
ABSTRACT
This study aimed to identify the role of lncRNA TUG1 with miR-221-3p on mice with lipopolysaccharide (LPS)-induced acute respiratory distress syndrome (ARDS). Animal model was established, and lung tissue histopathologic status and permeability were detected by hematoxylin-eosin (HE) or Evans blue dye assay respectively. Levels of inflammation cytokines, lncRNA TUG1, miR-221-3p, sprouty related EVH1 domain-containing 2 (SPRED2), and phosphorylated (p)-ERK1/2 were determined by ELISA, qRT-PCR or Western blot. Pulmonary impairment and apoptosis were examined by flow cytometry. We observed that LPS up-regulated levels of tumor necrosis factor-α (TNF-α), Interleukin-1β (1L-1β), and ERK1/2 phosphorylation, and reduced SPRED2 levels, which were rescued by overexpressed lncRNA TUG1. StarBase and dual-luciferase reporter assay verified that miR-221-3p was targeted by lncRNA TUG1. MiR-221-3p could reverse the effect of lncRNA TUG1 on cell apoptosis, levels of TNF-α, IL-1β, SPRED2, and p-ERK1/2. Therefore, overexpressed lncRNA TUG1 attenuated LPS-induced pulmonary impairment in ARDS mice via regulating miR-221-3p/SPRED2 axis.
GRAPHICAL ABSTRACT
Overexpressed TUG1 could alleviate LPS-induced up-regulation of miR-221-3p expression and RAW 264.7 macrophage apoptosis and inflammatory cytokine accumulation, which was reversed by overexpressed miR-221-3p.
Acute respiratory distress syndrome (ARDS) is related to diffuse alveolar damage and lung capillary endothelial injury [Citation1]. ARDS is a gradually progressed pathological disease rather than an independent one. The final stage of ARDS is often induced or combined with multiple organ dysfunction syndrome and even multiple organ failure, which too often cause a poor prognosis with mortality as high as 40% [Citation2]. Therefore, it is urgent to discover the molecular mechanisms of ARDS and develop a novel therapy for ARDS patients.
Long non-coding RNAs (lncRNAs) are a large class of transcripts (200 bases in length) but without protein-coding capacity [Citation3]. LncRNAs regulate expressions of genes and proteins in the cytoplasm and the nucleus, thus regulating the physiological functions of the organisms [Citation4]. Growing evidence found that lncRNAs play critical roles in regulating inflammation. Roux et al. [Citation5] proved that innate immune responses of lipopolysaccharide (LPS)-stimulated mouse RAW 264.7 macrophages are activated by up-regulation of 210 mouse lncRNAs; Du et al. [Citation6] found that expressions of inflammatory factors caused by LPS could be suppressed by lncRNA Mirt2 in tracheal epithelial cells and in hepatocytes, pointing to a positive role of lncRNA Mirt2 in inflammation suppression. LncRNA taurine upregulated gene 1 (TUG1) has been reported to act as a possible oncogene in osteosarcoma (OS) development [Citation7]. LncRNA TUG1 works as an inflammation inhibitory factor in several diseases [Citation8,Citation9], but the effects of lncRNA TUG1 on LPS-induced ARDS in characterizing respiratory inflammation are not clear.
MicroRNAs (miRNAs), are short RNA molecules with 19 to 25 nucleotides in length, regulate post-transcriptional silencing of target genes [Citation10]. Studies increasingly showed that miRNAs participate in the pathological conditions of a great number of diseases. The expressions of miRNA-466 family are reported increased in bronchoalveolar lavage fluid (BALF) of the ARDS mouse model [Citation11], and Ju et al. identified that miR-27a regulated the inflammatory response and apoptosis of LPS-treated RAW 264.7 cells through targeting toll-like receptor 4 (TLR4) [Citation12]. Moreover, the down-regulation of H19 suppresses the inflammation response in oxygenized low-density lipoprotein-treated RAW264.7 cells by up-regulating miR-130b [Citation13]. Moreover, lncRNAs act as a competing endogenous RNAs (ceRNAs) or endogenous sponge RNAs in modulating the miRNA expressions and their biological functions [Citation14]. LncRNA TUG1 enhances the chemosensitivity of non-small cell lung cancer by impairing miRNA-221-dependent phosphatase and tensin homolo (PTEN) inhibition [Citation15]. Nevertheless, whether lncRNA TUG1 had the similar effect on miR-221-3p in ARDS required further investigation.
The study explored the effect of lncRNA TUG1 on LPS-induced ARDS rats or macrophages and probed into the relationship between lncRNA TUG1 and miR-221-3p, hoping to provide a potential therapeutic method for treating ARDS.
Materials and methods
Ethics statement
Guidelines of the China Council on Animal Care and Use were strictly followed in all the animal experiments. The approval of the study was obtained from the Committee of Experimental Animals of Shenzhen Hospital, Southern Medical University (HXK20200203). Pain and discomfort to the animals were minimized. The animal experiments were performed in Shenzhen Hospital, Southern Medical University.
Animal model establishment
A LPS-induced ARDS mice model was established according to a previous study [Citation16]. Forty female BALB/c mice (aged 6 weeks old, weight: 20 ± 2 g) were purchased from CAVENS (Changzhou, China). The mice were given the same standard diet and housed at the temperature of 20 ± 5 °C under a light cycle of 12 h light/12 h dark in 40 ± 5% humidity. According to the aim of understanding the effect of lncRNA TUG1 on LPS-induced ARDS mice, the mice were divided into the following groups: control group, LPS group, LPS+NC (negative control) group, and LPS+TUG1 group, with 10 mice in a group. The mice in control group were injected with 2 mg/kg normal saline into their tracheas, and mice in LPS group were injected with 2 mg/kg LPS into their tracheas. In LPS+NC group and LPS+TUG1 group, the mice were respectively intravenously injected with 2 × 107 TU/mL blank lentivirus and lncRNA TUG1 overexpressed lentivirus before the injection of 2 mg/kg LPS into their tracheas. After 24 h, the LPS-treated mice were sacrificed with an overdose of anesthesia except for those in the survival experiment. At the same time, BALF and lung tissue samples were collected for subsequent experiments.
Cell culture and treatment
RAW 264.7 macrophages are widely used in the model of LPS-induced ARDS model [Citation17]. RAW 264.7 macrophages were purchased from the American Type Culture Collection (ATCC; Catalogue number: TIB-71; Manassas, VA, USA) and cultured in DMEM/F12 (Gibco BRL, Grand Island, NY, USA) with 10% FBS (C0227, Beyotime, China) at 37°C with 5% CO2. For LPS treatment, the cells were seeded in 12-well plates (1 × 106 cells/well) and incubated for 24 h at 37°C, followed by the treatment with different concentrations of LPS (0, 0.1, 1, 2 and 10 μg/mL) for 5 h.
BALF collection and analysis
The BALF was collected and centrifuged at 1500 rpm at 4°C for 10 min. For quantifying cytokine, the parted supernatant samples were reserved at −80°C. The sediments were re-suspended in Phosphate-Buffered Saline (PBS; 10010023, Thermo Fisher, Waltham, MA, USA) for counting cell number and neutrophils under a light microscope (BX53M, Olympus, Tokyo, Japan) and dyed by Wright-Giemsa method using a hemocytometer (Z359629-1EA, Sigma-Aldrich, USA).
Pathological observation of lung tissues with hematoxylin and eosin (H&E) staining
The right lower lobe lung samples were collected and flushed with frozen saline. After being sliced into 5 μm sections, the tissues were fixed with 10% paraformaldehyde for 24 h, and subjected to hematoxylin and eosin (H&E) staining. Subsequently, lung injury was observed under a light microscope (VHX-7000, KEYENCE, China) at ×200 magnification. And the injury degree was evaluated as previously described [Citation18].
Evaluation of lung permeability
Evans blue dye (DK001, Leagene, Beijing, China) was used to evaluate intraplaque microvessel permeability. Specifically, 0.5% Evans blue stain (4 mg/mL) diluted in normal saline was injected into the mice via tail vein before euthanization. After exposing the hearts and lungs of the mice, the upper and lower vena cava were clamped, and the pulmonary blood vessels were lavaged with physiological saline through the infusion needle until on bloody fluid in the left atrium then the left lung was collected. The lung lobes were incubated in formamide (3 mL/100 mg) at room temperature for 24 h, and the absorbance of the supernatant was assessed by a microplate reader (RNE90002, Reagent Technology Co., Ltd., Shenzhen, China) at 620 nm.
Enzyme-linked immunosorbent assay (ELISA)
The BALF was centrifuged at 800 × g for 6 min to obtain BALF supernatant. Concentrations of tumor necrosis factor-α (TNF-α) and Interleukin-1β (IL-1β) in BALF supernatant and RAW 264.7 macrophages supernatant were detected by ELISA (ab208348 for detecting TNF-α, Abcam, UK; E-EL-M0037c for detecting IL-1β, Elabscience, Wuhan, China). The absorbance at 450 nm was assessed by a microplate reader (RNE90002, Reagent Technology Co., Ltd., Shenzhen, China).
RNA extraction and real-time quantitative PCR (qRT-PCR)
The extraction of total RNAs from the lung tissues and cultured cells was conducted using the TRIzol reagent (R701, Vazyme Biotech, China) following the instructions. RNA concentration was quantified using a spectrophotometer (NanoDrop 1000, Nanodrop, USA). cDNAs were synthesized from a total of 2 μg RNA by RevertAid First Strand cDNA Synthesis Kit (K1622, Thermo Fisher Scientific, USA). The mRNA expression was measured by SYBR Premix Ex Taq™ kit (Takara, Dalian, China), with β-actin as an endogenous control. One Step PrimeScript® miRNA cDNA Synthesis Kit (Takara) was employed to detect miR-221-3p expression, which was then normalized to that of U6. qRT-PCR was performed in ABI 7300 Real-Time PCR machine (Applied Biosystems, Thermo Fisher Scientific, USA) under the following parameters: 95°C for 60 sec, and then 95°C for 15 sec for 40 cycles, 61°C for 15 sec and 72°C for 45 sec. The fold changes were calculated using the 2−ΔΔCt method [Citation19]. The primers used in this study are listed in .
Table 1. Primer for qRT-PCR
Transfection
MiR-221-3p mimic (#B02003, sequence: 5ʹ-AGCUACAUUGUCUGCUGGGUUUC-3ʹ) and its negative control (#B04001, sequence: 5ʹ-GCUAACUGUCUUUUCAUCUGGGG-3ʹ) were chemically synthesized by GenePharma (Shanghai, China). pcDNA3.1 overexpressed plasmid was purchased from (VT1010, YouBio, China). RAW 264.7 macrophages were transfected with miR-221-3p mimic, lncRNA TUG1 overexpressed plasmid, and their controls by Lipofectamine 2000 (Invitrogen). After 48-h transfection, the cells were exposed to 1 μg/mL LPS for 6 h and used in subsequent experiments.
Prediction of target gene and dual-luciferase reporter assay
Target gene and potential binding sites of lncRNA TUG1 and miR-221-3p were predicted by StarBase (http://www.starbase.sysu.edu.cn), and the predictions were then confirmed by dual-luciferase reporter assay.
PMIR-REPORT Luciferase vector (catalog number: AM5795; Thermo Fisher Scientific, USA) containing miR-221-3p sequences (wild-type or mutated) were cloned into the pMirGLO reporter vector (Promega, Madison, WI, USA) to form lncRNA TUG1-WT and lncRNA TUG1-MUT. For dual-luciferase assay, RAW 264.7 macrophages (5 × 103 cells/well) were incubated in 96-well plates and then co-transfected with luciferase reporter plasmids and mimic by Lipofectamine 2000 Transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA). The firefly luciferase activity was normalized to that of the corresponding Renilla luciferase activity.
Cell apoptosis assay
RAW 264.7 macrophages were centrifuged (300 × g) for 5 min at 4°C after 48-h transfection and the apoptosis was measured by Annexin V-FITC Apoptosis Kit (40302ES20, Yeasen Biotech Co., Ltd., Shanghai, China) following the instructions. Briefly, the macrophages were washed by cold PBS twice and centrifuged (300 × g) for 5 min each time. 3 × 105 cells were collected for the subsequent experiments. Later, the PBS was removed and added with 100 μL 1× Binding Buffer, 5 μL Annexin V-FITC and 10 μL PI Staining Solution. Then the macrophages were incubated at the room temperature for 10–15 min avoiding the light. Finally, 400 μL 1× Binding Buffer was added to the cells and gently shaken. The sample was placed on the ice, and the apoptotic cells were differentiated by the flow cytometer (ACEA NovoCyte, ACEA Biosciences, Inc., USA).
Western blot
Total proteins were lysed using RIPA buffer (9806S, Cell Signaling Technology, Danvers, MA, USA). Concentration of proteins was detected by bicinchoninic protein assay (BCA) kit (7780S, Cell Signaling Technology, Danvers, MA, USA), and then the protein extraction was electrophoresed by 10% SDS-PAGE (P0012A, Beyotime, China) and then transferred onto PVDF membranes. The membranes containing 20 mL sealing liquid (1 g non-fat milk powder, 20 mL PBS-Tween 20) and then incubated primary antibodies (anti-SPRED2 antibody (rabbit, ab153700, 1:500, Abcam, UK), anti-p-ERK1/2 antibody (rabbit, #4370, 1:2000, Cell Signaling Technology, Danvers, MA, USA), anti-ERK1/2 antibody (rabbit, ab36861, 1:1000, Abcam, UK), and anti-β-actin antibody (mouse, ab8226, 1 µg/mL, Abcam, UK)) at 4°C overnight. β-actin was as an internal control. The secondary horseradish peroxidase (HRP)-combined antibodies, goat anti-rabbit IgG H&L (HRP) (goat, ab205718, 1:2000, Abcam, UK) and goat anti-mouse IgG H&L (HRP) (goat, ab205719, 1:2000, Abcam, UK), were further incubated with the membranes at room temperature for 1 h and washed by tris-buffer saline tween (TBST) for three times. The relative amount of proteins was quantified using gel analysis software version 5.3 UNSCAN-IT gel (Silk Scientific, Orem, UT, USA) [Citation20].
Statistical analysis
Differences between groups were assessed using one-way analysis of variance followed by Student’s t-test using SPPS (version 22.0, Chicago, IL, USA). Values are expressed as the mean±standard deviation (SD). P-value was considered to be statistically significant when it was lower than 0.05.
Results
Overexpressed lncRNA TUG1 alleviated LPS-induced lung tissue impairment and capillary permeability and reduced total cell and neutrophil count
The total cells and neutrophils in BALF were collected and measured. “From” H&E staining in , control group showed normal lung tissue morphology. In LPS group lung tissues were obviously impaired, with alveolar congestion and bleeding, incrassation of alveolar wall, also inflammatory cells infiltrated blood vessel walls. However, lentivirus injection with lncRNA TUG1 overexpressed repaired the LPS-induced impairment of lung tissues. In addition, Evans Blue Dye demonstrated that Evans blue was increased in LPS group, indicating that LPS treatment promoted lung capillary permeability (, P < 0.01), but it was decreased in LPS+TUG1 group (, P < 0.01), which showed that lncRNA TUG1 overexpressed lentivirus injection reversed the function of LPS on capillary permeability. Besides, as shown in ,d, cells and neutrophils were increased by LPS treatment, which was reversed by lncRNA TUG1 overexpressed lentivirus injection (P < 0.01).
Figure 1. Overexpressed lncRNA TUG1 could attenuate Lipopolysaccharide (LPS)-induced ARDS mice pulmonary inflammation. (a) Lung tissue histological observation and impairment was evaluated by Hematoxylin-eosin (H&E) staining, wMagnification: 200×, scale bar = 200 µm. (b) Lung permeability was evaulated by Evans blue dye extravasation. (c,d) The numbers of total cells and neutrophils in bronchoalveolar lavage fluid (BALF) was measured by hemocytometer. (e,f) The expressions of tumor necrosis factor-α (TNF-α) and Interleukin-1β (IL-1β) in BALF were determined by enzyme-linked immunosorbent assay (ELISA). (g) The expression of lncRNA TUG1 in mice lung tissues (n = 10) was detected by quantitative real-time polymerase chain reaction (qRT-PCR). β-actin was used as an internal control. All experiments have been performed in triplicate and experimental data were expressed as mean ± standard deviation (SD). (***P < 0.001, vs. Control; ##P < 0.01, ###P < 0.001, vs. LPS+NC) NC: negative control. ANOVA was applied followed by Student’s t-test

Overexpressed lncRNA TUG1 reduced the function of LPS on proinflammatory cytokines
In order to further confirm that LPS could induce ARDS, levels of proinflammatory cytokines (IL-1β and TNF-α) were measured using ELISA in BALF. The data revealed that expressions of IL-1β and TNF-α were increased in LPS group but reduced in LPS+TUG1 group (, P < 0.01). Here, lncRNA TUG1 expression in mice lung tissues was detected by qRT-PCR, and the data demonstrated that lncRNA TUG1 expression was reduced by LPS treatment but increased by lncRNA TUG1 overexpressed lentivirus injection (, P < 0.001).
LncRNA TUG1 targeted miR-221-3p and reduced the expression of miR-221-3p
StarBase showed that lncRNA TUG1 was a target of miR-221-3p, with the target site located in the 3ʹ-UTR (). For confirming the online prediction, dual-luciferase reporter assay was carried out. Synthesized reporter plasmid with mutant and wild-type 3ʹ-UTR region of lncRNA TUG1 and miR-221-3p mimic were co-transfected into the RAW 264.7 cells, and mimic control (MC) group was set up at the same time. Dual-luciferase reporter revealed that luciferase activity in TUG1-WT-mimic group was reduced in comparison with MC group (, P < 0.001). Meanwhile, miR-221-3p expression in the lung tissues derived from LPS-induced ARDS mice injected with lncRNA TUG1 overexpressed lentivirus was detected by qRT-PCR. We discovered that miR-221-3p expression in LPS group was promoted when compared with control group (, P < 0.001), but in LPS+TUG1 group miR-221-3p expression was reduced in comparison with LPS+NC group, suggesting that miR-221-3p expression in ARDS mice model was down-regulated by lncRNA TUG1 overexpressed lentivirus injection.
Figure 2. LncRNA TUG1 was a direct target of miR-221-3p, and could reduce miR-221-3p expression in ARDS mice model. (a) StarBase (www.starbase.sysu.edu.cn/) was used to predict the binding sites of miR-221-3p and lncRNA TUG1. (b) The RAW 264.7 macrophagocyte were co-transfected with wild-type or mutant of lncRNA TUG1 (TUG1-WT; TUG1-MUT) and miR-221-3p mimic, and luciferase activity was measured by dual-luciferase reporter assay. (c) The miR-221-3p expression in lung tissues (n = 10) from LPS-induced ARDS mice injected with lncRNA TUG1 overexpressed lentivirus was detected by qRT-PCR. U6 was used an internal control. All experiments have been performed in triplicate and experimental data were expressed as mean ± standard deviation (SD). (***P < 0.001, vs. Control; ###P < 0.001, vs. LPS+NC; ^^^P < 0.001, vs. MC) MC: mimic control, NC: negative control. ANOVA was applied followed by Student’s t-test

LncRNA TUG1 reduced LPS-induced cell apoptosis and inflammation via miR-221-3p
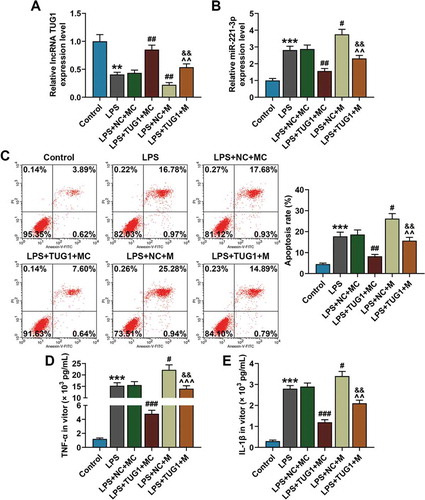
For understanding the effect of lncRNA TUG1 on the protective role of miR-221-3p in suppressing LPS-induced inflammatory response in the previous experiments, RAW 264.7 cells were co-transfected with miR-221-3p mimic and lncRNA TUG1 overexpressed plasmids, followed by 6-h exposure to 1 μg/mL LPS. First, the expressions of lncRNA TUG1 and miR-221-3p in RAW 264.7 cells of each group were detected by qRT-PCR, and it was clear that after the transfection miR-221-3p expression in the cells was evidently down-regulated by overexpressed lncRNA TUG1, and at the same time, lncRNA TUG1 expression in the cells was down-regulated by overexpressed miR-221-3p (, P < 0.05).
Figure 3. Overexpressed lncRNA TUG1 could alleviate LPS-induced up-regulation of miR-221-3p expression and RAW 264.7 macrophages apoptosis and inflammatory cytokines accumulation, which were reversed by overexpressed miR-221-3p. (a,b) RAW 264.7 macrophagocyte were divided into the following groups: Control, LPS, LPS+NC+mimic control, LPS+TUG1+ mimic control, LPS+NC+miR-221-3p, LPS+TUG1+ miR-221-3p. Expressions of lncRNA TUG1 and miR-221-3p in RAW 264.7 macrophages of each group were determined by qRT-PCR. β-actin and U6 were used as internal controls. (c) RAW 264.7 macrophages apoptosis rate in each group was detected by flow cytometry. (d,e) The expressions of inflammatory cytokines TNF-α and IL-1β in RAW 264.7 macrophages supernatant were detected by ELISA. All experiments have been performed in triplicate and experimental data were expressed as mean ± standard deviation (SD). (**P < 0.01, ***P < 0.001, vs. Control; #P < 0.05, ##P < 0.01, ###P < 0.001, vs. LPS+NC+MC; ^^P < 0.01, ^^^P < 0.01, vs. LPS+TUG1+ MC; &&P < 0.01, vs. LPS+NC+M). M: mimic, MC: mimic control, NC: negative control. ANOVA was applied followed by Student’s t-test

Flow cytometry was employed to verify the cell apoptosis rate in each group. When compared with the control group, cell apoptosis rate was accelerated by LPS treatment, while overexpressed lncRNA TUG1 could reverse such an effect of LPS on the apoptosis (, P < 0.05). Interestingly, the protective effect of overexpressed lncRNA TUG1 on cell apoptosis could be reversed by overexpressed miR-221-3p.
Next, TNF-α and IL-1β expressions in cell supernatant were detected by ELISA kits. The discoveries showed that in LPS group TNF-α and IL-1β expressions were enhanced as compared with the control group, while overexpressed lncRNA TUG1 could reduce the LPS-induced inflammatory cytokine accumulation (, P < 0.05). However, the inhibitory effect of overexpressed lncRNA TUG1 on the production of inflammatory cytokines was reversed by overexpressed by miR-221-3p.
LncRNA TUG1 reversed the function of LPS on SPRED2 and ERK1/2 via miR-221-3p in RAW 264.7 macrophages
The detection of expressions of SPRED2, phosphorylated(p)-ERK1/2, and ERK1/2 were performed by Western blot. Compared with the control group, SPRED2 expression in cells was decreased and the phosphorylation levels of ERK1 and ERK2 and the ratio of p-ERK/ERK were promoted in the LPS group, while ERK1 and ERK2 levels did not change (, P < 0.05). However, SPRED2 expression was up-regulated and the phosphorylation levels of ERK1 and ERK2 and the ratio of p-ERK/ERK were suppressed, while ERK1 and ERK2 levels did not change under overexpressed lncRNA TUG1. Interestingly, the regulatory function of overexpressed lncRNA TUG1 on SPRED2 expression and ERK1 and ERK2 phosphorylation levels was reversed by overexpressed miR-221-3p.
Figure 4. Overexpressed lncRNA TUG1 could up-regulate SPRED2 expression and down-regulate phosphorylation levels of ERK1 and ERK2, which were reversed by overexpressed miR-221-3p. (a-g) Expressions of SPRED2, phosphorylated (p)-ERK1/2 and ERK1/2 in RAW 264.7 macrophages were determined by Western blot. β-actin was used as an internal control. All experiments have been performed in triplicate and experimental data were expressed as mean ± standard deviation (SD). (*P < 0.05, ***P < 0.001, vs. Control; #P < 0.05, ##P < 0.01, ###P < 0.001, vs. LPS+NC+MC; ^P < 0.05, ^^P < 0.01, vs. LPS+TUG1+ MC; &&P < 0.01, &&&P < 0.001, vs. LPS+NC+M). M: mimic, MC: mimic control, NC: negative control, SPRED2: sprouty related EVH1 domain containing 2. ANOVA was applied followed by Student’s t-test

Discussion
ARDS is a result of a series of pathophysiologic changes related to early acute lung injury that leads to increased capillary leak and pulmonary edema [Citation21], and inflammatory response and endothelial barrier disruption are key factors in the development of the disease [Citation22].
In the past few decades, lncRNAs have been proven as promising key regulators of the inflammatory response through modulating the transcriptional control of inflammation-related mediators [Citation23]. LncRNA TUG1 promotes the development of osteosarcoma through RUNX2 [Citation24] as well as the proliferation of vascular smooth muscle cells and atherosclerosis through regulating miR-21/PTEN axis [Citation25]. In our study, we focused on exploring the role of lncRNA TUG1, and we speculated that it had an inhibitory effect on LPS-induced ARDS. The present study established an LPS-induced ARDS mice model. In line with the experimental data, overexpressed lncRNA TUG1 treatment alleviated the lung tissue impairment and pulmonary inflammation caused by LPS, suggesting that lncRNA TUG1 was an important factor in ARDS treatment. Studies increasingly revealed that lncRNA TUG1 play an anti-inflammatory role. For example, Zhao et al. [Citation26] demonstrated that lncRNA TUG1 attenuated TNF-α-induced inflammatory response in interstitial cells of Cajal by down-regulating miR-127 and then inactivating NF-κB and Notch pathways. Knocking down lncRNA TUG1 activates p38MAPK and JNK pathways and promotes inflammatory cytokine expression in human liver cells [Citation27], which proves that lncRNA TUG1 dose have an anti-inflammatory effect and this is consistent with our findings. TNF-α and IL-1β are proinflammatory cytokines [Citation28]. The present study discovered that TNF-α and IL-1β levels in BALF were enhanced by LPS treatment but alleviated by overexpressed lncRNA TUG1, which further supported the anti-inflammatory effect of lncRNA TUG1.
Currently, the roles of ceRNAs explain a novel regulatory mechanism of RNAs by seeing that lncRNAs has the ability to antagonize miRNA expression and work as sponges to bind specific miRNAs [Citation29]. MiRNAs are endogenous non-coding small RNAs and specific factors for silencing post-transcriptional genes [Citation30]. The discovery of miRNAs, identification of targets and further clarification of miRNA functions are key strategies for understanding the normal biological processes of miRNAs and their roles in disease development [Citation31]. miR-221 regulates CD44 in hepatocellular carcinoma through the PI3K-AKT-mTOR pathway [Citation32] and in asthma it participates in the airway epithelial cell injury via targeting SIRT1 [Citation33]. LncRNA TUG1 enhances chemosensitivity of non-small cell lung cancer cells by interrupting miR-221-dependent PTEN inhibition [Citation15]. Furthermore, in our study, lncRNA TUG1 was predicted and verified to sponge miR-221-3p by StarBase and dual-luciferase reporter assay. We discovered the role of lncRNA TUG1 on LPS-treated RAW 264.7 macrophages was realized via targeting miR-221-3p. On the basis of the experimental results from our study, overexpressed lncRNA TUG1 reduced LPS-induced RAW 264.7 macrophage apoptosis, which was reversed by miR-221-3p.
Studies found that SPRED2 is a target gene of miR-221-3p, and it is clearly expressed in the lung tissue and involved in the development of LPS-induced lung inflammation via negatively regulating the ERK1/2 pathway [Citation34]. The ERK1/2 pathway is activated mainly in response to mitogens and growth factors, and ERK1/2 can function as a pro-apoptotic factor in some conditions [Citation35,Citation36]. It has been reported that the ERK1/2 signaling pathway plays an important role in mediating inflammatory response [Citation37]. For example, Li et al. [Citation38] demonstrated that glucagon-like peptide-2 weakens LPS-induced inflammation in BV-2 cells via inhibiting ERK1/2, JNK1/2, and NF-κB signaling pathways. In addition, Yan et al. [Citation39] showed that vanillin treatment markedly inhibits LPS-induced inflammation via suppressing phosphorylation of p38, ERK1/2, and NF-κB p65. Meanwhile, in our study, according to the results of Western blot, overexpressed lncRNA TUG1 reduced inflammatory cytokines release of LPS-induced RAW 264.7 macrophages, and reversed the effects of LPS on SPRED2 level and phosphorylation levels of ERK1/2 in RAW 264.7 macrophages, which, however, were all reversed by miR-221-3p.
In summary, we found that LPS-induced ARDS mice overexpressed lncRNA TUG1 could attenuate pulmonary inflammation, and lncRNA TUG1 could attenuate LPS-caused RAW264.7 macrophages apoptosis, up-regulate SPRED2 expression and reduce phosphorylation level of ERK1/2 in RAW264.7 macrophages via targeting miR-221-3p. These data showed that lncRNA TUG1 and miR-221-3p expressions could serve as useful prognostic indicators for ARDS, and might be novel molecular targets in the development of ARDS therapy.
Authors’ contributions
Substantial contributions to conception and design: LH, HY
Data acquisition, data analysis and interpretation: JL
Drafting the article or critically revising it for important intellectual content: LH, HY
Final approval of the version to be published: All authors
Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of the work are appropriately investigated and resolved: All authors
Ethics approval and consent to participate
The approval of this study was obtained from the Committee of Experimental Animals of Shenzhen Hospital, Southern Medical University (HXK20200203). Every effort was made to minimize pain and discomfort to the animals. The animals’ experiments were performed in Shenzhen Hospital, Southern Medical University.
No human is involved in this research.
Disclosure statement
The authors declare no conflicts of interests.
References
- Cutts S, Talboys R, Paspula C, et al. Adult respiratory distress syndrome. Ann R Coll Surg Engl. 2017 Jan;99(1):12–16.
- Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. Jama. 2016 Feb 23;315(8):788–800.
- Kumar MM, Goyal R. LncRNA as a Therapeutic Target for Angiogenesis. Curr Top Med Chem. 2017;17(15):1750–1757.
- Ma Y, Zhang J, Wen L, et al. Membrane-lipid associated lncRNA: A new regulator in cancer signaling. Cancer Lett. 2018 Apr;10(419):27–29.
- Dai L, Zhang G, Cheng Z, et al. Knockdown of LncRNA MALAT1 contributes to the suppression of inflammatory responses by up-regulating miR-146a in LPS-induced acute lung injury. Connect Tissue Res. 2018 Nov;59(6):581–592.
- Du M, Yuan L, Tan X, et al. The LPS-inducible lncRNA Mirt2 is a negative regulator of inflammation. Nat Commun. 2017 Dec 11;8(1):2049.
- Xie C, Chen B, Wu B, et al. LncRNA TUG1 promotes cell proliferation and suppresses apoptosis in osteosarcoma by regulating miR-212-3p/FOXA1 axis. Biomed Pharmacothe. 2018 Jan;97:1645–1653.
- Zhang H, Li H, Ge A, et al. Long non-coding RNA TUG1 inhibits apoptosis and inflammatory response in LPS-treated H9c2 cells by down-regulation of miR-29b. Biomed Pharmacother. 2018 May;101:663–669.
- Su S, Liu J, He K, et al. Overexpression of the long noncoding RNA TUG1 protects against cold-induced injury of mouse livers by inhibiting apoptosis and inflammation. Febs J. 2016 Apr;283(7):1261–1274.
- Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018 Apr;141(4):1202–1207.
- Shikano S, Gon Y, Maruoka S, et al. Increased extracellular vesicle miRNA-466 family in the bronchoalveolar lavage fluid as a precipitating factor of ARDS. BMC Pulm Med. 2019 Jun 20;19(1):110.
- Ju M, Liu B, He H, et al. MicroRNA-27a alleviates LPS-induced acute lung injury in mice via inhibiting inflammation and apoptosis through modulating TLR4/MyD88/NF-κB pathway. Cell Cycle. 2018;17(16):2001–2018.
- Han Y, Ma J, Wang J, et al. Silencing of H19 inhibits the adipogenesis and inflammation response in ox-LDL-treated Raw264.7 cells by up-regulating miR-130b. Mol Immunol. 2018;93:107–114.
- Song P, Yin SC. Long non-coding RNA EWSAT1 promotes human nasopharyngeal carcinoma cell growth in vitro by targeting miR-326/-330-5p. Aging (Albany NY). 2016 Nov 3;8(11):2948–2960.
- Guo S, Zhang L, Zhang Y, et al. Long non-coding RNA TUG1 enhances chemosensitivity in non-small cell lung cancer by impairing microRNA-221-dependent PTEN inhibition. Aging (Albany NY). 2019 Sep 18;11(18):7553–7569.
- Wang T, Jiang L, Wei X, et al. Inhibition of miR-221 alleviates LPS-induced acute lung injury via inactivation of SOCS1/NF-kappaB signaling pathway. Cell Cycle. 2019 Aug;18(16):1893–1907.
- Li C, Yang D, Cao X, et al. LFG-500, a newly synthesized flavonoid, attenuates lipopolysaccharide-induced acute lung injury and inflammation in mice. Biochem Pharmacol. 2016;113:57–69.
- Arold SP, Mora R, Lutchen KR, et al. Variable tidal volume ventilation improves lung mechanics and gas exchange in a rodent model of acute lung injury. Am J Respir Crit Care Med. 2002;165(3):366–371.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001 Dec;25(4):402–408.
- Xu J, Yang J, Chen J, et al. Vitamin D alleviates lipopolysaccharide‑induced acute lung injury via regulation of the renin‑angiotensin system. Mol Med Rep. 2017;16(5):7432–7438.
- El-Haddad H, Jang H, Chen W, et al. Effect of ARDS severity and etiology on short-term outcomes. Respir Care. 2017 Sep;62(9):1178–1185.
- Middleton EA, Zimmerman GA. Early Returns in Vascular Inflammation in ARDS. Am J Respir Crit Care Med. 2018 Jun 15;197(12):1514–1516.
- Mathy NW, Chen XM. Long non-coding RNAs (lncRNAs) and their transcriptional control of inflammatory responses. J Biol Chem. 2017 Jul 28;292(30):12375–12382.
- Sheng K, Li Y. LncRNA TUG1 promotes the development of osteosarcoma through RUNX2. Exp Ther Med. 2019 Oct;18(4):3002–3008.
- Li FP, Lin DQ, Gao LY. LncRNA TUG1 promotes proliferation of vascular smooth muscle cell and atherosclerosis through regulating miRNA-21/PTEN axis. Eur Rev Med Pharmacol Sci. 2018 Nov;22(21):7439–7447.
- Zhao K, Tan JY, Mao QD, et al. Overexpression of long non-coding RNA TUG1 alleviates TNF-alpha-induced inflammatory injury in interstitial cells of Cajal. Eur Rev Med Pharmacol Sci. 2019 Jan;23(1):312–320.
- Huang L, Zhang X, Ma X, et al. Berberine alleviates endothelial glycocalyx degradation and promotes glycocalyx restoration in LPS-induced ARDS. Int Immunopharmacol. 2018 Dec;65:96–107.
- Martínez-Reza I, Díaz L, Barrera D, et al. calcitriol inhibits the proliferation of triple-negative breast cancer cells through a mechanism involving the proinflammatory cytokines IL-1 and TNF. J Immunol Res. 2019;2019:6384278.
- Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013 Mar 21;495(7441):384–388.
- Paladini L, Fabris L, Bottai G, et al. Targeting microRNAs as key modulators of tumor immune response. J Exp Clin Cancer Res. 2016 Jun;27(35):103.
- Liu B, Li J, Cairns MJ. Identifying miRNAs, targets and functions. Brief Bioinform. 2014 Jan;15(1):1–19.
- Kim J, Jiang J, Badawi M, et al. miR-221 regulates CD44 in hepatocellular carcinoma through the PI3K-AKT-mTOR pathway. Biochem Biophys Res Commun. 2017 Jun 3;487(3):709–715.
- Zhang H, Sun Y, Rong W, et al. miR-221 participates in the airway epithelial cells injury in asthma via targeting SIRT1. Exp Lung Res. 2018 Aug;44(6):272–279.
- Xu Y, Ito T, Fushimi S, et al. Spred-2 deficiency exacerbates lipopolysaccharide-induced acute lung inflammation in mice. PloS One. 2014;9(9):e108914.
- Zheng J, Liu P, Li C, et al. Umbilical cord-derived mesenchymal stem cells efficiently induced apoptosis of endometrial carcinoma cells. Eur J Gynaecol Oncol. 2019;40(5):839–845.
- Okada M, Yamane M, Yamamoto S, et al. SPRED2 deficiency may lead to lung ischemia-reperfusion injury via ERK1/2 signaling pathway activation. Surg Today. 2018 Dec;48(12):1089–1095.
- Hytti M, Andjelic S, Josifovska N, et al. CB2 receptor activation causes an ERK1/2-dependent inflammatory response in human RPE cells. Sci Rep. 2017 Nov 23;7(1):16169.
- Li N, Liu BW, Ren WZ, et al. GLP-2 Attenuates LPS-Induced Inflammation in BV-2 Cells by Inhibiting ERK1/2, JNK1/2 and NF-kappaB Signaling Pathways. Int J Mol Sci. 2016 Feb 4;17(2):190.
- Yan X, Liu DF, Zhang XY, et al. Vanillin protects dopaminergic neurons against inflammation-mediated cell death by inhibiting ERK1/2, P38 and the NF-kappaB signaling pathway. Int J Mol Sci. 2017 Feb 12;18(2).