
ABSTRACT
In this study, we investigated and compared characteristics of the bacterial community structures on hair (scalp hair) and scalp in 18 individuals. Significant differences were found between the sites, in terms of cell density, alpha and beta diversity, and relative abundance of the phyla, Firmicutes and Proteobacteria, whereas no difference was found in relative abundance of the phylum Actinobacteria. Bacteria of the genus Cutibacterium showed similar relative abundance at both sites, whereas those of genus Pseudomonas were highly abundant on hair, and those of genus Staphylococcus were significantly lesser in abundance on hair than on scalp. Statistical correlations between the sites were high for the individual relative abundance of five major operational taxonomic units (OTUs). This suggests that the bacterial community structure on hair is composed of hair-specific genus, Pseudomonas, and skin-derived genera, Cutibacterium and Staphylococcus, and is distinguishable from other human skin microbiomes.
GRAPHICAL ABSTRACT
The bacterial community structure on hair was specific and distinguishable from that on scalp, although major bacterial species were commonly present at body sites.
The entire human body surface, including hair and skin, is colonized by a wide variety of microorganisms, including bacteria, fungi, and viruses [Citation1–2Citation3]. Human skin, the largest organ of the human body, is colonized by 102–107 bacteria per cm2 [Citation4]. Some of these bacteria live in a symbiotic relationship with their host, and protect against invasion by pathogenic microorganisms [Citation5,Citation6]. The physical and chemical features of various parts of the skin form unique bacterial community structures, that are adapted to the niche that they inhabit [Citation7]. Colonization by bacteria is dependent on the physiology of the skin site, with specific bacteria being associated with their microenvironments [Citation8].
The scalp surface also provides a distinct microenvironment to the microbiome, primarily arising from the host skin’s physiological conditions, which include sebum content, moisture, pH, and topography of the hair. In general, skin sites are roughly classified into three groups, based on their microenvironments, which are: moist, dry, and sebaceous groups. Among these skin site groups, scalp belongs to the sebaceous group. Sebaceous glands of the scalp produce a large amount of oily sebum [Citation9]. Furthermore, several studies on scalp microbiome in various countries have revealed the association of dandruff with bacterial community structure [Citation10–13]. These studies showed that the major bacterial genera on the scalp are Cutibacterium (Propionibacterium) and Staphylococcus. When compared with normal scalp, the scalp with dandruff had a decreased population of Cutibacterium and an increased population of Staphylococcus. As dandruff is one of the disorders of the scalp, it is also a problem of the hair. Despite these facts, the relationship between the microbiomes on human hair (scalp hair) and scalp is yet unknown, and studies to elucidate this relationship and would be essential for a better understanding of both hair and scalp health.
Several studies have reported observations on the bacteria present on hair, using fluorescence light microscopy or scanning electron microscopy (SEM) [Citation14,Citation15]. Existence of bacteria was reported not only on hair shafts but also on hair follicles [Citation16]. Analysis of bacterial community structures on hair by terminal restriction fragment length polymorphism (T-RFLP) [Citation17] indicated that these bacterial community structures are specific for each individual, and when collected from any human body part including hand [Citation18], could be a tool for identifying a suspect. Bacterial community structures on specific parts of hair shaft and hair root were further compared by 16S rRNA gene amplicon sequencing analysis, and it was reported that the bacteria on hair shaft are indigenous and derived from the hair root, having similar number and structure from the top to the base parts of a long hair shaft [Citation19]. The report also suggested that bacterial community structures on scalp hair are distinct from those on other body sites, including various parts of the skin [Citation20]. However, the differences of bacterial community structures found on scalp hair and scalp were not studied, which would be essential for a better understanding of hair and scalp health, as well as bacterial ecology of hair.
In this study, we analyzed bacterial community structures, by 16S rRNA gene amplicon sequencing, to elucidate characteristics of the structures and to analyze the relationship between bacterial community structures of human scalp hair and human scalp within the same individual.
Materials and methods
This research was performed with permission from the research ethics committee of the Graduate School of Bioscience and Biotechnology at Kyushu University.
Samples and collection
Hair shaft samples and scalp swab samples were collected from 18 healthy Japanese and Chinese adults of both sexes (9 males and 9 females), ranging in age from 21 to 62 years, who consented to take part in this study (Supplemental Table1). None of the volunteers were taking any medication during the experimental period. All of the volunteers washed their hair 6 h prior to sample collection. Also, samples at both sites were collected on the same day. Samples of hair shafts and scalp swabs were collected using nitrile gloves. Scalp swab samples were directly taken from the crown of the head using cotton swabs (Mentip for hospital, Nihon Menbou Corporation, Saitama, Japan) pre-moistened with 50 μL of sterile distilled water. Cotton swabs were rubbed onto the scalp surface (between the hair strands) to cover a total surface area of 2.5 cm2. The head of each swab was cut from the handle and placed into an Eppendorf tube. Samples of hair shaft were cut using sterilized scissors, and chopped into pieces of 5 mm length with the scissors before use.
Table 1. Summary of differences in abundance and correlation of bacterial community between hair and scalp
Extraction of bacterial DNA from hair shaft and scalp swab samples
Bacterial DNA was extracted using the NucleoSpin® Tissue kit (MACHEREY-NAGEL, Düren, Germany) according to the manufacturer’s instructions, with a slight modification. First, samples of scalp swabs and hair shafts were immersed in 100 μL of lysozyme solution (20 mg/mL lysozyme derived from egg white [Wako Pure Chemical Industries, Osaka, Japan] in 20 mM Tris-HCl and 0.2 mM EDTA, pH 8.0) for 30 min at 37°C, as previously reported [Citation19], and the DNA extracts obtained (100 μL) were stored at ‒20°C until use.
Quantification of the bacterial cell number on hair and scalp by qPCR
The bacterial cell number on the hair and scalp of 18 volunteers was quantified by estimation of 16S rRNA gene copy number using real-time PCR (CFX Connect™ System, BIO-RAD Laboratories, Inc., CA, USA) with universal primers for a portion of the bacterial 16S rRNA gene. We have previously shown that the values estimated by quantitative PCR (qPCR) correspond well with the values obtained by the direct SEM observation [Citation19].
Each 10 μL reaction mixture consisted of 5 μL of KOD SYBR® qPCR Mix (TOYOBO Co., Ltd., Osaka, Japan), 0.1 μL of each primer [357 F (5′-CCT ACG GGA GGC AGC AG-3′) [Citation21] and 518 R (5′-ATT ACC GCG GCT GCT GG-3′) [Citation22]], and 2 μL of bacterial DNA. The amplification program included an initial denaturation step at 95°C for 5 min, followed by 40 cycles each of denaturation at 95°C for 5 s, annealing at 64°C for 20 s, and elongation at 72°C for 20 s. DNA extract from Escherichia coli DH5α was used as a standard to generate a calibration curve. After amplification, the copy numbers of the 16S rRNA genes per hair sample were calculated per cm of hair and converted to per cm2 of hair. For the calculation, the following equation was used:
Cells/cm2 = qPCR copies/hair length (cm) × hair diameter (cm)×π
The diameter of the hair was measured using a stereo microscope (Stemi 305, ZEISS, Oberkochen, Germany).
Analysis of the bacterial community structures on scalp and hair by 16S rRNA gene sequencing
To analyze the bacterial community structures of scalp and hair from 18 volunteers using the MiSeq™ platform (Illumina Inc., CA, USA), a three-step PCR method was performed using the extracted DNA samples. In the first-step PCR amplification, a universal primer set for the V4 region of the bacterial 16S rRNA gene (515 F, 5′-GTG CCA GCM GCC GCG GTA A-3′ and 806 R, 5′-GGA CTA CHV GGG TWT CTA AT-3′) [Citation23] was used. The 25 μL reaction mixture consisted of 12.5 μL of Kapa HiFi HotStart Ready Mix (Kapa Biosystems Inc., Wilmington, MA, USA), 0.5 μL of each primer (10 pM), and 11.5 μL of extracted bacterial DNA. The amplification program included an initial denaturation step at 95°C for 3 min, followed by 40 cycles each of denaturation at 98°C for 30 s, annealing at 56°C for 30 s, and elongation at 72°C for 30 s. After electrophoresis through a 1.5% (w/v) agarose gel, the targeted bands were excised from the gel with sterilized cutters, and the DNA was extracted using the FastGene® Gel/PCR Extraction Kit (NIPPON Genetics Co., Ltd., Tokyo, Japan), according to the manufacturer’s instructions. The DNA concentration was measured using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). In preparation of 16S rRNA amplicon sequencing with MiSeq, the templates are given tail, adapter, and index sequences in a two-step PCR. Therefore, long-tailed primers are required for the preparation, which makes the amplification difficult. We were unable to perform direct amplification in the two-step PCR, probably because the amount of bacterial DNA obtained from a 3 cm hair shaft is very small. Therefore, we first performed PCR using the universal primer set without any additional sequences. As a result, we succeeded in obtaining enough template fragments with a minimum number of reaction cycles.
For the second-step PCR, a universal primer set for the V4 region of the bacterial 16S rRNA gene and tailed sequences for MiSeq sequencing were used (1–515 F, 5′- TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG GTG CCA GCM GCC GCG GTA A-3′ and 1–806 R, 5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGG ACT ACH VGG GTW TCT AAT-3′) [Citation24]. Although it is reported that this primer set would poorly amplify Propionibacterium of human skin [Citation25], the results in this study showed good amplifications of Cutibacterium acnes (the previous name is Propionibacterium acnes) of not only the scalp but also hair using this primer set as the predominant species. The 25 μL reaction mixture consisted of 1.0 µL of each primer (5 µM) which was heat-shocked at 95°C for 5 min, 12.5 µL of Kapa HiFi HotStart Ready Mix, 12.5 ng of DNA obtained from the first-step PCR amplicon, and sterilized ultrapure water. The amplification program included an initial denaturation step at 95°C for 3 min, followed by 20 cycles of denaturation at 98°C for 30 s, annealing at 55°C for 30 s, and elongation at 72°C for 30 s. PCR products were purified using the FastGene® Gel/PCR Extraction Kit according to the manufacturer’s instructions.
For the third-step PCR, a primer set with flow cell adapter sequences, index sequences, and tailed sequences was used (Forward primer, 5′-AAT GAT ACG GCG ACC ACC GAG ATC TAC AC-Index sequence-TCG TCG GCA GCG TC-3′ and Reverse primer, 5′-CAA GCA GAA GAC GGC ATA CGA GAT-Index sequence-GTC TCG TGG GCT CGG-3′). The third-step PCR mixture (25 µL) was composed of 12.5 µL of Kapa HiFi HotStart Ready Mix, 0.5 µL of each primer (10 pM), 11.5 µL of the second-step PCR amplicon. The amplification program included an initial denaturation step at 95°C for 3 min, followed by 8 cycles of denaturation at 98°C for 30 s, annealing at 55°C for 30 s, and elongation at 72°C for 30 s. After electrophoresis in a 1.5% (w/v) agarose gel, the target bands were excised with sterilized cutters, and the DNA was extracted using the FastGene® Gel/PCR Extraction Kit as described above. The DNA concentration of the third-step PCR amplicons was quantified by using a Qubit™ dsDNA HS Assay Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA) according to the manufacturer’s instructions. The purified PCR products from each sample were mixed, denatured, and sequenced with an MiSeq System (Illumina) using MiSeq Reagent Kit v3 (300 bp ×2 cycles with pair-end; Illumina), according to the manufacturer’s instructions. We obtained Good’s coverage values (> 95%) for all hair samples using the DNA extraction kit and PCR conditions described above, indicating that the results obtained contain meaningful information (Supplemental Table 2). Good’s coverage values were estimated using QIIME™ 1.9.1 software [Citation26].
Bioinformatics and statistical analysis
The index and universal sequences of each read were checked, and reads with complete index sequences were selected as valid sequences. USEARCH V8.1.1861 [Citation27] software was used to merge paired-end reads and remove chimeric sequences. After the chimera check, the reads were grouped into operational taxonomic units (OTUs) at >97% similarity. Alpha diversity (observed OTUs and Shannon index) was evaluated at a 1% OTU distance using the QIIME™ software package [Citation26]. In the taxonomy-based analysis, representative sequences for each OTU were analyzed with the EzBioCloud platform [Citation28]. Statistical analysis in quantification of bacterial cell numbers was done using ANOVA, and statistical analysis in bacterial community structure was done using the Kruskal-Wallis test. Both statistical analyses were run under XLSTAT software ver. 2014 (http://www.xlstat.com/en/).
Accession number
Illumina raw read sequences and the top 13 most abundant OTU sequences were deposited in the DDBJ/ENA/GenBank database under accession numbers LC557428-LC557440.
Results
Quantification of bacterial cell number on hair and scalp
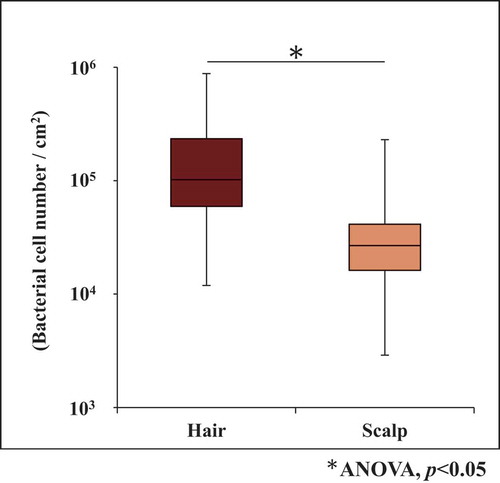
Bacterial cell number on hair and scalp from 108 samples derived from 18 volunteers was analyzed separately (Supplemental Table 1). The average bacterial cell number on a hair sample was 1.6 (±1.6) × 105 cells/cm2, while that on a scalp sample was 3.8 (±3.7) × 104 cells/cm2, which was lower than the number on hair by 1 order of magnitude (). In spite of that, analysis of the correlation between these numbers, at the level of an individual, showed that there was statistically no correlation between the numbers on both sites (Supplemental Figure 1) .
Figure 1. Bacterial cell number (copies/cm2) on hair and scalp
Alpha diversity of the bacterial community structures on hair and scalp
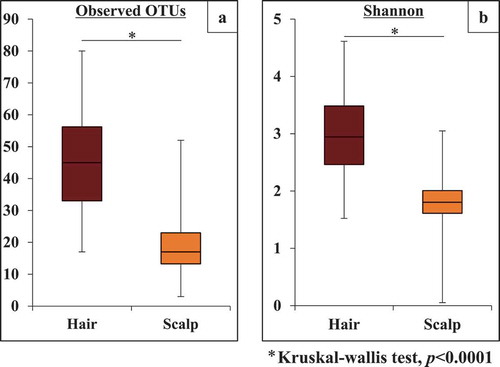
shows the two indexes of alpha diversity: observed OTUs, and the Shannon index. Both indexes were significantly higher (p< 0.0001) on hair than on scalp. It is noted that the average OTU numbers on hair and scalp were 46.2 and 19.2, respectively. On the other hand, there was weak correlation of observed OTUs, and no correlation of Shannon indexes at individual levels (Supplemental. and , R = 0.38, and −0.04, respectively). This suggests that the bacterial communities on hair and scalp were rather independent in alpha diversity formation.
Figure 2. Alpha diversity of bacterial community on the hair and scalp, based on (a) observed OTU and (b) Shannon index. The values are obtained from clustering of 1,000 reads per sample
Bacterial community structures on hair and scalp at phylum level
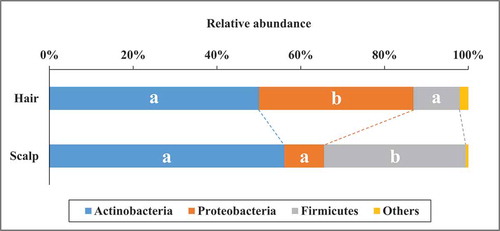
Three major phyla, Actinobacteria, Proteobacteria, and Firmicutes were commonly found on both sites, and abundances of the three phyla added up to 97.9% and 99.4% on hair and on scalp respectively (). The Kruskal-Wallis test revealed no statistically significant difference in the abundance of Actinobacteria between the sites. On the other hand, the abundance of Proteobacteria was significantly higher (p< 0.01) on hair (36.9 ± 20.1%) than on scalp (9.5 ± 9.4%), whereas the abundance of Firmicutes was significantly lower (p< 0.01) on hair (11.0 ± 9.3%) than on scalp (33.8 ± 12.1%). These results indicate that the bacterial communities on hair and on scalp had distinguishable structures at the phylum level. Correlation analysis at individual level showed that abundances of Firmicutes and Actinobacteria between the sites were correlated (R = 0.58 and 0.63 respectively), whereas the abundance of Proteobacteria between the sites was not correlated (R = 0.18) (Supplemental Figure 3) .
Figure 3. The average relative abundances of the major phyla in bacterial community structure on hair and scalp. The different alphabets mean that there are significant differences at comparison of each bacterial phyla between the sites (Kruskal-wallis test, p < 0.01)
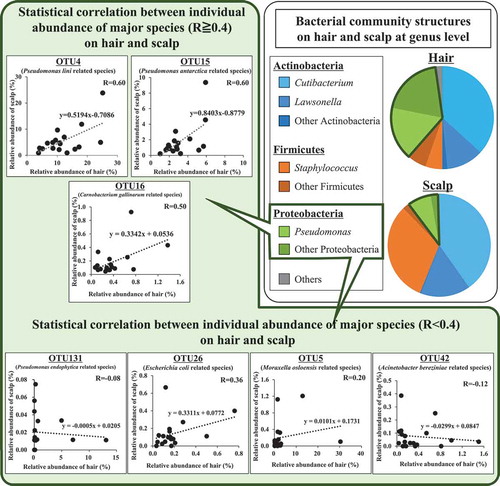
Bacterial community structures on hair and scalp at OTU level
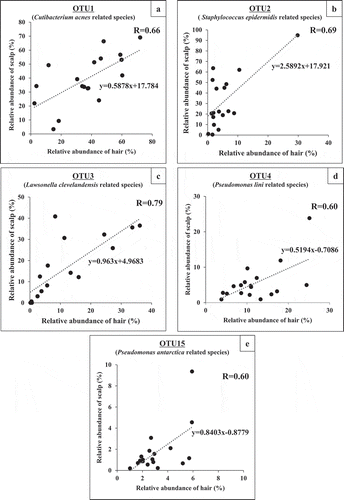
We extracted OTUs present in samples of all individuals, and found that 13 OTU sequences existed either on hair or scalp of each individual. Of these, eight OTUs (OTU 1, 2, 3, 4, 7, 9, 15, 16) were present on both sites in all individuals, and the other five OTUs (OTU 5, 18, 26, 42, 131) were present only on hair in some individuals. The total relative abundances of the eight common OTUs were 70.9% and 94.6% on hair and on scalp, respectively, being much lower on hair. This observation corresponded with the results of alpha diversity analysis ().
These 13 OTUs were assigned to a most closely related species (Phylum, pairwise similarity) (). The five major OTUs showing abundance higher than 5% were: OTU1 related to Cutibacterium acnes (Actinobacteria, 99.3%), OTU2 related to Staphylococcus epidermidis (Firmicutes, 99.3%), OTU3 related to Lawsonella clevelandensis (Actinobacteria, 99.3%), OTU4 related to Pseudomonas lini (Proteobacteria, 99.3%) and OTU15 related to Pseudomonas antarctica (Proteobacteria, 99.0%). Other three OTUs found in all individuals were OTU 7, 9, and 16. These show rather low total abundance at 3.2% and 1.0% in total on hair and on scalp, respectively. It is noticeable that the five OTUs (OTU 5, 18, 26, 42, 131), found only on hair, added up to 6.0% in total abundance, and were all assigned to phylum Proteobacteria.
Figure 4. Phylogenetic tree and abundance heatmap of the major 13 OTUs commonly found in all 18 individuals
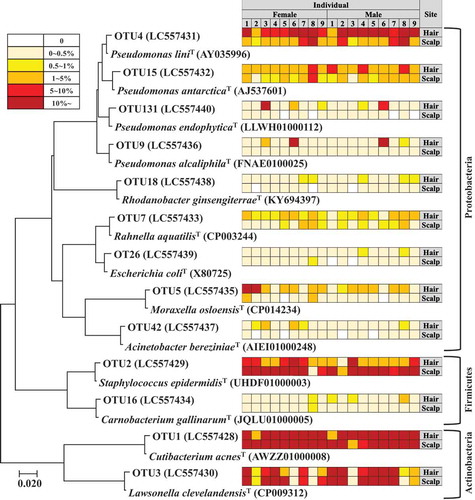
Relative abundances of the five major OTUs present at both sites, and their statistical correlations between hair and scalp are shown in and , respectively. OTU1 related to Cutibacterium acnes was the most predominant species on both sites. Its relative abundance was not significantly different between the sites (). On the other hand, its individual abundance was statistically correlated between the two sites (R = 0.66) (). OTU2 related to Staphylococcus epidermidis was less abundant on average, on hair than on scalp (), and its individual abundance was also statistically correlated between the two sites (R = 0.69) (). OTU3 related to Lawsonella clevelandensis showed no significant difference in its relative abundance between the sites (), while its individual abundance was statistically correlated between the two sites (R = 0.79) (). On the other hand, the relative abundances of OTU4 related to Pseudomonas lini and OTU15 related to Pseudomonas antarctica were higher on hair than on scalp (, ). These two, OTU4 and OTU15 showed correlation between individual abundance on hair and on scalp (R = 0.60, R = 0.60) (, ). The five OTUs (OTU 5, 18, 26, 42, 131), sometimes found only on hair showed relatively lower correlation between their abundance on the hair and on the scalp of the same individuals (Fig. S4). These results suggest that bacterial community structures on each site were partly formed with skin-resident bacteria including Cutibacterium, Staphylococcus, and Lawsonella, and partly formed with some hair-specific bacteria belonging to phylum Proteobacteria.
Figure 5. Comparison of relative abundance of most major five OTUs between hair and scalp. (a) relative abundance of OTU1 related to Cutibacterium acnes, (b) OTU2 related to Staphylococcus epidermidis, (c) OTU3 related to Lawsonella clevelandensis, (d) OTU4 related to Pseudomonas lini, and (e) OTU15 related to Pseudomonas antarctica.
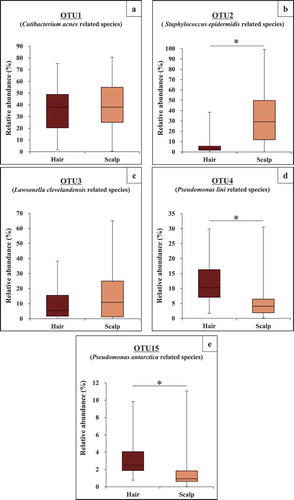
Figure 6. Correlation of relative abundance of most major five OTUs between hair and scalp. (a) OTU1 related to Cutibacterium acnes, (b) OTU2 related to Staphylococcus epidermidis, (c) OTU3 related to Lawsonella clevelandensis, (d) OTU4 related to Pseudomonas lini, and (e) OTU15 related to Pseudomonas antarctica.
Beta diversity of the bacterial community structures on hair and scalp
shows the beta diversity of bacterial community structures on hair and scalp samples obtained by principal coordinates analysis (PCoA) based on weighted UniFrac distances.
Figure 7. Beta diversity of bacterial community structure on hair and scalp in 108 samples from eighteen volunteers and their biplot analysis analysis at phylum level. (a) The PCoA plots based on the weighted UniFrac analysis are shown. (b) Comparison of weighted Unifrac distances within and between hair and scalp samples
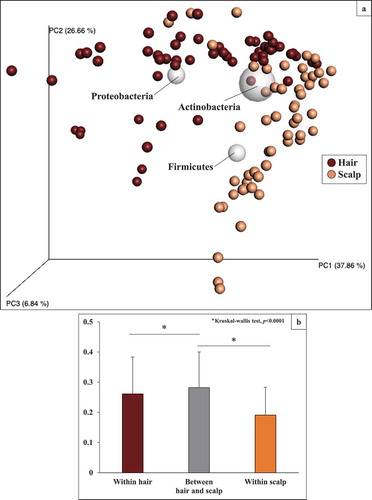
The plots were roughly grouped into two clusters on each site, although some plots were positioned at similar areas. We further performed biplot analysis at phylum level and the result corresponded well with the relative abundances of major phyla in bacterial community structures on each site shown in As seen in , biplot of Actinobacteria was located in the middle of the two clusters; biplot of Firmicutes was located in the scalp cluster. On the other hand, biplot of Proteobacteria was located in the hair cluster. This clustering tendency was confirmed by comparison of average-weighted UniFrac distance (): the distances within hair and within scalp were significantly lower (p< 0.0001) than those between hair and scalp. These results indicated that the bacterial community structure on hair was specified by a comparably higher abundance of Proteobacteria, although major OTUs were commonly present on both sites.
Discussion
Recent studies of bacterial community structures on scalp elucidated its relationships with health and disease of hair (scalp hair) and scalp [Citation9–13]. Also, relatively stable and individually unique bacterial communities were found on hair [Citation17,Citation19]. In this study, we provide the first overview of characteristics and relationship of the bacterial community structures on hair and scalp using real-time PCR and 16S rRNA amplicon sequencing. As a result, it was found that major bacterial species were commonly present at both sites, but the bacterial community structure on hair was specific and distinguishable from that on the scalp. Important relationships obtained are summarized in . Significant differences between the bacterial community structures on hair and scalp were found in terms of cell density, alpha diversity, and on relative abundances of Firmicutes and Proteobacteria, while no difference was found in terms of relative abundance of Actinobacteria. There was correlation between hair and scalp of an individual person in relative abundance of Actinobacteria and Firmicutes, while there was no correlation in terms of cell density, alpha diversity, and relative abundance of Proteobacteria. We will discuss each result one by one.
Average bacterial cell number on hair was significantly higher (p< 0.05) than on scalp (). Bacterial cell number on hair was similar to that seen in previous reports [Citation19,Citation29]. On the other hand, there were no correlation between hair and scalp on individual bacterial cell number. It should be notable that there was almost no variation in the three samples per person, showing that error due to sampling method was relatively low. In spite of that, taking into account that the sampling methods to collect bacterial DNA were different at each site (whole extraction for hair shaft and swabbing for scalp), further structural analysis was conducted by comparing relative abundances of a highlighted bacterial group, instead of the cell densities. Grice et al. [Citation30] reported that major OTUs were overlapping (97.2%) in bacterial skin samples collected using three different methods: swab, scrape, and punch biopsy.
We showed two indexes of alpha diversity on each site: observed OTUs and Shannon Index. Both indexes were significantly higher on hair than on scalp (). This could explain why the eight OTUs, commonly present on both sites of all individuals, show higher abundance on scalp (94.6%) than on hair (70.9%) (). These results suggest that various kinds of minor bacterial species inhabited only hair. It was reported that physical conditions affect the formation of skin bacterial communities, and thus, alpha diversity of bacterial community structures on sebaceous skin was lower than that on dry skin [Citation1,Citation12,Citation31]. In a previous study, we also reported that alpha diversity was higher on dry hair shaft than on sebaceous hair root [Citation19]. Higher hydrophobicity of hair would cause its higher alpha diversity, compared to a sebaceous scalp. On the other hand, there was no correlation between hair and scalp in terms of the individual alpha diversity of their bacterial community structures. This suggests that the bacterial community structures on the two sites are independent in alpha diversity formation.
At phylum level, average relative abundance of Actinobacteria were similar on hair and scalp (50.0 ± 22.5% and 56.1 ± 21.0%, respectively), but not similar for Firmicutes (11.0 ± 9.3% and 33.8 ± 12.1%), and Proteobacteria (36.9 ± 20.1% and 9.5 ± 9.4%) (). On the contrary, correlation between hair and scalp on individual relative abundances was found both in Actinobacteria and Firmicutes, but not in Proteobacteria. For more detailed analysis of bacterial community structure, we assigned the OTU sequences to the most closely related bacterial species. Four genera – Cutibacterium, Staphylococcus, Lawsonella, and Pseudomonas were present with more than 5% abundance in both sites for every sample (). These bacterial genera are also reported to be present in other human body sites. Cutibacterium is the major bacterial genus present in areas with sebaceous glands, such as the forehead [Citation32]. Staphylococcus widely inhabits various parts of human skin and the nasal area [Citation33]. In case of Lawsonella, the genus has been isolated from various kinds of human abscesses [Citation34]. Some of the common OTUs, Cutibacterium acnes (previous name is Propionibacterium acnes) and Staphylococcus epidermidis, were also found in other studies, as you indicated. These bacteria protect the host from pathogenic bacteria [Citation32]. They are considered to proliferate using sebum and sweat as nutrient, and playing role of barrier function on hair and scalp against pathogenic bacteria [Citation32]. On the other hand, the genus Proteobacteria generally inhabits the natural environment such as soil and river water, and has been recognized as being transient on human skin [Citation35]. Correlations between hair and scalp were found for individual relative abundances of five major OTUs () but not in some secondary OTUs related to Proteobacteria including Pseudomonas (Fig. S4). Our results indicated that Pseudomonas is the second major genus in hair, showing stable and non-transient habitation on hair. In a previous study, we reported that major bacterial OTUs including Pseudomonas were common on hair shaft and hair root, and it was possible that these bacteria on hair shaft were indigenous and not transient due to circumstances [Citation19]. Cutibacterium was the most abundant bacterial genus on hair, even though the physical condition of hair seems dry and different from other sebaceous human skin zones. Cutibacterium acnes carries the genes for biosynthesis of biotin, which is an essential nutrient for hair growth and scalp health [Citation11,Citation36,Citation37]. On the other hand, Pseudomonas, the second major bacterial genus present on hair, was not identified in a key role. Further detailed studies are required to clarify the bacterial ecology of hair.
Finally, we analyzed beta diversity and biplots at phylum level, to compare bacterial community structures between hair and scalp (). It was confirmed that phylum Proteobacteria (to which Pseudomonas lini and Pseudomonas antarctica belong) contributed to the formation of bacterial community structure specific for hair, and phylum Firmicutes (to which Staphylococcus epidermidis belongs) contributed to that on scalp. Although some reports analyzed bacterial community structures on scalp [Citation9–13], or on hair [Citation1,Citation19,Citation38], there was no study on the correlation between these sites at the same individual level. Also, Klerk et al. [Citation39] evaluated bacterial adherence and colonization on hair by SEM observation and reported that Pseudomonas aeruginosa (belonging to Proteobacteria) adhered to and colonized hair surfaces, while Staphylococcus epidermidis (belonging to Firmicutes) showed inhibited growth. This inhibition of S. epidermidis may be attributable to hair-derived antimicrobial proteins or peptides [Citation40]. Therefore, it was suggested that hair contained specific bacterial community structures, in that the relative abundances of Pseudomonas (Proteobacteria) was higher on hair than on other skin sites.
In conclusion, it was suggested that bacterial community structure on hair was formed from both hair-specific and skin-derived bacteria, which were different from other skin microbiomes, including forearms, nostrils, and forehead [Citation20]. In particular, Pseudomonas was one of the most hair-specific bacteria, but the genus has been recognized as being transient on skin and not much attention has been paid to it. Further studies on hair-specific bacteria, particularly genus Pseudomonas would clarify their roles and interaction with hair.
Author contribution
Kota Watanabe processed the experimental data, performed the analysis and wrote the manuscript. Azusa Yamada and Yuri Nishi carried out the part of experiments. Yukihiro Tashiro and Kenji Sakai supervised the project from experimental design to submission of the manuscript. All authors agreed and approved the manuscript to be published.
Supplemental_table_K.Watanabe.docx.docx
Download MS Word (20.1 KB)Supplemental_Figure_K.watanabe.pptx
Download MS Power Point (115.4 KB)Acknowledgments
The authors would like to acknowledge Center for Advanced Instrumental and Educational Supports, Faculty of Agriculture, Kyushu University for their support to use MiSeq. This work was supported by JSPS under the Grant-in-Aid for JSPS Fellows (Grant No. 20J12699).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Costello EK, Lauber CL, Hamady M, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326(5960):1694–1697.
- Findley K, Oh J, Yang J, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498(7454):367–370.
- Wylie KM, Mihindukulasuriya KA, Zhou Y, et al. Metagenomic analysis of double-stranded DNA viruses in healthy adults. BMC Biol. 2014;12(1):71.
- Fredricks DN. Microbial ecology of human skin in health and disease. J Invest Dermatol Symp Proc Proc. 2001;6(3):167–169.
- Montes LF. Anatomical location of normal skin flora. Arch Derm. 1970;101(2):145–159.
- Lange-Asschenfeldt B, Marenbach D, Lang C, et al. Distribution of bacteria in the epidermal layers and hair follicles of the human skin. Skin Pharmacol Physiol. 2011;24(6):305–311.
- Oh J, Byrd AL, Deming C, et al. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514(7520):59–64.
- Kong HH, Oh J, Deming C, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22(5):850–859.
- Clavaud C, Jourdain R, Bar-hen A, et al. Dandruff is associated with disequilibrium in the proportion of the major bacterial and fungal populations olonizing the scalp. PLoS One. 2013;8(3):e58203.
- Wang L, Clavaud C, Bar-Hen A, et al. Characterization of the major bacterial-fungal populations colonizing dandruff scalps in Shanghai, China, shows microbial disequilibrium. Exp Dermatol. 2015;24(5):381–400.
- Saxena R, Mittal P, Clavaud C, et al. Comparison of healthy and dandruff scalp microbiome reveals the role of commensals in scalp health. Front Cell Infect Microbiol. 2018;8:346.
- Xu Z, Wang Z, Yuan C, et al. Dandruff is associated with the conjoined interactions between host and microorganisms. Sci Rep. 2016;6(1):24877.
- Park T, Kim H-J, Myeong NR, et al. Collapse of human scalp microbiome network in dandruff and seborrhoeic dermatitis. Exp Dermatol. 2017;26(9):835–838.
- Mase K, Hasegawa T, Horii T, et al. Firm adherence of Staphylococcus aureus and Staphylococcus epidermidis to human hair and effect of detergent treatment. Microbiol Immunol. 2000;44(8):653–656.
- Yun A, Yang EJ, Lee YM, et al. Quantitative and qualitative estimation of bacteria contaminating human hairs. J Bacteriol Virol. 2010;40(1):11–18.
- Matard B, Meylheuc T, Briandet R, et al. First evidence of bacterial biofilms in the anaerobe part of scalp hair follicles: a pilot comparative study in folliculitis decalvans. J Eur Acad Derm Veneroeol. 2013;27(7):853–860.
- Nishi E, Watanabe K, Tashiro Y, et al. Terminal restriction fragment length polymorphism profiling of bacterial flora derived from single human hair shafts can discriminate individuals. Legal Med. 2017;25:75–82.
- Nishi E, Tashiro Y, Sakai K. Discrimination among individuals using terminal restriction fragment length polymorphism profiling of bacteria derived from forensic evidence. Int J Legal Med. 2015;129(3):425–433.
- Watanabe K, Nishi E, Tashiro Y, et al. Mode and structure of the bacterial community on human scalp hair. Microbes Environ. 2019;34(3):252–259.
- Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9(4):244–253.
- Fierer N, Jackson J, Vilgalys R. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl Environ Microbiol. 2005;71(7):4117–4120.
- Turner S, Pryer KM, Miao VP, et al. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryotic Microbiol. 1999;46(4):327–338.
- Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Nat Acad Sci U S A. 2011;108:4516–4522.
- Brown CT, Hug LA, Thomas BC, et al. Unusual biology across a group comprising more than 15% of domain bacteria. Nature. 2015;523(7559):208–211.
- Kuczynski J, Lauber CL, Walters WA, et al. Experimental and analytical tools for studying the human microbiome. Nat Rev Genet. 2012;13(1):47–58.
- Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):33–336.
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461.
- Chun J, Lee J-H, Jung Y, et al. EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol. 2007;57(10):2259–2261.
- Brinkac L, Clarke TH, Singh H, et al. Spatial and environmental variation of the human hair microbiota. Sci Rep. 2018;8(1):9017.
- Grice EA, Kong HH, Renaud G, et al. A diversity profile of the human skin microbiota. Genome Res. 2008;18(7):1043–1050.
- Gao Z, Tseng C-H, Pei Z, et al. Molecular analysis of human forearm superficial skin bacterial biota. Proc Nat Acad Sci. 2007;104(8):2927–2932.
- Chiller K, Selkin BA, Murakawa GJ. Skin microflora and bacterial infections of the skin. J Investig Dermatol Symp Proc. 2001;6(3):170–174.
- Eiff CV, Becker K, Machka K, et al. Nasal carriage as a source of Staphylococcus aureus bacteremia. N Engl J Med. 2001;344(1):11–16.
- Bell ME, Bernard KA, Harrington SM, et al. Lawsonella clevelandensis gen. nov., sp. nov., a new member of the suborder Corynebacterineae isolated from human abscesses. Int J Syst Evol Microbiol. 2016;66(8):2929–2935.
- Cogen AL, Nizet V, Gallo RL. Skin microbiota: A source of disease or defence? Br J Dermatol. 2008;158(3):442–455.
- Patel DP, Swink SM, Castelo-Soccio L. A review of the use of biotin for hair loss. Skin Append Dis. 2017;3(3):166–169.
- Chiu C-H, Huang S-H, Wang H-M. A review: hair health, concerns of shampoo ingredients and scalp nourishing treatments. Curr Pharm Biotechnol. 2015;16(12):1045–1052.
- Tridico SR, Murray DC, Addison J, et al. Metagenomic analyses of bacteria on human hairs: a qualitative assessment for applications in forensic science. Invest Gen. 2014;5(1):16.
- Kerk SK, Lai HY, Sze SK, et al. Bacteria display differential growth and adhesion characteristics on human hair shafts. Front Microbiol. 2018;9(9):2145.
- Adav SS, Subbaiaih RS, Kerk SK, et al. Studies on the proteome of human hair - Identification of histones and deamidated keratins. Sci Rep. 2018;8(1):1599.