
Abstract
The vascular endothelial growth factor receptor 2 (VEGFR2) is considered to be a pivotal target for anti-tumor therapy against angiogenesis of non-small cell lung cancer (NSCLC). However, effective and low-toxicity targeted therapies to inhibit VEGFR2 are still lacking. Here, biRGD–siVEGFR2 conjugate comprising murine VEGFR2 siRNA and [cyclo(Arg-Gly-Asp-D-Phe-Lys)-Ahx]2-Glu-PEG-MAL (biRGD) peptide which selectively binds to integrin αvβ3 receptors expressing on neovascularization endothelial cell was synthesized. The anti-tumor activity and renal toxicity of biRGD–siVEGFR2 or its combination therapy with low-dose apatinib were investigated on NSCLC xenografts. The immunogenicity of biRGD–siVEGFR2 was also evaluated in C57BL/6J mice. In vivo, intravenously injected biRGD–siVEGFR2 substantially inhibited NSCLC growth with a marked reduction of vessels and a down-regulation of VEGFR2 in tumor tissue. Furthermore, biRGD–siVEGFR2 in combination with low-dose apatinib achieved powerful anti-tumor effect with less nephrotoxicity compared with the regular dose of apatinib. Besides, no obvious immunogenicity of biRGD–siVEGFR2 was found. These findings demonstrate that biRGD–siVEGFR2 conjugate can be used as a new candidate for the treatment of NSCLC and its combination therapy with apatinib may also provide a novel strategy for cancer treatment in clinic.
1. Introduction
Non-small cell lung cancer (NSCLC) is the most common human malignancy in the world with increasing morbidity and mortality every year (Bray et al., Citation2018). Angiogenesis is an indispensable factor for NSCLC progression (Manzo et al., Citation2017). Currently, antiangiogenic therapy has been added to the standard treatment for patients with NSCLC (Hall et al., Citation2015). The vascular endothelial growth factor receptor 2 (VEGFR2, also known as KDR/Flk-1) plays a critical role in angiogenic process, during which VEGF binds to VEGFR2, then activates downstream angiogenic signaling pathways and results in proliferation and migration of endothelial vessels, consequently promoting angiogenesis and tumor proliferation (Carmeliet & Jain, Citation2011; Piperdi et al., Citation2014). Therefore, inhibition of VEGFR2 in neovascularization is an effective method for antiangiogenic therapy in NSCLC.
Apatinib, also known as YN968D1, is a novel oral tyrosine kinase inhibitor (TKI) that selectively inhibits VEGFR2, thus exerting an antiangiogenic effect and inhibiting tumor proliferation including NSCLC (Tian et al., Citation2011; Fan et al., Citation2020). Especially, apatinib has showed several notable characteristics, such as promoting tumor size shrinkage, inducing tumor regression and facilitating metastasis confinement, which are rarely found in other anti-angiogenic drugs. However, apatinib is also faced with the dilemma of short term of response, drug resistance and adverse effects including hypertension, proteinuria and hand-foot syndrome when it is used for a long time (Yang et al., Citation2020). Therefore, it is necessary to develop new targeted therapy to inhibit VEGFR2 in order to obtain better anti-tumor activity, fewer side effects and lower drug resistance.
RNA interference (RNAi) is a post-transcriptional gene regulatory process triggered by small interfering RNAs (siRNAs), which has the potential to treat tumor via silencing any disease-related genes in a sequence-specific manner (Barata et al., Citation2016; Shen et al., Citation2018). After a 20-year journey from its discovery, the United States Food and Drug Administration (FDA) and European Commission (EC) approved ONPATTRO® (patisiran, ALN-TTR02) achieved by Alnylam Pharmaceuticals (Cambridge, MA) as the first commercial siRNA therapeutic agent for the treatment of hereditary transthyretin amyloidosis (hATTR) with polyneuropathy in adults in 2018, which officially declared that RNAi therapy has come from theory to reality (Ruger et al., Citation2020). siRNA has innate advantages over small molecular therapeutics because siRNA executes its function by complete base pairing with mRNA, whereas small molecule drugs need to recognize the complicated spatial conformation of certain proteins, which confers the siRNA therapy with a shorter research and development span and a wider therapeutic area than small molecule drugs (Hu et al., Citation2020).
Even though siRNA technology has shown promising prospects in both preclinical and clinical trials, several barriers limit its clinical application (Ozcan et al., Citation2015). Intravenously injected siRNA is rapidly cleared without being taken up by target cells because it is a negatively charged macromolecule that has no bioavailability to cross cell membranes (Wittrup & Lieberman, Citation2015). Therefore, various chemically modified geometries and delivery systems such as nanoparticles and conjugates were established to assist the delivery of siRNA (Shen et al., Citation2017; Caillaud et al., Citation2020). Although nano-delivery systems show powerful delivery capabilities, they still have certain unavoidable disadvantages, such as burst release, toxicity risk associated with cationic materials and fast uptake by MPS cells (Caillaud et al., Citation2020). As an alternative to nano-delivery systems, researchers searched for a novel way to deliver siRNA. They obtained siRNA conjugates by directly linking siRNA covalently to different bioactive groups, such as lipids, aptamers and carbohydrates, which have been studied in preclinical and clinical trials (Lee et al., Citation2016; Weng et al., Citation2019). For example, GIVLAARI® (givosiran) from Alnylam (Cambridge, MA), a conjugate of N-acetylgalactosamine (GalNAc) to siRNA which enables specific binding to asialoglycoprotein receptor on hepatocytes, is approved to treat acute hepatic porphyria (AHP) in adults by FDA (Debacker et al., Citation2020). Currently, seven GalNAc–siRNA conjugates from Alnylam (Cambridge, MA) are undergoing clinical trials for liver genes, which show satisfactory progress (Hu et al., Citation2020). Similarly, in this study, we prepared a biRGD–siRNA conjugate by linking siRNA to the bivalent [cyclo(Arg-Gly-Asp-D-Phe-Lys)-Ahx]2-Glu-PEG-MAL peptide as our previous study shown (Cen et al., Citation2018).
Integrin αvβ3 receptors which play a key role in angiogenesis and tumor metastasis are significantly up-regulated in tumor neovascular endothelial cells and certain solid tumors such as glioblastoma, but not in quiescent endothelial cells and normal tissues (Beer & Schwaiger, Citation2008; Alday-Parejo et al., Citation2019). Several studies have confirmed that the arginine–glycine–aspartate peptide (RGD) can be specifically recognized by αvβ3 receptors and carry molecules into cells via αvβ3 receptor-mediated endocytosis (Liu et al., Citation2014a,b; Arosio & Casagrande, Citation2016; He et al., Citation2017; Cen et al., Citation2018). As of August 2020, more than 50 clinical trials of RGD-based tumor tracers or tumor-targeted drugs have been registered in the US FDA Clinical Trial Center. Therefore, RGD-based delivery systems have the potential to treat tumors.
This study intended to construct biRGD–siVEGFR2 conjugate delivery system which can silence VEGFR2 expression with targeting to neovascularization endothelial cells. Then we established an NSCLC model to observe the antitumor efficacy and renal toxicity of biRGD–siVEGFR2 monotherapy and its combination therapy with apatinib. Furthermore, we also evaluated the immunogenicity of biRGD–siVEGFR2 in C57BL/6J mice with intact immune system. To our knowledge, this is the first study to evaluate the antitumor activity of combination therapy with siRNA conjugate and small molecule TKI via co-inhibiting VEGFR2 gene.
2. Materials and methods
2.1. Cell culture
The luciferase-expressed A549 cell line (human non-small cell lung cancer cell line) was kindly provided by Guangzhou RiboBio Co., Ltd. (Guangzhou, China). MS1 cell line (the mouse islet endothelial cell line, Catalog No. CRL-2279) was purchased from ATCC (Manassas, VA). Cells were cultured using supplier recommended reagents and grown at 37 °C in a humidified atmosphere of 5% CO2 according to standard protocols and procedures.
2.2. Synthesis of biRGD–siRNA
In the preparation of biRGD–siRNA molecules, two 6-aminocaproic acids, one glutamic acid and two cRGD moieties underwent intermolecular dehydration condensation to form biRGD structure, which was covalently coupled to the 5′ end of siRNA sense strand via thiol-maleimide linker. The specific synthetic process and validation were performed as our previous description (Cen et al., Citation2018). The targeted murine VEGFR2 siRNA (siVEGFR2) and negative control siRNA (siNC) were synthesized by Guangzhou RiboBio Co., Ltd. (Guangzhou, China). Murine siVEGFR2 sequence refers to previous studies (Chen et al., Citation2013; Liu et al., Citation2014b). The details of siRNA sequences and modifications for this experiment were siVEGFR2 (sense strand: 5′-(mC)G(mG)AGAAGAAUGUGGU(mU)A(mA)dTdT-3′; anti-sense strand: 5′-(mU)(mU)AACCACAUUCU(mU)C(mU)C(mC)GdTdT-3′) and siNC (sense strand: 5′-(mC)(mG)(mU)GAUUGCGAGACUC(mU)(mG)(mA) dTdT-3′; anti-sense strand: 5′-(mU)(mC)(mA)GAGUCUCGCAAUC(mA)(mC)(mG)dTdT-3′). mN represented 2′-O-methyl (2′-OMe) sugar-modified RNA nucleosides.
2.3. Transfection experiments
Due to the lack of murine cell line with highly expressed integrin avβ3, we delivered biRGD–siVEGFR2 to MS1 cells, commonly used to study angiogenic signal transduction, with lipofectamine 3000 (Invitrogen, Carlsbad, CA). The cells were harvested for RT-qPCR and western blot analysis after being transfected for 48 hours and 72 hours, respectively.
2.4. RT-qPCR
Total RNA was extracted from MS1 cells or tumor samples using RNAiso Plus reagent (TaKaRa, Maebashi, Japan) and reverse transcribed into cDNA using PrimeScriptTMRT Reagent Kit with gDNA Eraser (TaKaRa, Maebashi, Japan) following the manufacturer’s protocol. Real-time monitoring of the PCR amplication of the cDNA was performed using the Applied Biosystems 7500 real-time PCR system (Life Technologies, Carlsbad, CA) with SYBR® Premix Ex TaqTM Kit (TaKaRa, Maebashi, Japan). qPCR amplication was conducted following the manufacturer’s protocol. Data were calculated using the 2−ΔΔCt method. The primers sequences were GAPDH (forward (5′–3′): GGCATTGCTCTCAATGACAA; reverse (5′–3′): TGTGAGGGAGATGCTCAGTG) and VEGFR2 (forward (5′–3′): GCCAATGAAGGGGAACTGAAGAC; reverse (5′–3′): TCTGGCTGCTGGTGATGCTGTC).
2.5. Western blot
Total protein was extracted from MS1 cells or tumor samples, subjected to electrophoresis in 8% polyacrylamide gel and transferred to a polyvinylidene fluoride membrane (Millipore Corporation, Billerica, MA). The bands were blocked in 5% BSA for two hours at room temperature. After washing for three times with Tris-buffered saline containing 0.1% Tween 20 (TBST) every 10 minutes, the membranes were incubated with primary anti-mouse VEGFR2 antibody (Cell Signaling Technology, Boston, MA) and β-actin antibody (Proteintech Group, Rosemont, IL) at 4 °C overnight. As described above, the membranes were washed and incubated with goat anti-rabbit secondary antibody (Proteintech Group, Rosemont, IL) for two hours and detected with ECL Chemiluminescence Substrate Reagent (Millipore Corporation, Billerica, MA) according to the manufacturer's instructions. The densities of bands were scanned and calculated by Image J software (Bethesda, MD).
2.6. Animal handing
C57BL/6J mice (female, 4–6 weeks, ∼20 g) and BALB/c nude mice (female, 4–6 weeks, ∼20 g) were purchased from the Experimental Animal Center of Sun Yat-sen University (Guangzhou, China) and raised in a specific-pathogen-free environment according to standardized animal care guidelines. The experiments were approved by the Southern Medical University animal experiments ethical committee and carried out according to national regulations.
2.7. Anti-tumor activity
BALB/c nude mice were subcutaneously inoculated with 6 × 106 luciferase-A549 cells to establish human NSCLC tumor model. When tumor volume reached 50–60 mm3, all tumor-bearing mice were randomly divided into six groups (n = 5). Saline group; biRGD–siNC (2 nmol/20 g) group; biRGD–siVEGFR2 (1 nmol/20 g) group; biRGD–siVEGFR2 (2 mol/20 g) group; apatinib (2.6 mg/20 g) group; biRGD–siVEGFR2 (1 nmol/20 g)+apatinib (1.3 mg/20 g) combination group. Tumor-bearing mice were intravenously injected with biRGD–siVEGFR2 for seven times with an interval of 48 hours between each injection or given apatinib intragastrically in the same interval. Tumor diameter was measured by a vernier caliper before each treatment and the volume was calculated using the formula: Volume = 1/2 × Length×(Width)2. Besides, tumor growth was also monitored by intratumoral fluorescence intensity via an in vivo imaging system (IVIS Lumina II, Caliper, Hopkinton, MA) on day 0, 5, 10 and 15 after treatment.
To further investigate whether biRGD–siVEGFR2 was dose-dependent, we reestablished a NSCLC xenograft tumor model as described above, increased the dose of biRGD–siVEGFR2 and compared its anti-tumor efficiency with that of apatinib. The apatinib-treated mice were administered at a dose of 2.6 mg/20 g body weight, which was converted from clinically regular dose. At the same time, a combination group containing biRGD–siVEGFR2 and gelofusine was added to alleviate the possible tubulointerstitial injury caused by high dose of biRGD–siRNA mentioned in our previous study (He et al., Citation2017; Cen et al., Citation2018). All the tumor-bearing mice were randomly divided into five groups (n = 5), when the tumor volume reached 100–150 mm3. Saline group; biRGD–siNC (5 nmol/20 g) group; biRGD–siVEGFR2 (5 nmol/20 g) group; biRGD–siVEGFR2 (5 nmol/20 g)+gelofusine (4 mg/20 g) group; apatinib (2.6 mg/20 g) group. Assays and analytical methods were the same as described above except for the different dosing regimen.
2.8. Immunohistochemistry analysis
Tumors isolated from all experimental groups were fixed immediately with 4% paraformaldehyde, embedded in paraffin and then cut into 4 μm thick sections. Primary antibodies including anti-CD31 antibody (catalog number: AF3628) from R&D Systems (Minneapolis, MN) and anti-Ki67 antibody (catalog number: ARG53222) from Arigo Biolaboratories (Shanghai, China) were used to label the positive expression of CD31 and Ki67 in tumor tissues in order to observe microvessel density and tumor cells proliferation.
2.9. Toxicity evaluation in vivo
Three days after the last administration, serum was collected from tumor-bearing mice. Aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatinine (Cr) and urea nitrogen (BUN) were detected by automatic Aeroset Chemistry Analyzer (Abbott, Abbott Park, IL) following the manufacturer’s instructions.
As described above, the kidneys were isolated, embedded and sectioned. All sections were stained with hematoxylin–eosin for histological examination according to standard clinical laboratory procedures.
2.10. Immunogenicity evaluation of biRGD–siVEGFR2 in vivo
C57BL/6J mice were randomly divided into saline group (n = 5) and biRGD–siVEGFR2 group (2 nmol/20 g, n = 5). Six hours after the administration, blood was collected and serum was separated to measure the levels of cytokines IFN-α, IFN-β, IL-6 and IL-12 using ELISA Kit (Bio-Swamp, Wuhan, China) according to manufacturer’s instructions.
2.11. Statistical analysis
All data were expressed as mean ± standard deviation (SD) and statistical analysis was performed using SPSS 20.0 statistical software (Chicago, IL). Independent-samples t-test was used to compare two groups. One-way analysis of variance (ANOVA) was used for multiple comparison. P<0.05 (two-tailed) was considered statistically significant different.
3. Results
3.1. Preparation and gene silencing efficiency of biRGD–siVEGFR2 in vitro
The diagram of biRGD–siVEGFR2 conjugate is shown in . The double-stranded siVEGFR2 (sense strand: 5′-(mC)G(mG)AGAAGAAUGUGGU(mU)A(mA)dTdT-3′; anti-sense strand: 5′-(mU)(mU)AACCACAUUCU(mU)C(mU)C(mC)GdTdT-3′) was selected as the representative siRNA drug, which was modified with 2′-OMe in recommended nucleotides so as to increase the stability and silencing potency and reduce the off-target effect and cytotoxicity. Here, the 5′-phosphate of sense strand of siVEGFR2 was linked to ([cyclo(Arg-Gly-Asp-D-Phe-Lys)-Ahx]2-Glu-PEG-MAL) peptide through the thiol-maleimide linker, as shown in our previous report (Cen et al., Citation2018).
Figure 1. The schematic depiction and characterization of biRGD–siRNA. (A) The diagram of biRGD–siRNA conjugate. The [cyclo(Arg-Gly-Asp-D-Phe-Lys)-Ahx]2-Glu-PEG-MAL (biRGD) peptide was conjugated to 5′-phosphate of sense strand of siRNA through thiol-maleimide linker. The backbone of siRNA strand was modified with 2′-O-methyl (2′-OMe) ribose in recommended nucleotides. (B, C) The high performance liquid chromatogram (HPLC) and mass spectrogram (MS) results of biRGD-conjugated sense stranded of VEGFR2 siRNA. (D, E) The high performance liquid chromatogram (HPLC) and mass spectrogram (MS) results of antisense stranded of VEGFR2 siRNA.
![Figure 1. The schematic depiction and characterization of biRGD–siRNA. (A) The diagram of biRGD–siRNA conjugate. The [cyclo(Arg-Gly-Asp-D-Phe-Lys)-Ahx]2-Glu-PEG-MAL (biRGD) peptide was conjugated to 5′-phosphate of sense strand of siRNA through thiol-maleimide linker. The backbone of siRNA strand was modified with 2′-O-methyl (2′-OMe) ribose in recommended nucleotides. (B, C) The high performance liquid chromatogram (HPLC) and mass spectrogram (MS) results of biRGD-conjugated sense stranded of VEGFR2 siRNA. (D, E) The high performance liquid chromatogram (HPLC) and mass spectrogram (MS) results of antisense stranded of VEGFR2 siRNA.](/cms/asset/81ec17ff-1b1e-4a64-a905-d6b44117f5b4/idrd_a_1937381_f0001_c.jpg)
The RP-HPLC results revealed that the purity of biRGD–siVEGFR2 sense strand and antisense strand were both >70%, as shown in . HPLC–MS results showed that the molecular mass of biRGD–siVEGFR2 sense strand was 8890.6 Da acceptably close to the predicted mass of 8891.8 Da and the molecular mass of biRGD–siVEGFR2 antisense strand was 6559.9 Da acceptably close to the predicted mass of 6558.1 Da (), which indicated that the molecular mass of synthesized biRGD–siVEGFR2 was consistent with its theoretical molecular mass.
To validate the silencing efficiency of 2′-OMe-modified siVEGFR2 sequence in the biRGD conjugate, we transfected biRGD–siRNA into MS1 cells with lipofectamine 3000 due to the lack of mouse-derived neovascularization endothelial cell line with highly expression of αvβ3 receptors. RT-qPCR analysis demonstrated that the VEGFR2 mRNA expression in MS1 cells transfected with 100 nM biRGD–siVEGFR2 was significantly decreased compared with that in the control group (the expression level was 42.68%, ). Furthermore, western blot analysis showed that the VEGFR2 protein expression was also significantly decreased after treatment with 100 nM biRGD–siVEGFR2 for 72 hours compared with that in the parallel control group (the expression level was 29.33%, ). In general, these data suggested that biRGD–siVEGFR2 was able to efficiently inhibit targeted gene expression.
3.2. Anti-non-small cell lung cancer activity of biRGD–siVEGFR2 and its combination with apatinib in vivo
A human NSCLC (A549-luc) xenograft expressing a luciferase reporter gene was established to investigate anti-tumor activity of biRGD–siVEGFR2 and its combination with apatinib in vivo. Briefly, nude mice bearing A549-Luc tumors were intravenously injected with biRGD–siVEGFR2 seven times over a 48 hours interval or given apatinib intragastrically in the same interval. The intratumoral fluorescence was visualized by bioimaging in and luciferase signal intensity was quantified and normalized in .
Figure 2. Anti-tumor activity and mechanism of biRGD–siVEGFR2 and its combination therapy with apatinib in NSCLC subcutaneous xenografts model. Six groups of NSCLC-bearing mice were treated as follows: Saline, biRGD–siNC (2 nmol/20 g), biRGD–siVEGFR2 (1 nmol/20 g), biRGD–siVEGFR2 (2 nmol/20 g), apatinib (2.6 mg/20 g) or biRGD–siVEGFR2 (1 nmol/20 g)+apatinib (1.3 mg/20 g). biRGD–siVEGFR2 was intravenously injected every 48 hours for seven times and apatinib was given intragastrically in the same interval. (A) IVIS luminescent imaging of NSCLC-bearing mice. (B) Tumor bioluminescence intensity of each group. Luminescence from tumors was measured and quantified by the IVIS imaging system. The data were normalized to saline group. (C) The tumor growth curve. Tumor volumes were measured before each treatment. (D) The image of ex vivo tumor from each group. Three days after the last treatment, tumors were isolated from mice and photographed. (E, F) Analysis of the expression of VEGFR2 in tumor tissues. Tumor tissues were collected to detect the expression levels of VEGFR2 mRNA and protein by RT-qPCR and western blot (normalized to β-actin level), respectively. *P<0.05, **P<0.01 vs. saline group.
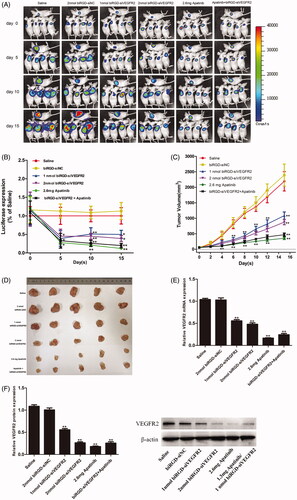
As seen in , these results demonstrated that all biRGD–siVEGFR2 treatment groups and apatinib groups significantly inhibited tumor growth after treating 15 days. Besides, the anti-tumor effects of biRGD–siVEGFR2 (1 nmol/20 g)/Apatinib (1.3 mg/20 g) combination group and high-dose apatinib (2.6 mg/20 g) group were more stronger than that of other groups, but there was no significant difference between biRGD–siVEGFR2 (1 nmol/20 g)/apatinib (1.3 mg/20 g) combination group and high-dose apatinib (2.6 mg/20 g) group.
More specifically, no significant difference was observed between saline and biRGD–siNC (2 nmol/20 g) groups, which indicated that biRGD–siNC did not have anti-tumor activity. Furthermore, on the fourth day after the first administration, compared with saline group, only biRGD–siVEGFR2 combination treatment group and apatinib (2.6 mg/20 g) group significantly inhibited tumor growth (P<0.05) and there was no significant difference between these two groups (P>0.05). Fifteen days after treatment, significant tumor growth inhibitions were observed in biRGD–siVEGFR2 (1 nmol/20 g), biRGD–siVEGFR2 (2 nmol/20 g), biRGD–siVEGFR2 (1 nmol/20 g)/apatinib (1.3 mg/20 g) combination group and apatinib (2.6 mg/20 g) alone group compared with that of saline and biRGD–siNC (2 nmol/20 g) administered groups (P<0.05). The order of their effects was as follows: biRGD–siVEGFR2 (1 nmol/20 g)/apatinib (1.3 mg/20 g) combination group ≈ apatinib (2.6 mg/20 g) alone group > biRGD–siVEGFR2 (2 nmol/20 g)>biRGD–siVEGFR2 (1 nmol/20 g).
These results suggested that biRGD–siVEGFR2 had the potential as an anti-NSCLC agent and tail vein injection at a dose higher than 1 nmol/20 g/48 h can effectively inhibit tumor growth. At the same time, biRGD–siVEGFR2 in combination with low-dose apatinib can achieve the same antitumor effect as the regular dose of apatinib. It is suggested that biRGD–siVEGFR2 can be used as an effective supplement for apatinib in clinical application.
Three days after the last injection, mice were euthanized and tumors were collected for further analysis. RT-qPCR and western blot results showed an obviously low expression of VEGFR2 mRNA and protein in all treatment groups compared with that in saline group ().
3.3. Anti-tumor activity of biRGD–siVEGFR2 monotherapy
As shown in , tumor growth was recorded by bio-imaging and luciferase signal intensity was quantified and normalized, which showed that intratumoral fluorescence intensity of biRGD–siVEGFR2 (5 nmol/20 g) group was significantly lower than that of saline and biRGD–siNC (5 nmol/20 g) groups. Tumor volume measurements further confirmed that biRGD–siVEGFR2 (5 nmol/20 g) significantly inhibited tumor growth and gelofusine did not affect its anti-tumor effect in combination therapy. Furthermore, there was no significant difference in inhibitory effect between biRGD–siVEGFR2 (5 nmol/20 g) and biRGD–siVEGFR2 (2 nmol/20 g) groups, suggesting that intravenous injection of biRGD–siVEGFR2 at a dose of 2 nmol/20 g/48 h may be a suitable administration dose because the cellular uptake of biRGD–siVEGFR2 at this dosage might be saturated.
Figure 3. Anti-tumor activity of biRGD–siVEGFR2 monotherapy. Five groups of NSCLC-bearing mice were treated with saline, biRGD–siNC (5 nmol/20 g), biRGD–siVEGFR2 (5 nmol/20 g), a mixture of biRGD–siVEGFR2 (5 nmol/20 g) and gelofusine (4 mg/20 g) or apatinib (2.6 mg/20 g). biRGD–siVEGFR2 or its gelofusine mixture was injected every 48 hours for seven times and apatinib was given intragastrically in the same interval. (A) IVIS luminescent imaging of NSCLC-bearing mice. (B) Tumor bioluminescence intensity of each group. (C) The tumor growth curve. (D) The image of ex vivo tumor from each group. (E, F) Analysis of the expression of VEGFR2 in tumor tissues. Tumor tissues were collected to detect the expression levels of VEGFR2 mRNA and protein by RT-qPCR and western blot (normalized to β-actin level), respectively. *P<0.05, **P<0.01 vs. saline group.
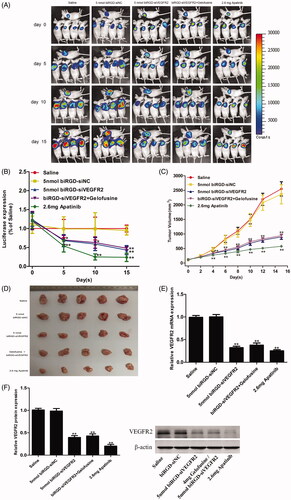
RT-qPCR and western blot results revealed that compared with saline group and biRGD–siNC (5 nmol/20g) group, the other treatment groups significantly inhibited the expression of VEGFR2 and gelofusine addition did not affect the silencing efficiency of biRGD–siVEGFR2 as shown in .
3.4. Evaluation of the effects of biRGD–siVEGFR2 on angiogenesis and tumor cells proliferation via immunohistochemistry assay
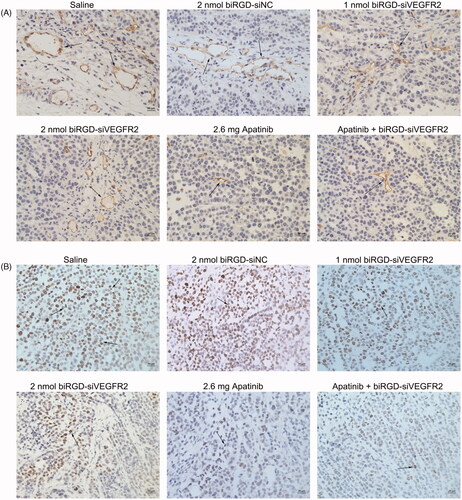
Immunohistochemistry assay showed that angiogenesis density and the number of proliferating cells in tumor tissues among all treatment groups were reduced to various extents compared with saline group and biRGD–siNC group (), which suggested that inhibition of tumor angiogenesis might negatively affect tumor growth via reducing supply of nutrition from blood to cancer cells.
Figure 4. The effect of biRGD–siVEGFR2 on angiogenesis and tumor proliferation. (A) Immunohistochemical staining with CD31 was used to detect neovascularization in tumor tissues at ×400 original magnification (indicated by arrows). (B) Immunohistochemical staining with ki67 was used to detect cell proliferation in tumor tissues at ×400 original magnification (indicated by arrows). Positive cells were brown. Bar = 20 μm.
3.5. The evaluation of liver and renal toxicity and immunogenicity of biRGD–siVEGFR2
Hematoxylin–eosin staining results indicated that co-injection of gelofusine and biRGD–siVEGFR2 could alleviate the renal toxicity caused by high-dose of biRGD–siVEGFR2. Long-term treatment with apatinib (2.6 mg/20 g) also produced significant renal toxicity, but a combination treatment of biRGD–siVEGFR2 and low-dose apatinib (1.3 mg/20 g) did not show significant renal toxicity (). Furthermore, we tested the concentrations of AST, ALT, Cr and BUN in the serum of tumor-bearing nude mice after long-term administration. Compared with saline group, all biochemical parameters had no significant changes in two low-dose biRGD–siVEGFR2 treatment groups. However, when the dosage of biRGD–siVEGFR2 was increased to 5 nmol/20 g, the concentrations of Cr and BUN in serum significantly increased while co-injection of gelofusine attenuated this trend. In addition, a significant increase was found in the concentrations of Cr and BUN in apatinib (2.6 mg/20 g) group, but the combination group of biRGD–siVEGFR2 (1 nmol/20g) and apatinib (1.3 mg/20 g) did not show significant changes ().
Figure 5. Toxicity and immunogenicity assessment of biRGD–siVEGFR2 in vivo. (A, B) Analysis of H&E stained sections of kidneys from in vivo anti-tumor assays. Black arrows indicate interstitial hyperemia. (C, D) Serum biochemical indicators from in vivo anti-tumor assays were used to assess potential liver and kidney toxicity. Three days after the last administration, blood serum was measured for AST, ALT, Cr and BUN in each group. (E) Analysis of IL-6, IL-12, IFN-β and IFN-α levels in the serum from C57BL/6J mice at six hours after the single injection of saline or biRGD–siVEGFR2 (2 nmol/20 g). **P<0.01, versus saline group, n = 5; bar = 20 μm.
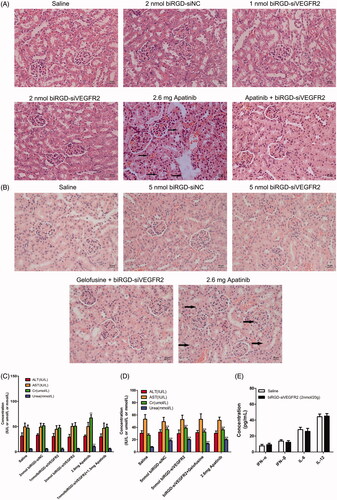
As shown in , there were no significant differences in serum levels of IFN-α, IFN-β, IL-6 and IL-12 between the treatment group and saline group in C57BL/6J mice with intact immune system, indicating that biRGD–siVEGFR2 did not elicit an immune response. All the results suggested that biRGD–siVEGFR2 had lower toxicity and higher safety compared with apatinib.
4. Discussion
In NSCLC, tumor angiogenic pathway has been identified as important therapeutic target, which is essential in the process of primary tumor growth, proliferation and metastasis (Hall et al., Citation2015). Vascular endothelial growth factor receptor 2 (VEGFR2) is a transmembrane protein that is the key tyrosine kinase receptor for angiogenic signaling in NSCLC (Pajares et al., Citation2012). In the current clinical application, targeting tumor angiogenesis has been approached through monoclonal antibodies such as bevacizumab that blocks VEGF–VEGFR binding or small molecule TKIs such as apatinib that selectively inhibits VEGFR2 phosphorylation (Mok et al., Citation2014; Fan et al., Citation2020). As an emerging gene-targeted therapeutic technology, siRNA interference technology, which provides a new therapeutic strategy for cancer therapy due to its powerful gene silencing mechanism, has attracted widespread attention in biomedical field (Setten et al., Citation2019; Weng et al., Citation2019). In addition to small molecular inhibitors or antibody drugs, RNAi technology to inhibit VEGFR2 is also an effective method for anti-angiogenesis and tumor suppression (Liu et al., Citation2014a,b).
In our study, we obtained an siRNA conjugate that can silence VEGFR2 expression by linking siVEGFR2 covalently to an optimized bivalent cyclic ([cyclo(Arg-Gly-Asp-D-Phe-Lys)-Ahx]2-Glu-PEG-MAL) peptide through the thiol-maleimide linker, which could specifically bind to integrin αvβ3 receptors overexpressing on the surface of tumor angiogenesis endothelial cells. Briefly, the specific method of siRNA conjugate synthesis was the same as our previous report (Cen et al., Citation2018). Our results showed that peptide–siRNA conjugates with accurate molecular weight were successfully obtained and they had no obvious immunogenicity in vivo. Meanwhile, our results demonstrated that biRGD–siVEGFR2 could substantially slow down NSCLC growth via inhibiting neovascularization and tumor cells proliferation.
In vivo antitumor pharmacodynamic studies in NSCLC A549 tumor xenograft model, we observed that biRGD–siVEGFR2 treatment groups significantly inhibited tumor growth in the early stage, but the inhibitory effect became weaker in the late stage of tumor development. Moreover, there was no obvious dose-dependent in a small range of dosage we tested. The following possible reasons account for this. (1) Limited number of αvβ3 receptors on the surface of neovascular endothelial cells. As biRGD–siVEGFR2 dosage increased, αvβ3 receptors gradually reached to saturation and the additional biRGD–siVEGFR2 molecules had no extra binding sites for endocytosis. (2) Moreover, lower escape capacity of biRGD–siVEGFR2 molecules in the endosome may be the another key factor for its RNAi activity in cytoplasma, which may be overcome by increasing functional structure to promote the release of biRGD–siVEGFR2 molecules from endosome.
Taken together, we selected a powerful siVEGFR2 sequence and constructed the biRGD–siVEGFR2 conjugate. Compared with nanoparticles, the biRGD–siVEGFR2 conjugate obtained by chemical synthesis has obvious advantages such as relatively small size, good pharmacokinetics, low immunogenicity and good reproduction, all of which make it potentially suit for clinical translation.
Apatinib, a specific anti-angiogenesis inhibitor targeting VEGFR2, has shown encouraging anticancer activity in a wide range of malignancies including non-small-cell lung cancer (Zhang, Citation2015). Nevertheless, some side effects were observed during its clinical treatment. The most common adverse reactions were urinary protein, hypertension and hand-foot syndrome (Xue et al., Citation2018). In this study, we administered apatinib intragastrically with clinically regular dosage and found its tumor inhibition effect was the most pronounced among all treatment groups during the treatment. However, a significant increase in the serum concentrations of Cr and BUN was observed in apatinib-treated mice. Additionally, a small amount of necrotic epithelial cells were deposited in the renal tubules, accompanied by renal interstitial hyperemia, indicating that apatinib had certain nephrotoxicity for long-term treatment.
It is undeniable that the levels of Cr and BUN in the serum were significantly increased and the renal pathological section was abnormal when biRGD–siVEGFR2 therapeutic dosage reached 5 nmol/20 g, suggesting slight nephrotoxicity. Yet, co-injection of gelofusine relieved the toxicity caused by high-dose of biRGD–siVEGFR2. Early studies have shown that gelofusine, acting as an effective renal protective agent, can reduce the reabsorption of biRGD–siRNA, thereby alleviating kidney damage (Cen et al., Citation2018; Liao et al., Citation2018). In addition, biRGD–siVEGFR2 had no obvious liver and kidney toxicity under 2 nmol/20 g therapeutic dosage.
Interestingly, although the antitumor effect of biRGD–siVEGFR2 was not as good as that of regular dosage of apatinib, its toxicity was much lower than that of apatinib. In the meanwhile, biRGD–siVEGFR2 in combination with low-dose apatinib could achieve the same antitumor effect as the regular dosage of apatinib without obvious nephrotoxicity. These reasons may explain the phenomenon. Apatinib, an effective small molecule inhibitor, could selectively bind to tyrosine kinase of VEGFR2 and quickly inhibit the activity of VEGFR2 protein, while biRGD–siVEGFR2 could directly silence the expression of VEGFR2 via RNAi and sustain anti-tumor activity for longer time. Moreover, biRGD–siVEGFR2 could be used as an effective supplementary inhibitor for tumor cells after apatinib resistance. The data in this study showed that the combination therapy with both biRGD–siVEGFR2 and lower-dosage apatinib targeted to mRNA and protein levels respectively could quickly, persistently and effectively inhibit tumor growth and reduce the toxicity of higher-dosage apatinib.
5. Conclusions
In this study, biRGD–siVEGFR2 conjugate was successfully constructed. The in vitro and in vivo data demonstrated that biRGD–siVEGFR2 could effectively knockdown VEGFR2 expression and substantially slow down NSCLC growth. Moreover, biRGD–siVEGFR2 could enhance the antitumor effect and reduce the nephrotoxicity of apatinib. In conclusion, biRGD–siVEGFR2 can effectively inhibit NSCLC growth and may provide a new treatment strategy in combination with low-dose apatinib for clinical anti-tumor therapy.
Supplemental Material
Download ()Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Alday-Parejo B, Stupp R, Ruegg C. (2019). Are integrins still practicable targets for anti-cancer therapy? Cancers (Basel) 11:978.
- Arosio D, Casagrande C. (2016). Advancement in integrin facilitated drug delivery. Adv Drug Deliv Rev 97:111–43.
- Barata P, Sood AK, Hong DS. (2016). RNA-targeted therapeutics in cancer clinical trials: current status and future directions. Cancer Treat Rev 50:35–47.
- Beer AJ, Schwaiger M. (2008). Imaging of integrin alphavbeta3 expression. Cancer Metastasis Rev 27:631–44.
- Bray F, Ferlay J, Soerjomataram I, et al. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424.
- Caillaud M, El Madani M, Massaad-Massade L. (2020). Small interfering RNA from the lab discovery to patients' recovery. J Control Release 321:616–28.
- Carmeliet P, Jain RK. (2011). Molecular mechanisms and clinical applications of angiogenesis. Nature 473:298–307.
- Cen B, Liao W, Wang Z, et al. (2018). Gelofusine attenuates tubulointerstitial injury induced by cRGD-conjugated siRNA by regulating the TLR3 signaling pathway. Mol Ther Nucleic Acids 11:300–11.
- Cen B, Wei Y, Huang W, et al. (2018). An efficient bivalent cyclic RGD-PIK3CB siRNA conjugate for specific targeted therapy against glioblastoma in vitro and in vivo. Mol Ther Nucleic Acids 13:220–32.
- Chen S, Liu X, Gong W, et al. (2013). Combination therapy with VEGFR2 and EGFR siRNA enhances the antitumor effect of cisplatin in non-small cell lung cancer lung cancer xenografts. Oncol Rep 29:260–8.
- Debacker AJ, Voutila J, Catley M, et al. (2020). Delivery of oligonucleotides to the liver with GalNAc: from research to registered therapeutic drug. Mol Ther 28:1759–71.
- Fan Y, Zhao J, Wang Q, et al. (2020). Camrelizumab plus apatinib in extensive-stage SCLC (PASSION): a multicenter, two-stage, phase 2 trial. J Thorac Oncol 16:299–309.
- Hall RD, Le TM, Haggstrom DE, et al. (2015). Angiogenesis inhibition as a therapeutic strategy in non-small cell lung cancer (NSCLC). Transl Lung Cancer Res 4:515–23.
- He S, Cen B, Liao L, et al. (2017). A tumor-targeting cRGD-EGFR siRNA conjugate and its anti-tumor effect on glioblastoma in vitro and in vivo. Drug Deliv 24:471–81.
- Hu B, Zhong L, Weng Y, et al. (2020). Therapeutic siRNA: state of the art. Sig Transduct Target Ther 5:1–25.
- Lee SH, Kang YY, Jang HE, et al. (2016). Current preclinical small interfering RNA (siRNA)-based conjugate systems for RNA therapeutics. Adv Drug Deliv Rev 104:78–92.
- Liao W, Qin Y, Liao L, et al. (2018). Protective effect of gelofusine against cRGD–siRNA-induced nephrotoxicity in mice. Ren Fail 40:187–95.
- Liu L, Liu X, Xu Q, et al. (2014a). Self-assembled nanoparticles based on the c(RGDfk) peptide for the delivery of siRNA targeting the VEGFR2 gene for tumor therapy. Int J Nanomedicine 9:3509–26.
- Liu X, Wang W, Samarsky D, et al. (2014b). Tumor-targeted in vivo gene silencing via systemic delivery of cRGD-conjugated siRNA. Nucleic Acids Res 42:11805–17.
- Manzo A, Montanino A, Carillio G, et al. (2017). Angiogenesis inhibitors in NSCLC. Int J Mol Sci 18:2021.
- Mok T, Gorbunova V, Juhasz E, et al. (2014). A correlative biomarker analysis of the combination of bevacizumab and carboplatin-based chemotherapy for advanced nonsquamous non-small-cell lung cancer: results of the phase II randomized ABIGAIL study (BO21015). J Thorac Oncol 9:848–55.
- Ozcan G, Ozpolat B, Coleman RL, et al. (2015). Preclinical and clinical development of siRNA-based therapeutics. Adv Drug Deliv Rev 87:108–19.
- Pajares MJ, Agorreta J, Larrayoz M, et al. (2012). Expression of tumor-derived vascular endothelial growth factor and its receptors is associated with outcome in early squamous cell carcinoma of the lung. J Clin Oncol 30:1129–36.
- Piperdi B, Merla A, Perez-Soler R. (2014). Targeting angiogenesis in squamous non-small cell lung cancer. Drugs 74:403–13.
- Ruger J, Ioannou S, Castanotto D, et al. (2020). Oligonucleotides to the (gene) rescue: FDA approvals 2017–2019. Trends Pharmacol Sci 41:27–41.
- Setten RL, Rossi JJ, Han SP. (2019). The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov 18:421–46.
- Shen J, Liu H, Mu C, et al. (2017). Multi-step encapsulation of chemotherapy and gene silencing agents in functionalized mesoporous silica nanoparticles. Nanoscale 9:5329–41.
- Shen J, Zhang W, Qi R, et al. (2018). Engineering functional inorganic–organic hybrid systems: advances in siRNA therapeutics. Chem Soc Rev 47:1969–95.
- Tian S, Quan H, Xie C, et al. (2011). YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci 102:1374–80.
- Weng Y, Xiao H, Zhang J, et al. (2019). RNAi therapeutic and its innovative biotechnological evolution. Biotechnol Adv 37:801–25.
- Wittrup A, Lieberman J. (2015). Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet 16:543–52.
- Xue JM, Astere M, Zhong MX, et al. (2018). Efficacy and safety of apatinib treatment for gastric cancer, hepatocellular carcinoma and non-small cell lung cancer: a meta-analysis. Onco Targets Ther 11:6119–28.
- Yang Q, Chen T, Wang S, et al. (2020). Apatinib as targeted therapy for advanced bone and soft tissue sarcoma: a dilemma of reversing multidrug resistance while suffering drug resistance itself. Angiogenesis 23:279–98.
- Zhang H. (2015). Apatinib for molecular targeted therapy in tumor. Drug Des Devel Ther 9:6075–81.