
Abstract
Platelet aggregation is a complicated process mediated by different signaling pathways. As the process is highly complex and apparently redundant, the relationships between these pathways are not yet fully known. The aim of this project was to study the interconnections among seven different aggregation pathways in a group of 53 generally healthy volunteers aged 20 to 66 years. Platelet aggregation was induced with thrombin receptor activating peptide 6 (TRAP), arachidonic acid (AA), platelet activating factor 16 (PAF), ADP, collagen, thromboxane A2 analogue U46619 or ristocetin (platelet agglutination) ex vivo in fasting blood samples according to standardized timetable protocol. Additionally, some samples were pre-treated with known clinically used antiplatelet drugs (vorapaxar, ticagrelor or acetylsalicylic acid (ASA)). Significant correlations among all used inducers were detected (Pearson correlation coefficients (rP): 0.3 to 0.85). Of all the triggers, AA showed to be the best predictor of the response to other inducers with rP ranging from 0.66 to 0.85. Interestingly, the antiplatelet response to ticagrelor strongly predicted the response to unrelated drug vorapaxar (rP = 0.71). Our results indicate that a response to one inducer can predict the response for other triggers or even to an antiplatelet drug. These data are useful for future testing but should be also confirmed in patients.
Plain Language Summary
What is the context?
• Platelet activation is a complicated process with multiple signaling cascades involved.
• A total of seven common platelet triggers (ADP, collagen, TRAP-6, PAF, arachidonic acid/AA/, ristocetin and U46619) were tested.
• The process is dependent on many factors including sex, age, concomitant disease(s), pharmacotherapy.
What is new?
• There were significant correlations between all tested aggregatory cascades.
• AA has the highest rate of response predictability in our heterogeneous generally healthy volunteer group.
• There was no correlation between impedance aggregometry in whole blood and turbidimetric measurement with platelet-rich plasma.
What is the impact?
• The effect of antiplatelet drugs can be assessed from the reaction to different trigger(s) at least in this group of healthy patients.
• Future studies must test these relationships in patients with different diseases.
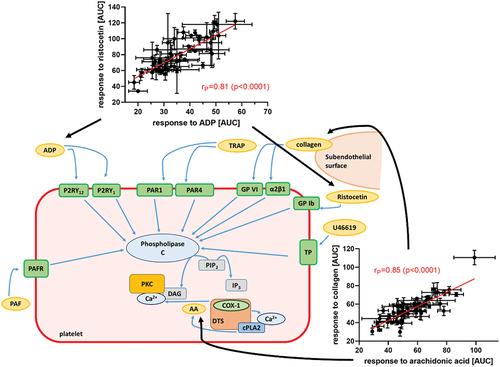
Graphical Abstract
Introduction
Blood hemostasis is a tightly regulated process in which complicated physiological pathways coordinate to avoid excessive blood loss upon endothelium rupture. The main players in this process are blood platelets, that are able to change their conformation after activation, secrete intracellular granules containing secondary mediators and associate with other activated platelets, forming a plug, which prevents bleeding.Citation1 Under normal circumstances, platelets are only activated upon endothelial injury, which leads to exposure of collagen from subendothelial layer. Collagen then interacts with platelets and von Willebrand factor (vWF) and this causes platelet adhesion and activation. The subsequent cascade is highly intricate and involves many receptors and signaling molecules.Citation2 All these cascades lead to the same final consequences: an increase in intracellular calcium, activation of glycoprotein IIb/IIIa receptor and clustering of platelets. Intracellular granule releases encompassing ADP, and thromboxane production seems to be a particularly crucial process, as they represent the targets of commonly used antiplatelet drugs.Citation3 These drugs, comprehending cyclooxygenase inhibitors (ASA), ADP antagonists and some novel drugs, such as thrombin antagonist vorapaxar, are used for prevention of possible serious thrombosis mainly in secondary prevention of cardiovascular diseases of ischemic origin.Citation4,Citation5
As different pathological conditions can induce or facilitate this activation even if the endothelial integrity has not been explicitly disrupted (e.g. atherosclerosis, diabetes mellitus, etc.), it is important to study different platelet activation pathways as these might be activated in cardiovascular diseases.Citation2 For triggering of certain aggregatory pathway, endogenous compounds can be used (e.g. ADP, arachidonic acid /AA/, platelet activating factor /PAF/ or collagen). However, some of these signaling molecules are unstable, too large or present multiple effects. Therefore, synthetic compounds mimicking platelet aggregation cascades are used (e.g. thromboxane analogue U-46619, vWF receptor binding activator ristocetin or thrombin sequence without enzymatic effect TRAP /thrombin receptor activating peptide/) in experimental conditions.
Most studies found in literature typically include one or two platelet triggers at most. However, platelet aggregation is a complicated process and the use of only some triggers may bias the results. We suppose that the use of more triggers can map the background of platelet aggregation process more accurately. Therefore, in this work we aimed to study the effect of seven relevant platelet activators (ADP, AA, collagen, PAF, TRAP, U-46619, and ristocetin) in blood samples of healthy patients (). In addition, blood samples of healthy individuals were treated with these activators alone, or in combination with clinically used antiplatelet drugs (ASA, ticagrelor, and vorapaxar), to study the interrelationships between them more extensively.
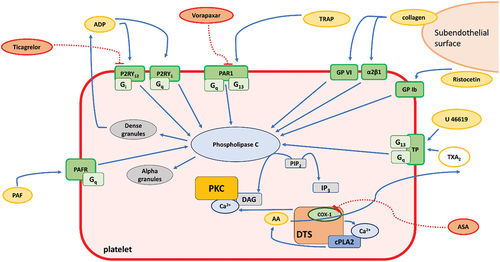
Figure 1. Platelet activation pathways of the different platelet triggers and antiplatelet compounds used. Tested inducers are depicted as yellow ellipses while tested antiplatelet drugs as red ellipses. Abbreviations: AA: arachidonic acid; ADP: adenosine diphosphate; ASA: acetylsalicylic acid; COX-1: cyclooxygenase-1; cPLA2: cytosolic phospholipase A2; DTS: dense tubular system; Gi: G protein α subunit i; GP Ib: glycoprotein Ib; GP VI: glycoprotein VI; Gq: G protein α subunit q; IP3: inositol triphosphate, P2RY1, P2RY12: two receptors for ADP, PAF: platelet activating factor; PAFR: PAF receptor; PAR1: protease activated receptor 1; PIP2: phosphatidylinositol 4,5-biphosphate; PKC: protein kinase C; TP: thromboxane receptor, TRAP: thrombin receptor-activating peptide; TXA2: thromboxane A2; α2β1: integrin α2β1.
Methods
Chemicals
Dimethyl sulfoxide (DMSO), ristocetin, PAF-16, ASA, ticagrelor and U-46619 were purchased from Sigma (St. Louis, MO, USA). AA, ADP, TRAP-6 were bought from Roche (Basel, Switzerland). Saline was purchased from B.Braun (Melsungen, Germany) while collagen was from Diagnostica, a.s. (Prague, Czech Republic).
Healthy donor enrollment
Details of the study were previously published.Citation6 Shortly, blood samples from a total of 53 generally healthy human donors were collected by venipuncture into plastic disposable tubes containing heparin sodium (170 IU/10 mL). Additional 13 patients were enrolled to perform complementary experiments on optical aggregometer and to compare the reactivity by impedance aggregometer using citrated and heparinized blood. The sampling was always performed in the morning (7:30–8:30 a.m.). The inclusion criteria were age ≥20 years and subjective good health. Exclusion criteria were the presence of heart disease, diabetes mellitus of either type, peripheral artery disease, a history of stroke, and the use of drugs which directly affect platelet aggregation (e.g. antiplatelet drugs, non-steroidal anti-inflammatory drugs). Smoking, administration of oral contraception, and controlled hypertension were acceptable for enrollment in this study, to enrich the variability of the group. Alcohol was not allowed 24 hours prior to blood collection. All donors signed an informed consent in line with the approval of ethics committee of the University Hospital in Hradec Králové (No. 202007S01P from 18 June 2020). All experiments conformed to the latest Declaration of Helsinki.
Aggregation experiments
Blood was carefully transferred under standard conditions to our laboratory for analysis. Aggregatory experiments were initiated precisely 30 minutes from blood draw. Whole heparinized or citrated blood was used for all measurements with impedance aggregometer Multiplate (Roche Diagnostic, Basel, Switzerland). The method has already been described elsewhere.Citation7 Shortly, 300 μL of whole blood was diluted with the same volume of preheated 0.9% sodium chloride, and platelet aggregation was induced with one of the following triggers: AA, collagen, PAF-16, ADP, U46619, TRAP-6, or ristocetin at desired concentration(s) and monitored for 6 min. For antiaggregatory experiments, a 3-minutes preincubation with the specified antiplatelet drugs at desired concentrations was performed, before triggering aggregation. A solvent (control) sample with DMSO at a final concentration of 0.8% was also included for each measurement.
Platelet-rich plasma (PRP) was prepared from citrated whole blood after centrifuging at 180 g for 10 min at room temperature (Eppendorf 5804 R, Hamburg, Germany). Platelet-poor plasma was prepared from the remaining blood at 2800 g (10 min, room temperature) and served as blank control for aggregation. These experiments were performed in optical aggregometer APACT 4S (Ahrensburg, Germany). Briefly, 200 μL of platelets set to a final concentration of 3 × 108 platelets/mL was incubated for a minute before triggering aggregation with the same compounds used in impedance aggregation experiments. The reaction was followed for a total of 6 minutes, after which area under the curve (AUC), maximal aggregation, and velocity of the aggregation were recorded.
Statistical analysis
GraphPad Prism 9.3.1 (GraphPad Software, San Diego, CA, USA) was used for performing all data analyses. Correlations were tested by the Pearson´s correlation test. In case of significant correlations, linear regression was performed. Differences in aggregation were compared by paired t-test or the Wilcoxon tests depending on data distribution pattern.
Results and discussion
Aggregatory responses obtained in whole heparinized blood after treatment with different inducers documented significant relationships among all triggers used (). Response to some of the used inducer significantly corresponded with a very high Pearson correlation coefficients (rP) to reaction on another platelet aggregation inducer. This suggests biologically relevant relationships between all investigated aggregatory pathways. A relatively high number of individual samples (53 donors) accentuate the biological relevance of the observed finding. The strongest relationships were found for the higher concentration of AA (200 μM) with all triggers with the exception of the lower concentration of collagen 0.16 μg/mL (, ). Interestingly, the lower concentration of AA used (60 μM) also provided significant correlations with other platelet aggregation triggers, but weaker than those at 200 μM. Similar outcomes were observed with collagen, although these correlations were always weaker than those of AA. The strongest association obtained was for the higher concentrations of both AA and collagen (rP = 0.85). These outcomes are understandable as collagen cascade is followed by a release of arachidonic acid with subsequent formation of thromboxane A2.Citation8 Collagen is one of the most studied platelet aggregation triggersCitation9 as it initiates the process of platelet aggregation. It exerts its activity through interaction with different surface receptors GP VI and GP Ia/IIa (α2β1).Citation10 Apparently, low concentration of collagen is only mildly proaggregatory and based on results from this study, higher concentrations collagen should be used in the experimentation.
Figure 2. The strongest associations between platelet aggregatory responses to inducers. (a): arachidonic acid (200 μM) and collagen (1 μg/mL), (d): arachidonic acid (200 μM) and U-46619 (80 nM), (c): arachidonic acid (200 μM) and TRAP (10 μM), (b): ADP (5 μM) and ristocetin (400 μM), (e): collagen (1 μg/mL) and acetylsalicylic acid (70 μM) + collagen (1 μg/mL), (f): ticagrelor 500 nM + ADP (5 μM) and vorapaxar (5 μM) and TRAP (10 μM). AUC: area under the curve measured by impedance aggregometry.
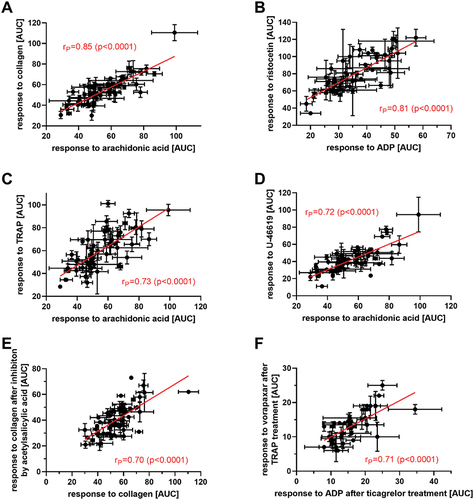
Table I. Relationships between responses to different aggregation inducers.
ADP, present in dense granules, is released after platelet stimulation with different inducers and further promotes platelet aggregation through interaction with platelet surface P2Y1 and P2Y12 receptors.Citation11 As vWF and collagen are involved in the first stages of the platelet aggregation process, significant correlations with ADP were expected.
Our results also showed a high correlation between ADP and AA. These pathways are, at first sight, unrelated. The observed correlation is likely consequence of AA-based thromboxane formation followed by stimulation of thromboxane receptors coupled with Gq, which stimulates phospholipase C. This leads to an increase in intracellular calcium concentration, which is a known trigger of platelet granule release encompassing ADP release. Similarly, thromboxane, thrombin and PAF activate receptors coupled with Gq. Strong correlations among platelet responses to these compounds with ADP were observed too. These results indicate that intracellular calcium increase with a subsequent release of ADP is at least partly mediating the response to thromboxane, thrombin, and PAF. The situation is more complicated with thromboxane and thrombin as their receptors couple both with Gq and G13. The fact that AA is needed for synthesis of thromboxane can explain the strong correlation between AA and U-44619 (rP = 0.75), a stable thromboxane A2 analogue. The strongest correlation of ADP was that with ristocetin (rP = 0.81; ). Ristocetin, originally an antibiotic,Citation12 is employed as a useful pharmacological tool mediating activation of vWF leading to platelet agglutination. Ristocetin provided a higher correlation with ADP than with collagen. This might point out that vWF activation is linked with a more intense release of ADP than collagen itself.
PAF is a lipid-like molecule with complex effects including an ability as a potent platelet trigger.Citation13 Its synthesis is a complicated process via two main pathways (de novo and remodeling) not only in platelets but also in leukocytes, endothelial and mast cells.Citation14 PAF is able to activate platelets even at picomolar concentrations and has an important role in inflammation, recruiting phagocytes, leukocytes and macrophages. This molecule interacts with its specific receptor coupled with Gq on platelet surface. According to our results, it correlated significantly with all triggers while the highest correlation was obtained with ristocetin. This is a novel finding since no common signaling pathways are known for these triggers. Whereas ristocetin causes platelet agglutination through interaction with GP Ib, PAF has a different signalization pathway mediated by mentioned Gq protein. Correlations with thromboxane, TRAP and ADP pathways might be based on the fact, that all of them activate Gq-coupled receptors and hence have the same consequences, although there are some differences as described above.
TRAP is another synthetic molecule, acting directly on thrombin receptors PAR1 and PAR4.Citation15 In our study, we chose this molecule instead of thrombin, as it does not activate blood coagulation, since it is free of the enzymatic activity but retains the ability to activate thrombin receptors on platelets. The strongest correlations were observed, quite surprisingly, with AA and collagen. The connection is not clear as there is not a direct connection among these signaling pathways. Regardless, it is possible that the found correlations are consequence of a higher platelet reactivity, which can be reflected in all aggregation pathways.
Since differences in platelet response to a given trigger can vary among the different aggregatory methods available, complementary experiments were performed. No correlation between platelet aggregation measured in whole citrated blood and PRP prepared from citrated blood by impedance and turbidimetric aggregometry, respectively, was found (Table S1). Only a nearly significant correlation was observed for AA (p = .055). There are two likely explanations: a) other blood cells can impact the process of platelet aggregation and/or b) PRP preparation affects platelet reactivity, e.g. by partly activating them. The latter can be supported by the fact that optical aggregation using PRP resulted in numerically lower platelet aggregation compared to whole blood impedance aggregation with the exception of ADP (Figure S1-S2). Another aspect investigated was the impact of used anticoagulants to drawn blood samples (heparin and sodium citrate). In impedance experiments, a significant correlation was observed solely for AUC in relation to collagen. Additionally, an almost significant correlation was observed for U-46619 (p = .051). This means, that solely collagen and partly U-46619 triggered platelet aggregation are giving results which are predictable by using citrate or heparinized blood. Moreover, when considering maximal aggregation values, no correlation was observed at all. Such results were unexpected. Heparin blocks coagulation via interaction with antithrombin III whereas citrate chelates free calcium. A previous study reported that both citrated and heparinized blood are not completely stable in time in relation to platelet count, therefore, this was likely not the reason for the observed differences in our results.Citation16 For ADP, PAF and U-46619, no significant differences between platelet reactions in citrated and heparinized blood were observed. For the remaining triggers, the measured reaction, either as AUC and/or maximal aggregation, was stronger with citrated blood samples. It is known that heparin also interacts with von Willebrand factor, although the clinical relevance of this aspect is not clear.Citation17 Regardless, AUC but not maximal aggregation after ristocetin was significantly lower in case of heparinized blood (Figure S1-S2). Whether this fact can explain the lower reaction to other inducers is not clear.
In the last step, the inhibitory capacity of antiplatelet drugs in heparinized blood was correlated with platelet aggregatory responses to the different triggers used. Observed correlations () can facilitate future research by assessing the response to particular drugs as it is not possible to test all aggregatory pathways in every patient. Inhibitor-trigger pairing with inducers were chosen according to their activation pathways and only clinically relevant drugs ASA (aspirin), ticagrelor and vorapaxar, were selected. The mechanism of action of ASA is well known, through irreversible blockade of platelet cyclooxygenase 1. The other two drugs target different cellular structures. Vorapaxar is a thrombin receptor antagonist (PAR1) whereas ticagrelor targets P2Y12 receptor, preventing agonist binding.Citation18 Our data provided significant correlations in the response to antiplatelet drug pre-treatment to most other inhibitors and inducers emphasizing a high degree of predictability even in unrelated cascades. An example of an expectable effect is the intensity of the platelet response to both low and high concentrations of collagen-induced aggregation, which was highly predictive to the effect of ASA 70 μM. Interestingly, the response to ticagrelor in ADP-triggered aggregation was also highly predictable to the response obtained after treatment with vorapaxar (5 µM) and TRAP (, ). This correlation was unexpected, as they are antagonists at two different receptors. Although one type of ADP receptor (P2Y1) is Gq, like the TRAP receptor, this is not likely the explanation since ticagrelor selectively antagonizes the P2Y12 (Gi) receptor for ADP. P2Y12 antagonists have been reported to prevent thrmoboxane A2 synthesis and this could explain the effect on other cascades.Citation19
Table II. Relationships between responses to different platelet aggregation inhibitors and platelet aggregation inducers.
To our best knowledge, no previous scientific work so far has studied and compared the different activity of such a wide number of triggers. The main outcome is the fact that the magnitude of response to one platelet aggregation trigger can predict the response of another platelet signaling pathway as well as that of antiplatelet treatment. Our study has both strengths and limitations. The most important ones are: 1) data from 53 different volunteers were included, 2) the volunteers were of a wide age range, 3) on the other hand, a limitation of our study is the inclusion of healthy donors, 4) extrapolation of our data needs to be confirmed in cardiovascular patients
Availability of data and materials
Raw data from patients cannot be shared but are available upon reasonable request to the corresponding author.
Ethics approval
This study was performed in accordance to the Ethics Committee approval from the Faculty of Pharmacy, Charles University and the University Hospital Hradec Králové.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Sang Y, Roest M, de Laat B, de Groot PG, Huskens D. Interplay between platelets and coagulation. Blood Rev. 2021 Mar;46:100733. doi:10.1016/j.blre.2020.100733.
- Jurk K, Kehrel BE. Platelets: physiology and biochemistry. Semin Thromb Hemost. 2005 Aug;31(4):381–6. (in English). doi:10.1055/s-2005-916671.
- Vilahur G, Gutierrez M, Arzanauskaite M, Mendieta G, Ben-Aicha S, Badimon L. Intracellular platelet signalling as a target for drug development. Vascul Pharmacol. 2018 Dec;111:22–5. doi:10.1016/j.vph.2018.08.007.
- Leonardi S, Tricoci P, Becker RC. Thrombin receptor antagonists for the treatment of atherothrombosis: therapeutic potential of vorapaxar and E-5555. Drugs. 2010 Oct 1;70(14):1771–83. doi:10.2165/11538060-000000000-00000.
- Wijeyeratne YD, Heptinstall S. Anti-platelet therapy: ADP receptor antagonists. Br J Clin Pharmacol. 2011 Oct;72(4):647–57. doi:10.1111/j.1365-2125.2011.03999.x.
- Carazo A, Hrubša M, Konečný L, Skořepa P, Paclíková M, Musil F, Karlíčková J, Javorská L, Matoušová K, Krčmová LK, et al. Sex-related differences in platelet aggregation: a literature review supplemented with local data from a group of generally healthy individuals (in English). Semin Thromb Hemost. 2022 Oct 7;49(5):488–506. doi:10.1055/s-0042-1756703.
- Applova L, Karlíčková J, Warncke P, Macáková K, Hrubša M, Macháček M, Tvrdý V, Fischer D, Mladěnka P. 4-methylcatechol, a flavonoid metabolite with potent antiplatelet effects. Mol Nutr Food Res. 2019 Aug 7;63(20):e1900261. doi:10.1002/mnfr.201900261.
- Badimon L, Vilahur G, Rocca B, Patrono C. The key contribution of platelet and vascular arachidonic acid metabolism to the pathophysiology of atherothrombosis. Cardiovasc Res. 2021 Jul 27;117(9):2001–15. doi:10.1093/cvr/cvab003.
- Wilner GD, Nossel HL, Leroy EC. Aggregation of platelets by collagen (in English). J Clin Invest. 1968;47(12):2616–. doi:10.1172/Jci105944.
- Nakashima D, Onuma T, Tanabe K, Kito Y, Uematsu K, Mizutani D, Enomoto Y, Tsujimoto M, Doi T, Matsushima-Nishiwaki R, et al. Synergistic effect of collagen and CXCL12 in the low doses on human platelet activation. PLoS One. 2020;15(10):e0241139. doi:10.1371/journal.pone.0241139.
- Rivera J, Lozano ML, Navarro-Nunez L, Vicente V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica. 2009 May;94(5):700–11. doi:10.3324/haematol.2008.003178.
- Coller BS, Gralnick HR. Studies on the mechanism of ristocetin-induced platelet agglutination. effects of structural modification of ristocetin and vancomycin. J Clin Invest. 1977 Aug;60(2):302–12. doi:10.1172/JCI108778.
- Rao GHR, White JG. Platelet activating factor (paf) causes human-platelet aggregation through the mechanism of membrane modulation (in English). Prosta Leukotr Med. 1982;9(4):459–72. doi:10.1016/0262-1746(82)90102-0.
- Lordan R, Tsoupras A, Zabetakis I. Phospholipids of animal and marine origin: structure, function, and anti-inflammatory properties. Molecules. 2017 Nov 14;22(11):1964. doi:10.3390/molecules22111964.
- Olivier CB, Weik P, Meyer M, Weber S, Anto-Michel N, Diehl P, Zhou Q, Geisen U, Bode C, Moser M, et al. TRAP-induced platelet aggregation is enhanced in cardiovascular patients receiving dabigatran. Thromb Res. 2016 Feb;138:63–8. (in English). doi10.1016/j.thromres.2015.10.038.
- Fox SC, Burgess-Wilson M, Heptinstall S, Mitchell JR. Platelet aggregation in whole blood determined using the ultra-flo 100 platelet counter. Thromb Haemost. 1982 Dec;48(3):327–9. doi:10.1055/s-0038-1657292.
- Ranger A, Gaspar M, Elkhatteb A, Jackson T, Fox S, Aw TC, Vipond L, Cotterill J, Ghori A, Laffan M, et al. The heparin-von Willebrand factor interaction and conventional tests of haemostasis – the challenges in predicting bleeding in cardiopulmonary bypass. Br J Haematol. 2021 Mar;192(6):1073–81. doi:10.1111/bjh.17263.
- Gelbenegger G, Jilma B. Clinical pharmacology of antiplatelet drugs. Expert Rev Clin Pharmacol. 2022 Oct;15(10):1177–97. doi:10.1080/17512433.2022.2121702.
- Armstrong PC, Leadbeater PD, Chan MV, Kirkby NS, Jakubowski JA, Mitchell JA, Warner TD. In the presence of strong P2Y12 receptor blockade, aspirin provides little additional inhibition of platelet aggregation. J Thromb Haemost. 2011;9(3):552–61. doi:10.1111/j.1538-7836.2010.04160.x.