Then the cells were stained with 50 μg/mL propidium iodide (PI) solution according to a method described previously [Citation25]. After incubation for 30 min at 4°C in the dark, the apoptosis of HUVECs was detected by flow cytometry (Beckman Coulter, Inc., USA). The experiment was performed in triplicate.
Western blot analysis
After the treatments, cells were collected, washed three times with ice-cold PBS and lysed in RIPA lysis buffer with 1% PMSF and protease inhibitors (Merk) to obtain protein extracts. The protein concentration of the lysates was quantified by a BCA kit (Beyotime) according to the manufacturer’s instructions. Equal amounts of protein lysates were subjected to SDS-PAGE and transferred to PVDF membranes (Merck Millipore Corporation, Darmstadt, Germany). The membranes were blocked with 5% nonfat dry milk for 2 h, and then incubated with primary antibodies overnight at 4°C, followed by three washes with Tris-buffered saline containing Tween 20. The blots were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10,000; Beyotime) for 1 h at room temperature. Finally, the protein bands were detected by the enhanced chemiluminescent (ECL) system. The protein levels were quantified by Image J software.
Statistical analysis
Statistical analysis was conducted with SPSS 21.0 software (IBM Corporation, New York, USA). The values are expressed as the mean ± SEM. Data were analyzed by one-way ANOVA. P < 0.05 was considered statistically significant.
Results
Establishment of the H2O2-induced HUVECs injury model
Oxidative stress is an important factor that causes cardiovascular diseases and excessive ROS may lead to endothelial cell dysfunction [Citation26]. Therefore, HUVECs induced by H2O2 were selected as the model of oxidative stress injury to examine the protective effects of ALA on vascular endothelial cells.
To determine the appropriate H2O2 concentration for the H2O2-induced HUVECs injury model, the cells were divided into seven groups (control and different concentrations of H2O2). The MTT assay showed that the cell survival rate gradually decreased with the increase in H2O2 concentration, and it decreased to about 44.26% ± 5.83% (P < 0.01) after 48 h of exposure to 250 μmol/L H2O2, indicating an obvious oxidative stress injury to HUVECs caused by H2O2 treatment. Therefore, 250 μmol/L H2O2 was designed for the injury model in the subsequent experiments ()).
Figure 1. Effects of H2O2 and ALA on HUVECs cell viability. (a) Effects of H2O2 on the viability of HUVECs. HUVECs were either not treated (control) or treated with different concentrations of H2O2 for 48 h. Cell viability was evaluated by the MTT. Data are mean ± SEM. N = 6. ##P < 0.01 vs control. (b) Effects of ALA on survival rate of injuried cells induced by H2O2. In H2O2 injury model, HUVECs were treated with H2O2 at 250 μmol/L for 48 h. In ALA groups, HUVECs were co-incubated with H2O2 (250 μmol/L) and ALA at 100 μmol/L, 200 μmol/L and 400 μmol/L for 48 h. Cell viability was evaluated by the MTT assay. Data are mean ± SEM. N = 6. ## P < 0.01 vs control, *P < 0.05 vs model, **P < 0.01 vs model.

ALA improves the viability of H2O2-induced HUVECs
The selected experimental concentrations for the low, medium and high-dose ALA groups were 100, 200 and 400 μmol/L, respectively. After co-incubation with 250 μmol/L H2O2 and different concentrations of ALA for 48 h, the MTT assay was performed to assess cell viability. As shown in ), the cell survival rate of the H2O2 injury group was significantly reduced (P < 0.01) compared with the control group. However, after co-incubation with ALA, the cell survival rate increased from 46.75% ± 6.31% to 57.09% ± 6.37%, 68.17% ± 7.28% and 78.10% ± 6.58%, respectively. The differences among the ALA treatment groups were significant (P < 0.05). These results confirmed that ALA protected HUVECs from injury induced by H2O2 ()).
ALA reduces LDH release from H2O2-induced cells
LDH is considered to be a biomarker for assessing cellular injury [Citation27,Citation28]. To further investigate ALA’s protective effects on H2O2-induced HUVECs, LDH release to the culture medium was measured. As expected, the LDH activity in the model group was significantly higher than that of the control group (P < 0.01). However, after co-incubation with ALA (100, 200 or 400 μmol/L) for 48 h, the release of LDH decreased in a dose-dependent manner compared with the model group (P < 0.05; ).
Figure 2. Effects of different concentrations of ALA on the LDH activity in H2O2-induced HUVECs. The LDH activity was measured by LDH kit. In H2O2 injury model, HUVECs were treated with H2O2 at 250 μmol/L for 48 h. In ALA groups, HUVECs were co-incubated with H2O2 (250 μmol/L) and ALA at 100 μmol/L, 200 μmol/L and 400 μmol/L for 48 h. Values are expressed as mean ± SEM, n = 6 independent experiments. ##P < 0.01 vs control, *P < 0.05 vs model, **P < 0.01 vs model.

ALA downregulates the expression of Nox4
In HUVECs, ROS is produced mainly by Nox4 [Citation29]. In this paper, ALA’s effect on Nox4 protein expression was measured. The Nox4 level in the H2O2 model group was remarkably high compared with the control group. However, co-incubation with ALA resulted in obvious downregulation of Nox4 protein expression in a dose-dependent manner (P < 0.05; ).
Figure 3. The expression of Nox4 protein in different treatment groups. HUVECs were exposed to H2O2 (250 μmol/L) while treated with or without ALA (100, 200, 400 μmol/L) for 48 h. (a) Western blotting analysis of Nox4 protein. (b) The mean ± SEM relative protein levels of Nox4 were normalized to GADPH (internal control). All the experiments were executed three times independently. ##P < 0.01 vs control, **P < 0.01 vs model.

ALA inhibits H2O2-induced activation of the NF-κB signaling pathway
NF-κB as a master transcription factor induces expression of many inflammation-related genes. It has been shown that NF-κB is involved in oxidative stress-mediated endothelial cell dysfunction [Citation30].
The protein levels of NF-κB p65 from HUVECs were analyzed to assess their translocation from cytoplasm to nucleus. By western blotting, we observed that the expression of nuclear p65 was remarkably increased after stimulation with H2O2. In a parallel comparison, the cytoplasmic levels of p65 decreased. More importantly, we noticed that the H2O2-enhanced nuclear translocation of p65 was alleviated by treatment with different concentrations of ALA ().
Figure 4. Effects of ALA on NF-κB activation in HUVECs. HUVECs were exposed to H2O2 (250 μmol/L) while treated with or without ALA (100, 200, 400 μmol/L) for 48 h. (a) Western blotting analysis of NF-κB p65 in the cytoplasm and nucleus. (b) Mean ± SEM relative protein levels of cytoplasmic NF-κB p65 were normalized to β-actin. (c) Mean ± SEM relative protein levels of nuclear NF-κB p65 were normalized to Histone H3. All the experiments were executed three times independently. ##P < 0.01 vs control, **P < 0.01 vs model.

The results suggest that ALA could suppress the activation of NF-κB signal pathway.
ALA protects HUVECs from H2O2-induced apoptosis
It has been revealed that injury and apoptosis in the vascular endothelium play a critical role in atherosclerotic plaque formation [Citation31], In our study, the apoptosis rate was detected by flow cytometry to investigate whether ALA has an effect on apoptosis induced by H2O2. As shown in , compared with the control group (24.83% ± 2.27%), there was an obvious increase in the apoptosis rate in the H2O2 model group (72.20% ± 5.79%, P < 0.01). However, after treatment with different concentrations of ALA, this rate reduced to 59.00% ± 3.95%, 52.73% ± 5.01% and 41.10% ± 4.37%, respectively. The difference between the H2O2 model group and the ALA groups was statistically significant (P < 0.01). These results illustrated that ALA significantly reduced the H2O2-induced apoptosis rate.
Figure 5. Cell apoptotic rate evaluation by flow cytometry. HUVECs were exposed to H2O2 (250 μmol/L) while treated with or without ALA (100, 200, 400 μmol/L) for 48 h. The apoptosis rate was detected by flow cytometry. (a) Control group. (b) H2O2 (250 μmol/L) group. (c) ALA (100 μmol/L) group. (d) ALA (200 μmol/L) group. © ALA (400 μmol/L) group. (f) The mean ± SEM percentage of apoptotic cells of three individual experiments. ##P < 0.01 vs control, **P < 0.01 vs model.

Effects of ALA on the protein expression of Bcl-2, Bax, cleaved caspase-3 and caspase-9
To study the potential mechanism by which ALA regulates H2O2-induced apoptosis, the expression changes in apoptosis-related proteins in HUVECs were evaluated by western blotting.
and ) shows that in comparison to the control group, the expression level of Bax was significantly higher (P < 0.01) and that of Bcl-2 was obviously decreased in the H2O2 group (P < 0.01). In contrast, when the cells were co-incubated with ALA, Bax expression was downregulated in a dose-dependent manner (P < 0.01) and Bcl-2 expression was upregulated (P < 0.01). As previously reported, the ratio between Bcl-2 and Bax protein expression is a key factor in apoptosis regulation [Citation32–34]. Statistical analysis showed that the Bcl-2/Bax ratio increased in association with the ALA dose ()).
Figure 6. Effects of ALA on the expression of apoptosis-related proteins in HUVECs. (a) Western blotting analysis of Bax, Bcl-2 and the cleaved caspase-3 and caspase-9. The protein expression levels of Bax (b), Bcl-2 (c), cleaved caspase-3 © and cleaved caspase-9 (f) were analyzed, respectively. GAPDH was used to normalize the data. The Bcl-2/Bax ration (d) was calculated. Results are expressed as mean ± SEM (n = 3 per group). ##P < 0.01 vs control, **P < 0.01 vs model.

We also found that the expression of cleaved caspase-3 and caspase-9 in HUVECs was significantly elevated (P < 0.01) after H2O2 stimulation alone. On the contrary, ALA co-incubation with H2O2 markedly reduced the expression levels of these caspases (P < 0.01; and )).
These results suggest that H2O2 stimulation led to obvious apoptosis, whereas ALA reversed this effect. The anti-apoptotic effect of ALA may be mediated by the regulation of Bcl-2 and Bax protein expression and/or the inhibition of the activation of caspase-3 and caspase-9.
ALA suppresses the upregulation of iNOS, VCAM-1 and ICAM-1 in H2O2-induced HUVECs
Inflammation usually occurs with the development of oxidative stress. To assess whether ALA has an anti-inflammatory effect on H2O2-stimulated HUVECs, we examined the expression of iNOS, which is closely linked to the inflammatory response [Citation35]. In addition, we examined the changes in the protein expression of VCAM-1 and ICAM-1 ()). Western blot analysis revealed that compared with the control group, the protein expression levels of iNOS, VCAM-1 and ICAM-1 in the injury model group were significantly upregulated (P < 0.01). Notably, co-incubation with ALA attenuated this upregulation. The differences between the groups were significant (P < 0.05; -)).
Figure 7. Effects of ALA on the expression of iNOS, VCAM-1 and ICAM-1 in HUVECs. (a) The protein levels of iNOS, VCAM-1 and ICAM-1 in different treatment groups were determined by Western blot analysis. (b), (c), (d) The relative levels of iNOS, VCAM-1 and ICAM-1 were normalized to GAPDH. Values are presented as mean ± SEM from 3 independent experiments. ##P < 0.01 vs control, **P < 0.01 vs model.

Discussion
In the present study, our results demonstrated that ALA regulates the cardiovascular system by inhibiting apoptosis and reducing the release of inflammatory-related factors to protect against H2O2 mediated HUVECs injury.
Under normal conditions, the proliferation rate and apoptosis rate of endothelial cells are very low. ROS produced in the cells during metabolism causes oxidative damage to the vascular endothelium and leads to apoptosis. HUVECs apoptosis induced by oxidative stress plays an important role in the pathogenesis of cardiovascular diseases, thus eliminating excessive ROS and inhibiting apoptosis may provide beneficial intervention in the treatment of cardiovascular diseases [Citation36]. Lipid peroxidation is one of the main events in free radical-mediated cell damage [Citation37]. H2O2 causes lipid peroxidation, markedly increasing cell permeability and LDH leakage. Therefore, an in vitro model of H2O2-induced endothelial cells injury has been applied to mimic the oxidative stress in endothelial cells. Previous reports have suggested that H2O2 increases cell permeability, impairs the cellular antioxidant defense and induces endothelial cell apoptosis [Citation38]. Our results showed that H2O2 damaged cell, decreased cell viability, increased the ratio of apoptotic cells and LDH activity. Furthermore, we also shown that ALA significantly reduced H2O2-induced HUVECs injury in a dose-dependent manner () and ). Namely, ALA protected HUVECs against oxidative damage induced by H2O2, as assessed by cell viability and LDH activity. The results of the current study are consistent with previous reports that ALA significantly reduced LDH release from HUVECs induced by hypoxia/reoxygenation or ox-LDL [Citation39,Citation40]. The present results suggest that ALA can suppress LDH release from HUVECs induced by H2O2 in a dose-dependent manner. It seems that ALA can promote HUVECs repair and protect the endothelial cell. The experimental results showed that ALA appears to have the potential to reduce excessive ROS production, thereby protecting HUVECs from peroxide toxicity.
There is evidence that the expression of Nox4 in endothelial cells is significantly higher than that of other Nox members and that Nox4 plays a crucial role in endothelial redox reactions and in functional regulation of endothelial cells physiology [Citation41]. H2O2 inhibit the proliferation of vascular endothelial cells, activate the NADPH oxidase pathway, and cause the cells to be oxidized stress, NADPH oxidase can transfer NADPH electrons to molecular oxygen to produce superoxide anions, indicating that ROS production is excessive, the balance of the oxidative stress system is disturbed, and eventually damages cells or tissues. In our study, ALA reduced Nox4-mediated ROS generation in HUVECs and further inhibited the NF-κB signaling pathway. Furthermore, ALA downregulated the expression of Nox4 induced by H2O2 in HUVECs () and )). Thus, we propose that this pathway is one of the mechanisms by which ALA reduces oxidative stress in endothelial cells. So, ALA may exert its antioxidant effects by regulating Nox4 protein expression and play a protective role in cardiovascular disease.
Oxidative stress mediates mitochondrial apoptosis in endothelial cells [Citation42]. The mitochondrial pathway regulated by the Bcl-2 family is one of the most important pathways [Citation43]. Bcl-2 and Bax have opposing functions and play important roles in the mitochondrial pathway of apoptosis. Previous study has reported that H2O2 downregulated Bcl-2 protein expression [Citation38], which is consistent with our research. In contrast, the present study indicated that ALA promoted Bcl-2 protein expression and inhibited Bax expression in the H2O2-induced HUVECs, showing the anti-apoptotic effects of ALA on H2O2-induced HUVECs ( and )). These results suggest that ALA regulates the balance between the antiapoptotic protein, Bcl-2, and the proapoptotic protein, Bax.
Caspases play an important role in apoptosis induced by different stimuli [Citation44]. Caspases can selectively cleave certain proteins and break down intracellular related substrate proteins, resulting in changes in cell structure and metabolism, thus causing apoptosis. It is the condensation point of multiple apoptotic pathways and the final route of apoptosis. Caspase-3 mediates the apoptosis of exogenous and endogenous pathways, during which it is cleaved and activated. It has been reported that Bcl-2 is an antiapoptotic protein that inhibits apoptosis induced by the mitochondria-dependent caspase-9 pathway [Citation43,Citation45]. In the present study, ALA co-treatment reduced the expression of cleaved caspase-3 and inhibited apoptosis compared with the untreated HUVECs induced by H2O2. Consistent with the results that ALA suppressed the H2O2-induced expression of Bax/Bcl-2 and caspase-3,9, we observed that ALA protected HUVECs from oxidative damage and significantly decreased the number of apoptotic cells (-)). This suggests that ALA protects vascular endothelial cells by inhibiting or blocking the mitochondrial apoptosis pathway.
In oxidative stress-induced cell damage, researchers are concerned about the apoptotic pathway of mitochondrial damage, such as decreased cell membrane potential, caspase-3 activation, cytochrome c release, and Bax and Bcl-2 expression. Studies have shown that oxidative stress may be involved in cell damage via the common central NF-κB [Citation46,Citation47]. NF-κB regulates the expression of cell adhesion molecules such as VCAM-1, ICAM-1 and pro-inflammatory mediator iNOS. In this study, the expression of NF-κB p65 and iNOS was significantly upregulated during oxidative stress, but this expression was significantly downregulated when the cells were co-treated with ALA, indicating that ALA reduced the expression of NF-κB p65 and iNOS, and finally the inflammation of HUVECs induced by oxidative stress was suppressed. We also found that ALA can significantly reduce the expression of VCAM-1, ICAM-1 in H2O2-induced HUVECs (-)). This suggests that ALA can inhibit the expression of VCAM-1, ICAM-1 by interfering with mononuclear cell adhesion. Thus, we believe that the protective effects of ALA on HUVECs are achieved by inhibiting NF-κB activation and reducing the release of inflammatory-related factors. ALA is a powerful antioxidant that removes ROS [Citation48]. Therefore, the activation of the NF-κB pathway caused by oxidative stress is inhibited by ROS scavenging, and then the release of inflammatory-related factors such as VCAM-1, ICAM-1 and pro-inflammatory mediator iNOS are reduced.
In summary, HUVECs from oxidative stress can cause metabolic dysfunction and oxidative damage to proteins, carbohydrates, DNA, RNA, mtDNA and lipids in the cell membrane, thereby activating the apoptotic pathway. On the basis of the changes in protein expression and the results of this study, we propose the following mechanism of ALA protection against H2O2-induced apoptosis in HUVECs: H2O2 stimulation induces the binding of Bax and Bcl-2, which activate caspase-9 that finally activates the apoptotic executive molecule, caspase-3. Cell opening programmed death mode. Co-treatment with ALA inhibits the apoptosis pathway in HUVECs induced by H2O2 mainly through the mitochondrial pathway. ALA suppresses inflammation induced by oxidative stress by downregulating the NF-κB pathway. Furthermore, ALA inhibits the expression of ICAM-1 and VCAM-1, affects monocyte adhesion and ameliorates atherosclerosis ().
Figure 8. Potential molecular signaling pathways that ALA protects against H2O2-induced HUVECs damage.

Authors contributions
LAY mainly designed the research. WW and ALP performed the research and analyzed the data. LYF, AR and BZ modified the research. LWZ and SQH help us in writing and manuscript. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Ildikó K. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;4:353.
- Madamanchi NR, Runge MS. Redox signaling in cardiovascular health and disease. Free Radic Biol Med. 2013 Aug;61:473–501.
- Roquer J, Segura T, Serena J, et al. Endothelial dysfunction, vascular disease and stroke: the ARTICO study. Cerebrovasc Dis. 2009;27(Suppl 1):25–37.
- D’Agnillo F, Wood F, Porras C, et al. Effects of hypoxia and glutathione depletion on hemoglobin- and myoglobin-mediated oxidative stress toward endothelium. Biochim Biophys Acta. 2000 Feb 2;1495(2):150–159.
- Skibska B, Goraca A. The protective effect of lipoic acid on selected cardiovascular diseases caused by age-related oxidative stress. Oxid Med Cell Longev. 2015;2015:313021.
- Lee YJ, Kang IJ, Bunger R, et al. Mechanisms of pyruvate inhibition of oxidant-induced apoptosis in human endothelial cells. Microvasc Res. 2003 Sep;66(2):91–101.
- Drummond GR, Selemidis S, Griendling KK, et al. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10(6):453–471.
- Armoza A, Haim Y, Bashiri A, et al. Tomato extract and the carotenoids lycopene and lutein improve endothelial function and attenuate inflammatory NF-κB signaling in endothelial cells. J Hypertens. 2013 Mar;31(3):521–529. discussion 529.
- Warren MC, Bump EA, Medeiros D, et al. Oxidative stress-induced apoptosis of endothelial cells. Free Radic Biol Med. 2000;29(6):537–547.
- Choi YJ, Kang JS, Park JH, et al. Polyphenolic flavonoids differ in their antiapoptotic efficacy in hydrogen peroxide-treated human vascular endothelial cells. J Nutr. 2003 Apr;133(4):985–991.
- Fotis L, Agrogiannis G, Vlachos IS, et al. Intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 at the early stages of atherosclerosis in a rat model. In Vivo. 2012 Mar-Apr;26(2):243–250.
- Marzolla V, Armani A, Mammi C, et al. Essential role of ICAM-1 in aldosterone-induced atherosclerosis. Int J Cardiol. 2017 Apr 1;232:233–242.
- Smith JD, Dansky HM, Breslow JL. Genetic modifiers of atherosclerosis in mice. Ann N Y Acad Sci. 2001 Dec;947:247–252. discussion 252-3.
- Fu Y, Hu X, Cao Y, et al. Saikosaponin a inhibits lipopolysaccharide-oxidative stress and inflammation in Human umbilical vein endothelial cells via preventing TLR4 translocation into lipid rafts. Free Radic Biol Med. 2015;89:777–785.
- Chen J, Gong F, Chen M-F, et al. In vitro vascular-protective effects of a tilapia by-product oligopeptide on angiotensin II-induced hypertensive endothelial injury in HUVEC by Nrf2/NF-κB pathways. Mar Drugs. 2019;17(7):431.
- Ghibu S, Richard C, Vergely C, et al. Antioxidant properties of an endogenous thiol: alpha-lipoic acid, useful in the prevention of cardiovascular diseases. J Cardiovasc Pharmacol. 2009 Nov;54(5):391–398.
- Ismawati, Mukhyarjon, Asni E, et al. The effect of alpha-lipoic acid on expression of VCAM-1 in type 2 diabetic rat. Anat Cell Biol. 2019 Jun;52(2):176–182.
- Wollin PJHJ SD. Alpha-lipoic acid and cardiovascular disease. J Nutr. 2003;133(11):3327–3330.
- Sungmi P. Physiological effect and therapeutic application of alpha lipoic acid. Curr Med Chem. 2014;32(21):3636–3645.
- Lee WJ, Lee IK, Kim HS, et al. Alpha-lipoic acid prevents endothelial dysfunction in obese rats via activation of AMP-activated protein kinase. Arterioscler Thromb Vasc Biol. 2005 Dec;25(12):2488–2494.
- Jones W, Li X, Qu ZC, et al. Uptake, recycling, and antioxidant actions of alpha-lipoic acid in endothelial cells. Free Radic Biol Med. 2002 Jul 1;33(1):83–93.
- Rochette L, Ghibu S, Richard C, et al. Direct and indirect antioxidant properties of alpha-lipoic acid and therapeutic potential. Mol Nutr Food Res. 2013 Jan;57(1):114–125.
- Shen J, Zhu Y, Huang K, et al. Buyang Huanwu Decoction attenuates H2O2-induced apoptosis by inhibiting reactive oxygen species-mediated mitochondrial dysfunction pathway in human umbilical vein endothelial cells. BMC Complement Altern Med. 2016 May 31;16:154.
- Kim EJ, Park WH, Ahn SG, et al. 5ʹ-nitro-indirubinoxime inhibits inflammatory response in TNF-alpha stimulated human umbilical vein endothelial cells. Atherosclerosis. 2010 Jul;211(1):77–83.
- Tang X, Yang X, Peng Y, et al. Protective effects of lycopene against H2O2-induced oxidative injury and apoptosis in human endothelial cells. Cardiovasc Drugs Ther. 2009 Dec;23(6):439–448.
- Hort MA, Straliotto MR, de Oliveira J, et al. Diphenyl diselenide protects endothelial cells against oxidized low density lipoprotein-induced injury: involvement of mitochondrial function. Biochimie. 2014;105:172–181.
- Song L, Kang C, Sun Y, et al. Crocetin inhibits lipopolysaccharide-induced inflammatory response in human umbilical vein endothelial cells. Cell Physiol Biochem. 2016;40(3–4):443–452.
- Xu J, Zhou L, Weng Q, et al. Curcumin analogues attenuate Aβ25-35-induced oxidative stress in PC12 cells via Keap1/Nrf2/HO-1 signaling pathways. Chem Biol Interact. 2019;305:171–179.
- Konior A, Schramm A, Czesnikiewicz-Guzik M, et al. NADPH oxidases in vascular pathology. Antioxid Redox Signal. 2014 Jun 10;20(17):2794–2814.
- Jiawei Chen JLM. Role of oxidative stress in coronary heart disease. Indian Heart. 2004;56(02):163–173.
- Deng H, Chu X, Song Z, et al. MicroRNA-1185 Induces Endothelial Cell Apoptosis by Targeting UVRAG and KRIT1. Cell Physiol Biochem. 2017;41(6):2171–2182.
- Brenner C, Cadiou H, Vieira HLA, et al. Bcl-2 and Bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene. 2000;19(3):329–336.
- Renault TT, Manon S. Bax: addressed to kill. Biochimie. 2011 Sep;93(9):1379–1391.
- Ferreira Cda S, Maganhin CC, Simoes Rdos S, et al. Melatonin: cell death modulator. Revista da Associacao Medica Brasileira (1992). 2010 Nov-Dec;56(6):715–718.
- Choi WS, Jeong JW, Kim SO, et al. Anti-inflammatory potential of peat moss extracts in lipopolysaccharide-stimulated RAW 264.7 macrophages. Int J Mol Med. 2014 Oct;34(4):1101–1109.
- Luo Y, Lu Y. 2ʹ,4ʹ-dihydroxy-6ʹ-methoxy-3ʹ,5ʹ-dimethylchalcone inhibits apoptosis of MIN6 cells via improving mitochondrial function. Pharmazie. 2012 Sep;67(9):798–803.
- Fang WT, Li HJ, Zhou LS. Protective effects of prostaglandin E1 on human umbilical vein endothelial cell injury induced by hydrogen peroxide. Acta Pharmacol Sin. 2010 Apr;31(4):485–492.
- Hao XL, Kang Y, Li JK, et al. Protective effects of hyperoside against H2O2-induced apoptosis in human umbilical vein endothelial cells. Mol Med Rep. 2016 Jul;14(1):399–405.
- Zhang J, Deng H, Liu L, et al. Alpha-Lipoic acid protects against hypoxia/reoxygenation-induced injury in human umbilical vein endothelial cells through suppression of apoptosis and autophagy. Mol Med Rep. 2015 Jul;12(1):180–186.
- Liu YX, Han GZ, Wu T, et al. Protective effect of alpha-lipoic acid on oxidized low density lipoprotein-induced human umbilical vein endothelial cell injury. Pharmacol Rep. 2011;63(5):1180–1188.
- Petry A, Djordjevic T, Weitnauer M, et al. NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid Redox Signal. 2006 Sep-Oct;8(9–10):1473–1484.
- Surico D, Farruggio S, Marotta P, et al. Human chorionic gonadotropin protects vascular endothelial cells from oxidative stress by apoptosis inhibition, cell survival signalling activation and mitochondrial function protection. Cell Physiol Biochem. 2015;36(6):2108–2120.
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59.
- Onishi Y, Kawamoto T, Ueha T, et al. Transcutaneous application of carbon dioxide (CO2) induces mitochondrial apoptosis in human malignant fibrous histiocytoma in vivo. PloS One. 2012;7(11):e49189.
- Antonsson B. Mitochondria and the Bcl-2 family proteins in apoptosis signaling pathways. Mol Cell Biochem. 2004;256-257(1–2):141–155.
- McCabe C, Samali A, O’Brien T. beta cell cytoprotective strategies: establishing the relative roles for iNOS and ROS. Biochem Biophys Res Commun. 2006 Apr 21;342(4):1240–1248.
- Baldwin AS Jr. Series introduction: the transcription factor NF-kappaB and human disease. J Clin Invest. 2001 Jan;107(1):3–6.
- Balkis Budin S, Othman F, Louis SR, et al. Effect of alpha lipoic acid on oxidative stress and vascular wall of diabetic rats. Rom J Morphol Embryol. 2009;50(1):23–30.