
Abstract
Pharmacologic inhibitors of cellular hydroxylase oxygen sensors are protective in multiple preclinical in vivo models of inflammation. However, the molecular mechanisms underlying this regulation are only partly understood, preventing clinical translation. We previously proposed a new mechanism for cellular oxygen sensing: oxygen-dependent, (likely) covalent protein oligomer (oxomer) formation. Here, we report that the oxygen sensor factor inhibiting HIF (FIH) forms an oxomer with the NF-κB inhibitor β (IκBβ). The formation of this protein complex required FIH enzymatic activity and was prevented by pharmacologic inhibitors. Oxomer formation was highly hypoxia-sensitive and very stable. No other member of the IκB protein family formed an oxomer with FIH, demonstrating that FIH-IκBβ oxomer formation was highly selective. In contrast to the known FIH-dependent oxomer formation with the deubiquitinase OTUB1, FIH-IκBβ oxomer formation did not occur via an IκBβ asparagine residue, but depended on the amino acid sequence VAERR contained within a loop between IκBβ ankyrin repeat domains 2 and 3. Oxomer formation prevented IκBβ from binding to its primary interaction partners p65 and c-Rel, subunits of NF-κB, the master regulator of the cellular transcriptional response to pro-inflammatory stimuli. We therefore propose that FIH-mediated oxomer formation with IκBβ contributes to the hypoxia-dependent regulation of inflammation.
Introduction
Decreased oxygen availability (hypoxia) and inflammation frequently co-occur with mutual functional effects.Citation1 Cells adapt to hypoxia via cellular oxygen sensors, including prolyl-4-hydroxylase domain (PHD) proteins 1–3 and factor inhibiting HIF (FIH).Citation2 FIH was first described to catalyze the hydroxylation of an asparagine residue within α subunits of the heterodimeric hypoxia-inducible factor (HIF),Citation3–6 inhibiting the interaction of HIF with the transcriptional co-activators p300 and CBP histone acetyl transferases.Citation7
Pharmacologic hydroxylase inhibitors (HIs) are protective in animal models of inflammation, including inflammatory bowel disease, sepsis, lung inflammation, skin abscess, chronic kidney disease, bladder infection and more.Citation8–10 However, the underlying molecular mechanisms of the HI’s protective effects are scarcely understood.Citation11–13 FIH has recently been linked to the regulation of inflammation in vivo. Treatment with the FIH-selective pharmacologic agent N-oxalyl-D-phenylalanine (NOFD) was protective against irradiation-induced injuries of the gastrointestinal tract and FIH deletion in the colon epithelium attenuated chronic colitis.Citation14,Citation15 We previously demonstrated that the enzymatic activities of FIH and PHD1 attenuate the major pro-inflammatory IL-1β signaling pathway.Citation12 Nonetheless, the general relevance and the detailed FIH-dependent regulation of inflammation remains elusive.
FIH enzymatic activity is not limited to asparagine hydroxylation or HIF-α subunits.Citation16 The first FIH substrates identified outside the HIF pathway were IκBα and p105, both members of the inhibitor of NF-κB (IκBs) protein family.Citation17 The IκB protein family can be separated into three different groups: typical (IκBα, IκBβ and IκBε), precursor (p105/IκBγ and p100/IκBδ) and atypical (nuclear) IκBs (Bcl-3, IκBζ, IκBNS, and IκBη).Citation18,Citation19 In the absence of a stimulus, NF-κB is retained in the cytoplasm by interaction with typical IκB proteins.Citation18 Following an inflammatory stimulus, typical IκBs are phosphorylated and degraded, releasing NF-κB which then translocates into the nucleus and upregulates the transcription of specific genes.Citation20,Citation21 In addition to the hydroxylation of IκBα and p105, IκBε has previously been shown to interact with FIH.Citation22 Of note, all members of the IκB protein family contain ankyrin repeat domains (ARDs) and proteins with ARDs represent the largest group among the known FIH substrates.Citation17,Citation22,Citation23 Thus, there is strong link between FIH and IκB proteins, although the hydroxylation of IκB proteins is apparently not affecting IκBs’ function.Citation17,Citation24
We recently reported that the deubiquitinase (DUB) ovarian tumor domain-containing ubiquitin aldehyde binding 1 (OTUB1) is a substrate for FIH-mediated hydroxylation of asparagine residue N22.Citation12,Citation25 In addition, FIH and OTUB1 form an oxygen-dependent denaturation-resistant (likely covalent) protein-protein complex catalyzed by FIH activity, which we refer to as “oxygen-dependent stable protein oligomer” (oxomer).Citation16,Citation26,Citation27 FIH-OTUB1 oxomer formation is highly hypoxia sensitive and regulates OTUB1 enzymatic function.Citation26 A mass spectrometry-based analysis identified 12 additional putative stable protein complexes formed by FIH, including ubiquitin-like modifier-activating enzyme 1 (UBA1) and IκBβ.Citation26 Interestingly, we had previously identified IκBβ as interaction partner of FIH together with oxidation of aspartate residue 195, indicating a potential hydroxylation of IκBβ by FIH.Citation12
Here, we investigated if FIH forms oxomer complexes with other proteins. Due to the existing functional links between FIH and inflammation, we focused on IκB proteins as putative substrates for oxomer formation. The identification of additional oxomer complexes and their characterization will increase our understanding of this novel type of oxygen-dependent signaling. Moreover, characterization of the functional interaction of FIH with proteins of pro-inflammatory signaling pathways will contribute to our understanding of the molecular mechanism(s) underlying the protective effect of pharmacologic FIH inhibition and genetic deletion in inflammatory diseases in vivo.
Results
FIH forms an oxomer with IκBβ but not with other IκB protein family members
Following the previous identification of 12 potential substrate proteins for oxomer formation with FIH,Citation26 we aimed to analyze these candidates in more detail and focused on IκBβ and UBA1. IκBβ was of special interest, as members of its protein family had been shown to be hydroxylated and/or to interact with FIH.Citation17,Citation22,Citation23 Moreover, we had previously identified IκBβ as interactor of FIH and we found a putative hydroxylation site within IκBβ.Citation12 UBA1 was selected for further investigation because it plays a pivotal role in protein ubiquitination as E1 enzyme, initiating the enzymatic ubiquitination cascade.Citation28,Citation29 We thus assessed the oxomer formation between ectopically expressed FIH and IκBβ () and between FIH and UBA1 (Figure S2) by immunoblotting. Oxomer formation between FIH and OTUB1 served as positive control ().Citation26 Ectopic co-expression of UBA1 or IκBβ with FIH led to the formation of denaturing condition-resistant (SDS-PAGE) complexes at molecular weights that were consistent with the covalent addition of UBA1 or IκBβ, respectively, to FIH ( and Figure S2). Moreover, a stable FIH-IκBβ complex was also formed in HepG2, Hep3B and MCF7 cells (), suggesting a general occurrence of the observed oxomer formation. Interestingly, the ratio between the FIH-IκBβ oxomer and IκBβ monomer differed between the cell lines with the highest relative FIH-IκBβ oxomer levels being observed in MCF-7 cells ().
Figure 1. Selective oxomer formation between FIH and IκBβ. (A) Immunoblot analysis of the oxomer formation between ectopically expressed FIH-V5 and V5-IκBβ in HEK293 cells, using FIH-OTUB1 oxomer formation via ectopic expression of FIH-V5 and FLAG-OTUB1 as control. (B) Immunoblot analysis of FIH-IκBβ oxomer formation in HepG2, Hep3B and MCF7 cells with ectopic FIH-V5 and IκBβ-FLAG expression. (C) Immunoprecipitation of endogenous IκBβ from HEK293 cells. A signal was detected at a molecular weight consistent with a FIH-IκBβ oxomer (highlighted by *). This complex was absent in the presence of the hydroxylase inhibitor DMOG (1 mM for 24 h) and in FIH knockout (KO) cells. (D) Immunoblot analysis of oxomer formation between FIH-V5 and the Indicated members of the IκB protein family following transient transfection of HEK293 cells with corresponding vectors for ectopic expression. Predicted molecular weights of putative FIH oxomers (without post-translational protein modifications): FIH-IκBα, 76 kDa; FIH-IκBβ, 78 kDa; FIH-Bcl-3, 88 kDa; FIH-IκBδ, 90 kDa; FIH-IκBε, 78 kDa; FIH-p105, 145 kDa; FIH-p100, 137 kDa. Data are representative of three (A, B, D) and two (C) independent experiments. F, FIH-V5; I, V5-IκBβ; F-O, FIH-OTUB1 oxomer; F-I, FIH-IκBβ oxomer; interm., intermediate; exp, exposure; DMOG, dimethyloxalylglycine; ns, non-specific.
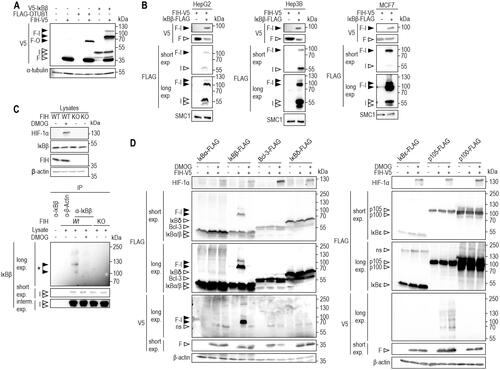
Next, we assessed if FIH-IκBβ oxomer formation was also observable between endogenous proteins and whether it was dependent on FIH enzymatic function. Following IP of endogenous IκBβ, a signal was detected at a molecular weight that corresponded to the predicted FIH-IκBβ oxomer ( and Figure S3A). This signal was abolished following FIH inhibition by DMOG treatment or FIH gene deletion ( and Figure S3A). Thus, also endogenous FIH and IκBβ form a denaturing condition-resistant oxomer which depends on FIH enzymatic activity.
The successful validation of FIH-IκBβ oxomer formation raised the question whether other IκB family members also served as FIH substrates for oxomer formation. To investigate this possibility, FIH was ectopically co-expressed with either IκBα, IκBβ, Bcl-3, IκBδ, IκBε, p105 or p100 and it was assessed if any higher molecular weight signals could be detected that were sensitive to pharmacologic FIH inhibition () or FIH deletion (Figure S3B). Although several IκB family members had previously been shown to serve as hydroxylation substrates and/or interaction partners of FIH,Citation17,Citation22,Citation23 none of the other IκB proteins formed an oxomer with FIH ( and Figure S3B), demonstrating that FIH-mediated oxomer formation is highly selective for IκBβ.
FIH-IκBβ oxomer formation is highly sensitive to hypoxia
To investigate the relevance of FIH enzymatic activity for FIH-IκBβ oxomer formation in detail, HEK293 cells were incubated in hypoxia (0.2% O2; pan-hydroxylase inhibition) or treated with pan- (DMOG, DFX), PHD- (FG-4592) or FIH-selective (DM-NOFD) hydroxylase inhibitors.Citation27,Citation30 Formation of the FIH-IκBβ oxomer was abrogated by hypoxia, DMOG and DFX but not by FG-4592 (roxadustat) () in agreement with the reported selectivity of this inhibitor for PHDs.Citation27,Citation31 Interestingly, DM-NOFD did not prevent FIH-IκBβ oxomer formation (), like we previously observed for FIH-OUTB1 oxomer formation.Citation27
Figure 2. FIH-IκBβ oxomer formation depends on FIH enzymatic activity and is highly hypoxia-sensitive. (A) FIH-IκBβ oxomer formation is prevented by hypoxia (0.2% O2), DMOG (1 mM) and DFX (0.1 mM), but not by the PHD-selective FG-4592 (0.1 mM) or the FIH-selective DM-NOFD (1 mM). Treatments were performed for 16 h in HEK293 cells. (B, C) Genetically inactivated FIH (H199A or L340R loss of function mutations) prevented FIH-IκBβ oxomer formation. (D) Hypoxia sensitivity of HIF-1α stabilization and FIH-IκBβ oxomer formation after 24 h at the indicated oxygen levels. (E) Quantification of the results shown in (D) and calculation of the corresponding oxygen sensitivities. Data are shown as mean ± SEM. F, FIH-V5; I, IκBβ-FLAG; Nx, normoxia; Hx, hypoxia (0.2% O2); WT, wild-type. Data are representative of three (A-C) and four (D, E) independent experiments.
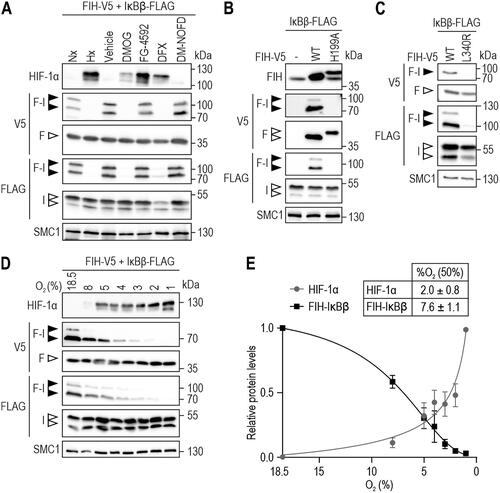
To investigate the effect of genetic inhibition of FIH activity, cells were used with ectopic expression of FIH carrying a point mutation of the active site aa residue histidine 199 to alanine (H199A).Citation4 This point mutation prevented FIH-IκBβ oxomer formation (). Moreover, also the mutation of FIH lysine 340 to arginine, disrupting FIH dimerization and thereby FIH hydroxylase activity,Citation32 prevented FIH-IκBβ oxomer formation (). Overall, these results demonstrated that FIH enzymatic activity is required for FIH-IκBβ oxomer formation.
Because FIH-IκBβ oxomer formation required FIH activity, the sensitivity of FIH-IκBβ oxomer formation to different oxygen availability (18.5–1% O2) was investigated. Interestingly, FIH-IκBβ oxomer formation was highly sensitive to hypoxia, with an EC50 of 7.6% O2 compared to an EC50 for HIF-1α stabilization of 2.0% O2 within the same cells ().
The FIH-IκBβ oxomer is an extraordinarily stable protein complex
Given the resistance of the FIH-IκBβ oxomer against the denaturing conditions during immunoblotting, we next characterized the stability of this oxomer in more detail. Recently, a new type of covalent bond between a lysine and a cysteine residue has been described, called a NOS bridge.Citation33 The NOS bridge can be disrupted under very strong reducing conditions (1–5 mM dithiothreitol, DTT). To investigate if FIH and IκBβ are linked through a NOS bridge within the observed oxomer, samples were treated with different reducing agents at even higher concentrations than those used for disrupting the NOS bridge (). None of the used concentrations of DTT or β-mercaptoethanol disrupted the FIH-IκBβ oxomer, indicating that the linkage between FIH and IκBβ is not a NOS bridge or any other type of bond that is susceptible to the reducing conditions described.
Figure 3. FIH-IκBβ oxomer stability. (A) Immunoblot analysis of the FIH-IκBβ oxomer upon treatment of cell lysates with the indicated reducing agents followed by incubation for 5 min at 95 °C. (B) FIH-IκBβ oxomer formation in HEK293 cells following the indicated transfection and treatment with the proteasome inhibitor MG132 (10 μM). (C) HEK293 cells were transfected with vectors encoding FIH and IκBβ for 22 h to form the FIH-IκBβ oxomer. Subsequently, oxomer stability was analyzed in hypoxia (0.2% O2), inhibiting FIH and thus preventing any additional oxomer formation. (D) Quantification of the results shown in (C), presented as mean ± SEM. F, FIH-V5; I, IκBβ-FLAG; F-I, FIH-IκBβ oxomer; DTT, dithiothreitol; interm., intermediate; exp., exposure. *P < 0.05 by Student’s t test. Data are representative of three independent experiments.
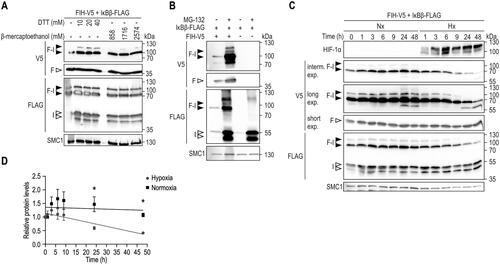
Next, we analyzed the degradation and stability of the FIH-IκBβ oxomer. To assess if the oxomer was degraded by the proteasome, cells were allowed to form the oxomer for 24 h and subsequently treated with the proteasome inhibitor MG132 for 21 h (). Proteasome inhibition strongly increased FIH, IκBβ and the oxomer levels (), also demonstrating that the oxomer is degraded via the proteasome. In a subsequent experiment, the stability of the oxomer was analyzed. Following oxomer formation for 22 h, cells were exposed to normoxia or hypoxia (0.2% O2) for the indicated time periods to prevent any additional oxomers to be formed (). In normoxia, no change of the oxomer protein level was observed over the entire 48 h (). In hypoxia, a first significant reduction of the oxomer was detected only 24 h after the start of the hypoxia exposure and reached approximately 50% of the initial levels at around 48 h following the onset of hypoxia ().
The IκBβ loop between ankyrin repeat domains 2 and 3 is necessary for oxomer formation
We next aimed to identify the IκBβ amino acid targeted by FIH for oxomer formation. FIH is known to hydroxylate asparagine residues and the FIH-OTUB1 oxomer is formed through asparagine 22 (N22) of OTUB1.Citation4,Citation26 IκBα has so far been the best characterized FIH target protein outside the HIF pathway,Citation17,Citation34 it is hydroxylated by FIH on the two asparagine residues Asn-244 and Asn-210 and it is closely related to IκBβ.Citation17 We therefore aimed to compare the IκBα and IκBβ structures to obtain insights into the potential part of IκBβ that might be targeted by FIH enzymatic activity. However, to the best of our knowledge, the protein structure of human IκBβ has not been experimentally solved to date. We therefore used AlphaFold to predict the structure of IκBβ and superimposed the obtained structure with the structure of IκBα.Citation35–37 highlights the asparagine residues of IκBα that are hydroxylated by FIH and which are located within a loop between ARDs 2 and 3.Citation17 Interestingly, one IκBβ asparagine residue (N266) was positioned in a comparable area of IκBβ as the hydroxylated asparagine residues in IκBα (). Although this indicates that N266 may be the targeted amino acid residue within IκBβ, we decided to point mutate all of the five IκBβ asparagine residues to alanine. In addition, one aspartate residue was also point mutated to alanine (D195A), as previous mass spectrometry analyses had shown its oxidation and because FIH also hydroxylates aspartate residues.Citation12,Citation38 AlphaFold-mediated structure prediction indicated that the introduced point mutations do not alter the structure of IκBβ (Figure S4A). Unexpectedly, none of the introduced IκBβ point mutations prevented FIH-IκBβ oxomer formation (). The higher molecular weight signal of the oxomer doublet showed some variability within its intensity (), but this was likely due to differences in the ectopic expression efficiency of the mutated IκBβ (). Thus, in contrast to the asparagine residue in the FIH-OTUB1 oxomer, FIH appears to utilize a different aa residue for oxomer formation with IκBβ.
Figure 4. Analysis of potential IκBβ amino acid residues as substrates for FIH-IκBβ oxomer formation. (A) Overlay of protein structures of IκBα (shown in cyan, identifier AF-P25963-F1) and IκBβ (shown in green, identifier AF-Q15653-F1) predicted by AlphaFold Protein Structure Database. Asparagine residues of IκBα previously demonstrated to be hydroxylated by FIHCitation17 are indicated in dark blue, all asparagine residues of IκBβ are highlighted in red. An aspartic acid residue that had previously been found to be oxidized is highlighted in magenta.Citation12 (B) Separate point mutations were introduced in IκBβ-FLAG, changing each asparagine residue and the highlighted aspartic acid residue to alanine. Oxomer formation was assessed in HEK293 cells by immunoblotting. (C) Analysis of oxomer formation between FIH and truncated IκBβ (ARD 1–3, 1–166 amino acids; ARD 4–6, 167–356 amino acids of full length IκBβ). (D) Analysis of oxomer formation between FIH and the indicated C-terminally truncated IκBβ fragments. (E) Analysis of oxomer formation between FIH and IκBβ containing the indicated amino acid deletions. Amino acids identified to be involved in oxomer formation (122–126) are highlighted in red. F, FIH-V5; I, IκBβ-FLAG; WT, wild-type; ARD, ankyrin repeat domain; *FIH-IκBβ oxomer with truncated IκBβ forms; trunc., truncated; exp., exposure. Predicted molecular weights of proteins without tags (kDa): ARD 1–3: 21, ARD 4–6: 25; predicted molecular weights of oxomers without tags (kDa): FIH-IκBβ ARD 1–3: 61, FIH-IκBβ ARD 4–6: 65. Data are representative of three (B), one (C and E) and two (D) independent experiments.
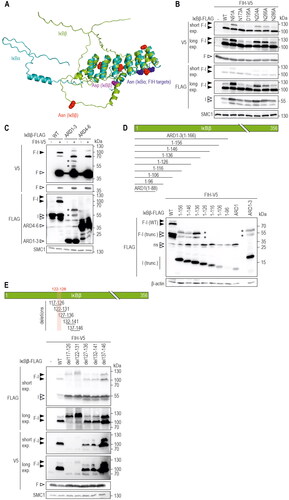
Next, we analyzed whether IκBβ is hydroxylated by FIH via mass spectrometry (MS), because for OTUB1 the same asparagine residue that is involved in oxomer formation is also hydroxylated in monomeric OTUB1.Citation26 We identified IκBß peptides containing four of the five asparagine residues. Despite detecting numerous oxidations on methionines, none of the asparagines were deemed to be hydroxylated (Supplemental Table S1). The one unidentified asparagine resides in the N-terminal region and was not accessible by standard MS workflows because the tryptic digest generated a peptide that was too large for MS identification. Nevertheless, it is highly likely that IκBß is not hydroxylated as none of the sequences surrounding any of the IκBß asparagines matches the well-characterized FIH consensus motif Lx(6)[VI]N.Citation39 Therefore, we systematically screened for the IκBβ moiety that was involved in FIH-IκBβ oxomer formation. First, we assessed the N-(ARDs 1–3, aa residues 1–166) or C-terminal (ARDs 4–6, aa residues 167–356) parts of IκBβ (). According to AlphaFold predictions, these truncations largely maintained the corresponding protein structures (Figure S4B). While IκBβ ARDs 1–3 formed an oxomer with FIH with the predicted molecular weight, there was no oxomer formed with ARDs 4–6 ().
To identify the IκBβ residues involved in oxomer formation, vectors were constructed containing IκBβ ARD 1–3 with truncations of 10 aa increments from the C-terminal end (). Truncated IκBβ fragments containing the N-terminal 126 aa or less were no longer able to form an oxomer with FIH (). Next, IκBβs with consecutive deletions were generated in otherwise full-length IκBβ with overlapping steps of 5 aa, surrounding the identified area (). While deletion of IκBβ aa 127–136, 132–141 and 137–146 did not interfere with oxomer formation, the absence of IκBβ aa 117–126 and 122–131 abrogated oxomer formation (). Therefore, the IκBβ aa 122–126 (VAERR) region, common to both deletion constructs, is necessary for oxomer formation between FIH and IκBβ. These residues are located in the loop between the IκBβ ARDs 2 and 3 (Figure S4C and D). Of note, the IκBβ peptide containing VAERR was detected in the MS analyses, but no relevant amino acid residue was deemed to be hydroxylated within this peptide. Overall, the IκBβ VAERR sequence is necessary for FIH-IκBβ oxomer formation, but it remains unclear if any of the included amino acid residues is directly targeted by FIH enzymatic activity.
FIH-IκBβ oxomer formation prevents IκBβ from binding the major NF-κB subunits p65 and c-Rel
It has previously been suggested that IκBα can scavenge FIH from HIF-1α, reducing or preventing FIH-mediated hydroxylation of HIF-1α, and thus increasing HIF-1 transcriptional activity.Citation17 Therefore, we aimed to investigate a potential effect of IκBβ on FIH activity through oxomer formation by employing a HIF-dependent firefly luciferase reporter gene assay.Citation40,Citation41 HEK293 cells were transfected with various IκB proteins for 24 h and treated with FG-4592 for an additional 24 h to specifically inhibit the PHDs to increase HIF activity without pharmacologically affecting FIH.Citation27 FG-4592 treatment increased HIF-dependent firefly luciferase activity (). Ectopic expression of IκBα and IκBε significantly increased HIF activity likely by reducing FIH activity towards HIF (), as previously described.Citation17,Citation22 Interestingly, p105 had no additional effect compared to FG-4592 treatment alone (), although p105 is hydroxylated by FIH.Citation17 No other IκB protein family member, including IκBβ, affected HIF activity. These results indicate that the effect of IκBα and IκBε towards HIF activity is selective and not a general effect of all FIH target proteins or all ARD-containing FIH substrates.
Figure 5. FIH-IκBβ oxomer formation prevents IκBβ from binding NF-κB subunits. (A) HEK293 cells were transiently cotransfected with a HIF-driven luciferase plasmid (pH3SVL) together with a constitutive Renilla luciferase plasmid (pRL-SV40) as well as plasmids expressing the indicated IκB protein family member 24 h prior to treatment. FG-4592 treatment was applied for 24 h in a concentration of 0.1 mM. Efficient transfection and FG-4592 treatment were determined by immunoblotting (Figure S5). Significance was assessed by two-way ANOVA with Tukey’s correction; *P < 0.05, **P < 0.01. (B) Immunoprecipitation (IP) analysis of the interaction of IκBβ and the FIH-IκBβ oxomer with the NF-κB subunits c-Rel and p65. (C) Quantification of the results shown in B. The p65 and c-Rel levels were normalized to the levels of total bait/FLAG pulldown (IκBβ or IκBβ + oxomer), respectively. Statistical analysis was performed by Student’s t test. *P < 0.05; F, FIH-V5; I, IκBβ-FLAG. Data are shown as mean ± SD. Data are representative of three independent experiments.
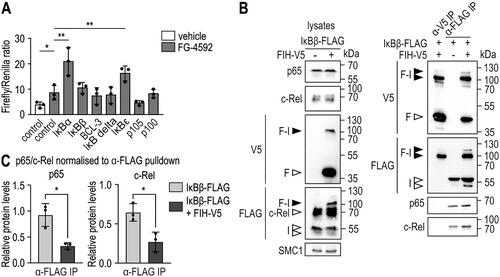
The main function of IκBβ is to bind to NF-κB transcription factor dimers, which sequesters them in the cytosol and regulates NF-κB-mediated gene expression.Citation20,Citation21 Thus, we investigated whether IκBβ retains its capability to bind NF-κB proteins within the oxomer. Specifically, the interaction between IκBβ and p65 as well as c-Rel was assessed, as these NF-κB subunits are most commonly bound by IκBβ.Citation42 IκBβ-FLAG was ectopically expressed in HEK293 cells alone or in combination with FIH-V5, and immunoprecipitated using anti-V5 or anti-FLAG antibodies, respectively. The FLAG IP pulled down the FIH-IκBβ oxomer as well as monomeric IκBβ, whereas the V5 IP pulled down the oxomer as well as FIH but no monomeric IκBβ (), indicating that FIH was bound as homodimer to IκBβ as observed within the FIH-OTUB1 oxomer.Citation26 p65 and c-Rel were both co-immunoprecipitated together with IκBβ-FLAG, but not in the FIH-V5 IP (), which contained IκBβ only as oxomer. These results suggest that oxomer formation of IκBβ with FIH prevents IκBβ from binding p65 and c-Rel and therefore removes IκBβ from the functionally relevant pool of IκB proteins for the regulation of NF-κB.
Discussion
Over the last years, it has become apparent that a tight functional connection exists between hypoxia and inflammation.Citation1,Citation8,Citation11,Citation43–46 Tissue hypoxia is commonly observed in inflamed tissue areas and an inflammatory response can occur in hypoxic tissues.Citation9,Citation46 Furthermore, pharmacologic hydroxylase inhibitors demonstrated anti-inflammatory effects in numerous different inflammatory diseases in mice.Citation8–10 Our understanding of the molecular mechanisms underlying this mutual interplay, however, is in its infancy. FIH may be part of the regulatory network connecting hypoxia and inflammation, as FIH alters IL-1β-induced NF-κB activity and is relevant for the inflammatory response in the gut in vivo.Citation12,Citation14,Citation15 Nonetheless, the extent of contribution of FIH activity to inflammation as well as the molecular mechanisms that connect FIH and inflammation are unclear.
Previously, we reported that FIH forms an oxomer with OTUB1.Citation26 Here, we demonstrate that FIH forms a similar oxomer specifically with IκBβ. FIH enzymatic activity was necessary for the FIH-IκBβ complex formation, which was prevented by pan-hydroxylase inhibitors and FIH mutations. The oxomer was formed in several different cell lines and by endogenous proteins. The endogenous oxomer was observed at relatively low levels compared to the IκBβ monomer. However, this assessment was performed in HEK293 cells and may differ in other cell lines. Interestingly, we observed (with ectopically expressed proteins) that the oxomer level relative to monomeric IκBβ is higher in MCF7 than in HEK293, Hep3B and HepG2 cells, indicating that there might be cell types in which oxomer formation is favored. Moreover, following a pro-inflammatory stimulus, IκBβ is degraded and re-expressed.Citation47,Citation48 We have previously reported that oxomer formation occurs co-translationally.Citation26 Thus, following a pro-inflammatory stimulus, (endogenous) oxomer formation may be increased due to the strong induction of IκBβ expression and translation. This will be analyzed in the future.
Interestingly, IκBβ was the only protein of the IκB protein family that was targeted by FIH for oxomer formation, although all members contain ARDs which are prominent FIH substrates.Citation17,Citation22,Citation23 Within our initial screen for FIH target proteins for oxomer formation, IκBβ was the only identified ARD-containing protein.Citation26 Together, these results demonstrate that FIH-mediated oxomer formation is highly selective and indicate that ARDs are not a defining characteristic of substrate proteins for FIH-dependent oxomer formation.
The FIH-mediated FIH-IκBβ oxomer formation demonstrated a higher hypoxia sensitivity than the PHD-mediated stabilization of HIF-1α. Moreover, this sensitivity was even higher than the sensitivity of FIH-mediated HIF-1α hydroxylation, which is known to be less susceptible to hypoxia than PHD-mediated hydroxylation.Citation49 These results are remarkable and comparable to our previous findings regarding the hypoxia sensitivity of the FIH-OTUB1 oxomer formation.Citation26 This further strengthens the hypothesis that oxomer formation serves as an alternative cellular mechanism to fine-tune the adaptation to hypoxia. Moreover, it indicates that the hypoxia sensitivity of oxomer formation is largely defined by yet unknown characteristics of FIH and less by its protein substrates. In addition, the FIH-IκBβ oxomer was very stable and only started to noticeably decline 24 h after the onset of hypoxia. These results are consistent with the FIH-OTUB1 oxomer and support the conclusion that FIH-dependent oxomers may serve for signaling long-term rather than short-term fluctuations in local oxygen availability.Citation26
As previously observed for the FIH-OTUB1 oxomer,Citation27 the FIH-selective inhibitor DM-NOFD failed to prevent FIH-IκBβ oxomer formation. Of note, both oxomers showed a higher hypoxia sensitivity than FIH-mediated HIF-α asparagine hydroxylation,Citation26 indicating that the sensitivity of FIH for co-substrate binding depends on the protein substrate. This may also extend to co-substrate mimetics, such as DM-NOFD. To test this hypothesis, it would be necessary to assess the DM-NOFD sensitivity of FIH-dependent HIF-α asparagine hydroxylation and oxomer formation within the same lysates. However, a reliable antibody detecting HIF-α asparagine hydroxylation is currently not commercially available.
The connection between FIH and IκBβ in the FIH-IκBβ oxomer resisted high concentrations of reducing agents and boiling, suggesting a likely covalent bond, and indicating that the oxomer is formed independently of cysteine residues. FIH hydroxylates asparagine residuesCitation4 and the FIH-OTUB1 oxomer is formed through asparagine 22 (N22) of OTUB1. However, FIH is known to be promiscuousCitation16 and hydroxylates aspartate,Citation38 histidine,Citation50 and tryptophan residues as well.Citation51 Interestingly, we observed that point mutations of each asparagine within IκBβ did not interfere with oxomer formation, demonstrating that the FIH-IκBβ oxomer was not formed via an IκBβ asparagine residue. IκBβ-targeted MS analysis did not identify any potential hydroxylation of IκBβ, indicating that the previously identified oxidation of D195 likely occurred by random reaction with oxygen during sample preparation and not by enzymatic activity.Citation12 In addition, IP of FIH pulled down the FIH-IκBβ oxomer, but not the IκBβ monomer. Together, these results strongly indicate that IκBβ is not hydroxylated by FIH. Previously, in an in vitro decarboxylation assay using purified FIH and IκBβ, no FIH-dependent activity toward IκBβ was detected,Citation17 further supporting that IκBβ is not a hydroxylation substrate of FIH. Nonetheless, the results obtained by the in vitro assay are not contradicting our observation of the FIH-IκBβ oxomer formation, because such oxomers are likely cotranslationally formed.Citation26 Assuming that also the FIH-IκBβ oxomer is formed cotranslationally, such a formation would not be detected by an assay that utilizes fully folded proteins.
Analyzing possible functional effects of the FIH-IκBβ oxomer, the FIH-dependent regulation of HIF transactivation activity appeared to be unaffected in a HIF-dependent reporter gene assay. In the same experiment, ectopic expression of IκBα and IκBε increased HIF-dependent firefly luciferase activity, which is in agreement with a previous report that IκBα can inhibit FIH activity toward HIF-1α.Citation52 To our knowledge, this is the first report that also IκBε – but no other IκB protein family member – affects HIF activity.
The main function of IκBβ is to bind to NF-κB transcription factor dimers (mostly containing c-Rel and p65) and to retain them in the cytosol, preventing gene expression enhancement by these specific NF-κB dimers.Citation42 It is therefore of interest that IκBβ complexed with FIH as oxomer did not retain its ability to interact with the NF-κB subunits p65 and c-Rel. Consequently, oxomer formation may increase NF-κB-dependent transcription by preventing p65 and c-Rel binding to IκBβ. Such a regulation would be specific to NF-κB dimers involving p65 and/or c-Rel and would thus not necessarily affect the overall NF-κB-dependent gene expression, as 13 different NF-κB dimers have (so far) been demonstrated to exist.Citation53 Further investigations will be necessary to determine the extent of the contribution of FIH-IκBβ oxomer formation to the regulation of pro-inflammatory gene expression via inhibition of IκBβ binding to NF-κB dimers.
Recently, it has been reported that FIH expression is increased in inflamed renal tissue during chronic kidney disease (CKD).Citation54 It would therefore be of interest to test for the existence and potential regulation of the FIH-IκBβ oxomer in healthy and diseased renal tissues, as an enhanced FIH-IκBβ oxomer formation may contribute to increased inflammation. However, it is currently unclear to which extent oxomers exist in healthy or diseased tissues.
In summary, FIH forms a previously unknown oxomer with IκBβ in a likely covalent manner and with high hypoxia sensitivity. FIH-mediated sequestration of IκBβ within an oxomer does not affect the total FIH enzymatic activity present within a cell, but it prevents the complexed IκBβ from inhibiting specific NF-κB dimers. Therefore, the FIH-IκBβ oxomer formation and/or its hypoxic inhibition may contribute to the interplay between hypoxia/FIH and inflammation.
Materials and Methods
Cell culture and transient transfection
Human HEK293 (embryonic kidney), HepG2 and Hep3B (hepatocellular carcinoma), and MCF7 (breast adenocarcinoma) cell lines were cultivated in high glucose Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, USA) with supplementation of 100 U/mL penicillin and 100 μg/mL streptomycin (Sigma-Aldrich) as well as 10% heat-inactivated fetal calf serum (Gibco, USA). Transient transfection of plasmids was performed with lipofectamine 2000 reagent according to the manufacturers’ protocol (Invitrogen, USA).
RNA isolation and RT-qPCR
RNA was extracted by the guanidinium thiocyanate-acid phenol-chloroform method as described earlier.Citation55 Complementary DNA (cDNA) was obtained by reverse transcription (RT) with AffinityScript transcriptase (Agilent, Santa Clara, CA, USA) and quantitated using the SYBR Green qPCR reagent kit (Kapa Biosystems, London, UK) and a MX3000P light cycler (Agilent) using standard curves. Transcript levels were normalized to the levels of human ribosomal protein L28 mRNA. Primer sequences are listed in Supplemental Table S2.
Cell treatment
Cells were treated with the following compounds dissolved in dimethylsulfoxide (DMSO, Sigma-Aldrich)Citation56 and the final concentrations indicated: dimethyloxalylglycine (DMOG; Frontier Scientific, Logan, UT, USA; 1 mM), FG-4592 (roxadustat; Selleckchem, Houston, TX, USA; 0.1 mM) and dimethyl N-oxalyl-D-phenylalanine (DM-NOFD; Sigma-Aldrich; 1 mM). Desferrioxamine (DFX; Sigma-Aldrich; 0.1 mM) was dissolved in bidistilled water. MG132 (Sigma-Aldrich; 10 μM) was dissolved in ethanol (Sigma-Aldrich). To test the sensitivity of the oxomer against reducing agents, cell lysates were treated with dithiothreitol (DTT; Agilent Technologies) or β-mercaptoethanol (Sigma-Aldrich).
Incubations in hypoxia were performed using the InvivO2 400 humidified cell culture workstation (Baker Ruskinn, Bridgend, South Wales, UK) operated with 0.2% O2 or 1% O2, 5% CO2 at 37 °C as previously described, or in humidified oxygen-regulated cell culture incubators (Binder, Tuttlingen, Germany) operated with 2–8% O2 and 5% CO2 at 37 °C.Citation57 ”normoxia” refers to the air oxygen level in the gas phase within a humidified cell culture incubator at 500 m altitude (18.5% O2).Citation58
Reporter gene assays
HEK293 cells were cotransfected with pH3SVL, encoding the firefly (Photinus pyralis) luciferase reporter gene under the control of a SV40 promoter and concatamerized transferrin-derived hypoxia response elements (HREs) (total of six HIF binding sites) and pRL-SV40, encoding a constitutively expressed Renilla (Renilla reniformis) luciferase reporter construct driven by the SV40 promoter, in combination with a member of the IκB protein family (IκBα, IκBβ, Bcl-3, IκBδ, IκBε, p105, p100) or empty vector.Citation40,Citation41 Twenty-four hours after transfection, cells were treated with 0.1 mM prolyl hydroxylase inhibitor FG-4592 (roxadustat) or vehicle (DMSO) for 24 h. Dual-Luciferase Reporter Assay was used to determine firefly and Renilla luciferase bioluminescence according to the manufacturers’ instruction (Promega, Madison, Wisconsin, USA). In brief, cells were washed with PBS and lysed with Passive Lysis Buffer (Promega) for 10 min at room temperature. Lysates were mixed with equal volumes of Luciferase Assay Reagent II and luminescence was measured by a microplate luminometer (Berthold Technologies, Bad Wildbach, Germany). Subsequently, freshly combined Stop & Glo reagent (Promega) was added and the activity of Renilla luciferase was determined.
Protein structure prediction
Predictions of protein structures were created with ColabFold using the default settings, which resulted in five structure predictions, ranked by Predicted Aligned Error (PAE).Citation59 Rank 1 model was used for further analysis and representation (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb).
Statistical analysis
For the analysis of the significance of difference between two data points, Student’s t test was applied. For comparison of more than two data points, one-way or two-way ANOVA followed by Tukey’s post-test was applied. P values < 0.05 were considered statistically significant. The sample sizes for each experiment have been indicated in the corresponding figure legends.
Supplemental Material
Download Zip (3.5 MB)Acknowledgments
We thank Prof. Daniel J. Peet (Department of Molecular and Biomedical Science, University of Adelaide, Australia) for the gifts of plasmids and Dr. Anja Bremm (Institute of Biochemistry II, Goethe-University, Frankfurt am Main, Germany) for providing FIH KO HEK293 cells.
Disclosure statement
The authors report there are no competing interests to declare.
Data availability statement
All underlying raw data have been made available on https://dataverse.harvard.edu/. The data has obtained the DOI 10.7910/DVN/XYXNPA and is accessible under https://dataverse.harvard.edu/privateurl.xhtml?token=29762520-1072-418d-873a-5a110caa04cf. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDECitation60 partner repository with the dataset identifier PXD047442.
Additional information
Funding
References
- Taylor CT, Scholz CC. The effect of HIF on metabolism and immunity. Nat Rev Nephrol. 2022;18:573–587. doi:10.1038/s41581-022-00587-8.
- Bishop T, Ratcliffe PJ. Signaling hypoxia by hypoxia-inducible factor protein hydroxylases: a historical overview and future perspectives. Hypoxia (Auckl). 2014;2:197–213. doi:10.2147/HP.S47598.
- Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686. doi:10.1101/gad.924501.
- Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–1471. doi:10.1101/gad.991402.
- Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277:26351–26355. doi:10.1074/jbc.C200273200.
- Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858–861. doi:10.1126/science.1068592.
- Kaelin WG, Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi:10.1016/j.molcel.2008.04.009.
- Chan MC, Holt-Martyn JP, Schofield CJ, Ratcliffe PJ. Pharmacological targeting of the HIF hydroxylases-A new field in medicine development. Mol Aspects Med. 2016;47-48:54–75. doi:10.1016/j.mam.2016.01.001.
- Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol. 2017;17:774–785. doi:10.1038/nri.2017.103.
- Faivre A, Scholz CC, de Seigneux S. Hypoxia in chronic kidney disease: towards a paradigm shift? Nephrol Dial Transplant. 2021;36:1782–1790. doi:10.1093/ndt/gfaa091.
- Eltzschig HK, Bratton DL, Colgan SP. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov. 2014;13:852–869. doi:10.1038/nrd4422.
- Scholz CC, Cavadas MAS, Tambuwala MM, Hams E, Rodríguez J, von Kriegsheim A, Cotter P, Bruning U, Fallon PG, Cheong A, et al. Regulation of IL-1beta-induced NF-kappaB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc Natl Acad Sci U S A. 2013;110:18490–18495. doi:10.1073/pnas.1309718110.
- Tambuwala MM, Cummins EP, Lenihan CR, Kiss J, Stauch M, Scholz CC, Fraisl P, Lasitschka F, Mollenhauer M, Saunders SP, et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology. 2010;139:2093–2101. doi:10.1053/j.gastro.2010.06.068.
- Schützhold V, Gravemeyer J, Bicker A, Hager T, Padberg C, Schäfer J, Wrobeln A, Steinbrink M, Zeynel S, Hankeln T, et al. Knockout of Factor-Inhibiting HIF (Hif1an) in Colon Epithelium Attenuates Chronic Colitis but Does Not Reduce Colorectal Cancer in Mice. J Immunol. 2022;208:1280–1291. doi:10.4049/jimmunol.2100418.
- Yang YW, Meng X, Meng YY, Tang HK, Cheng MH, Zhang ZQ, Xu WQ, Long W. ceRNA regulatory network of FIH inhibitor as a radioprotector for gastrointestinal toxicity by activating the HIF-1 pathway. Mol Ther Nucleic Acids. 2021;25:173–185. doi:10.1016/j.omtn.2021.05.008.
- Volkova YL, Pickel C, Jucht AE, Wenger RH, Scholz CC. The Asparagine Hydroxylase FIH: A Unique Oxygen Sensor. Antioxid Redox Signal. 2022;37:913–935. doi:10.1089/ars.2022.0003.
- Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML, Coles CH, Yu X, Hay RT, Ley SC, et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc Natl Acad Sci U S A. 2006;103:14767–14772. doi:10.1073/pnas.0606877103.
- Mulero MC, Huxford T, Ghosh G, kappaB NF. IkappaB, and IKK: integral components of immune system signaling. Adv Exp Med Biol. 2019;1172:207–226. doi:10.1007/978-981-13-9367-9_10.
- Hinz M, Arslan SC, Scheidereit C. It takes two to tango: IkappaBs, the multifunctional partners of NF-kappaB. Immunol Rev. 2012;246:59–76. doi:10.1111/j.1600-065X.2012.01102.x.
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi:10.1146/annurev.immunol.16.1.225.
- DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–1304. doi:10.1128/MCB.16.4.1295.
- Cockman ME, Webb JD, Kramer HB, Kessler BM, Ratcliffe PJ. Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol Cell Proteomics. 2009;8:535–546. doi:10.1074/mcp.M800340-MCP200.
- Cockman ME, Webb JD, Ratcliffe PJ. FIH-dependent asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Ann N Y Acad Sci. 2009;1177:9–18. doi:10.1111/j.1749-6632.2009.05042.x.
- Devries IL, Hampton-Smith RJ, Mulvihill MM, Alverdi V, Peet DJ, Komives EA. Consequences of IkappaB alpha hydroxylation by the factor inhibiting HIF (FIH). FEBS Lett. 2010;584:4725–4730. doi:10.1016/j.febslet.2010.10.060.
- Scholz CC, Rodriguez J, Pickel C, Burr S, Fabrizio JA, Nolan KA, Spielmann P, Cavadas MA, Crifo B, Halligan DN, et al. FIH regulates cellular metabolism through hydroxylation of the deubiquitinase OTUB1. PloS Biol. 2016;14:e1002347. doi:10.1371/journal.pbio.1002347.
- Pickel C, Günter J, Ruiz-Serrano A, Spielmann P, Fabrizio J-A, Wolski W, Peet DJ, Wenger RH, Scholz CC. Oxygen-dependent bond formation with FIH regulates the activity of the client protein OTUB1. Redox Biol. 2019;26:101265. doi:10.1016/j.redox.2019.101265.
- Sulser P, Pickel C, Günter J, Leissing TM, Crean D, Schofield CJ, Wenger RH, Scholz CC. HIF hydroxylase inhibitors decrease cellular oxygen consumption depending on their selectivity. FASEB J. 2020;34:2344–2358. doi:10.1096/fj.201902240R.
- Groen EJN, Gillingwater TH. UBA1: at the crossroads of ubiquitin homeostasis and neurodegeneration. Trends Mol Med. 2015;21:622–632. doi:10.1016/j.molmed.2015.08.003.
- Kumbhar R, Vidal-Eychenié S, Kontopoulos DG, Larroque M, Larroque C, Basbous J, Kossida S, Ribeyre C, Constantinou A. Recruitment of ubiquitin-activating enzyme UBA1 to DNA by poly(ADP-ribose) promotes ATR signalling. Life Sci Alliance. 2018;1:e201800096. doi:10.26508/lsa.201800096.
- Chan MC, Ilott NE, Schödel J, Sims D, Tumber A, Lippl K, Mole DR, Pugh CW, Ratcliffe PJ, Ponting CP, et al. Tuning the transcriptional response to hypoxia by inhibiting hypoxia-inducible factor (HIF) prolyl and asparaginyl hydroxylases. J Biol Chem. 2016;291:20661–20673. doi:10.1074/jbc.M116.749291.
- Yeh TL, Leissing TM, Abboud MI, Thinnes CC, Atasoylu O, Holt-Martyn JP, Zhang D, Tumber A, Lippl K, Lohans CT, et al. Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials. Chem Sci. 2017;8:7651–7668. doi:10.1039/c7sc02103h.
- Lancaster DE, McNeill LA, McDonough MA, Aplin RT, Hewitson KS, Pugh CW, Ratcliffe PJ, Schofield CJ. Disruption of dimerization and substrate phosphorylation inhibit factor inhibiting hypoxia-inducible factor (FIH) activity. Biochem J. 2004;383:429–437. doi:10.1042/BJ20040735.
- Wensien M, von Pappenheim FR, Funk LM, Kloskowski P, Curth U, Diederichsen U, Uranga J, Ye J, Fang P, Pan KT, et al. A lysine-cysteine redox switch with an NOS bridge regulates enzyme function. Nature. 2021;593:460–464. doi:10.1038/s41586-021-03513-3.
- Linke S, Hampton-Smith RJ, Peet DJ. Characterization of ankyrin repeat-containing proteins as substrates of the asparaginyl hydroxylase factor inhibiting hypoxia-inducible transcription factor. Methods Enzymol. 2007;435:61–85. doi:10.1016/S0076-6879(07)35004-0.
- Anonymous. AlphaFold Protein Structure Database. Available. From: https://alphafold.ebi.ac.uk/.
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi:10.1038/s41586-021-03819-2.
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50:D439–D444. doi:10.1093/nar/gkab1061.
- Yang M, Ge W, Chowdhury R, Claridge TD, Kramer HB, Schmierer B, McDonough MA, Gong L, Kessler BM, Ratcliffe PJ, et al. Asparagine and aspartate hydroxylation of the cytoskeletal ankyrin family is catalyzed by factor-inhibiting hypoxia-inducible factor. J Biol Chem. 2011;286:7648–7660. doi:10.1074/jbc.M110.193540.
- Rodriguez J, Pilkington R, Garcia Munoz A, Nguyen LK, Rauch N, Kennedy S, Monsefi N, Herrero A, Taylor CT, von Kriegsheim A. Substrate-trapped interactors of PHD3 and FIH cluster in distinct signaling pathways. Cell Rep. 2016;14:2745–2760. doi:10.1016/j.celrep.2016.02.043.
- Rolfs A, Kvietikova I, Gassmann M, Wenger RH. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J Biol Chem. 1997;272:20055–20062. doi:10.1074/jbc.272.32.20055.
- Wanner RM, Spielmann P, Stroka DM, Camenisch G, Camenisch I, Scheid A, Houck DR, Bauer C, Gassmann M, Wenger RH. Epolones induce erythropoietin expression via hypoxia-inducible factor-1 alpha activation. Blood. 2000;96:1558–1565.
- Kerr LD, Inoue J, Davis N, Link E, Baeuerle PA, Bose HR, Jr., Verma IM. The rel-associated pp40 protein prevents DNA binding of Rel and NF-kappa B: relationship with I kappa B beta and regulation by phosphorylation. Genes Dev. 1991;5:1464–1476. doi:10.1101/gad.5.8.1464.
- Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol. 2008;586:4055–4059. doi:10.1113/jphysiol.2008.157669.
- Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7:281–287. doi:10.1038/nrgastro.2010.39.
- Scholz CC, Taylor CT. Hydroxylase-dependent regulation of the NF-kappaB pathway. Biol Chem. 2013;394:479–493. doi:10.1515/hsz-2012-0338.
- Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–665. doi:10.1056/NEJMra0910283.
- Thompson JE, Phillips RJ, Erdjument-Bromage H, Tempst P, Ghosh S. I kappa B-beta regulates the persistent response in a biphasic activation of NF-kappa B. Cell. 1995;80:573–582. doi:10.1016/0092-8674(95)90511-1.
- Weil R, Laurent-Winter C, Israël A. Regulation of IkappaBbeta degradation. Similarities to and differences from IkappaBalpha. J Biol Chem. 1997;272:9942–9949. doi:10.1074/jbc.272.15.9942.
- Tian YM, Yeoh KK, Lee MK, Eriksson T, Kessler BM, Kramer HB, Edelmann MJ, Willam C, Pugh CW, Schofield CJ, et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J Biol Chem. 2011;286:13041–13051. doi:10.1074/jbc.M110.211110.
- Yang M, Chowdhury R, Ge W, Hamed RB, McDonough MA, Claridge TD, Kessler BM, Cockman ME, Ratcliffe PJ, Schofield CJ. Factor-inhibiting hypoxia-inducible factor (FIH) catalyses the post-translational hydroxylation of histidinyl residues within ankyrin repeat domains. FEBS J. 2011;278:1086–1097. doi:10.1111/j.1742-4658.2011.08022.x.
- Kang J, Chun YS, Huh J, Park JW. FIH permits NAA10 to catalyze the oxygen-dependent lysyl-acetylation of HIF-1alpha. Redox Biol. 2018;19:364–374. doi:10.1016/j.redox.2018.09.002.
- Shin DH, Li SH, Yang SW, Lee BL, Lee MK, Park JW. Inhibitor of nuclear factor-kappaB alpha derepresses hypoxia-inducible factor-1 during moderate hypoxia by sequestering factor inhibiting hypoxia-inducible factor from hypoxia-inducible factor 1alpha. FEBS J. 2009;276:3470–3480. doi:10.1111/j.1742-4658.2009.07069.x.
- Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell. 2017;168:37–57. doi:10.1016/j.cell.2016.12.012.
- Faivre A, Dissard R, Kuo W, Verissimo T, Legouis D, Arnoux G, Heckenmeyer C, Fernandez M, Tihy M, Rajaram RD, et al. Evolution of hypoxia and hypoxia-inducible factor asparaginyl hydroxylase (FIH) regulation in chronic kidney disease. Nephrol Dial Transplant. 2023;38:2276–2288. doi:10.1093/ndt/gfad075.
- Ruiz-Serrano A, Monné Rodríguez JM, Günter J, Sherman SPM, Jucht AE, Fluechter P, Volkova YL, Pfundstein S, Pellegrini G, Wagner CA, et al. OTUB1 regulates lung development, adult lung tissue homeostasis, and respiratory control. FASEB J. 2021;35:e22039. doi:10.1096/fj.202100346R.
- Pickel C, Taylor CT, Scholz CC. Genetic Knockdown and Pharmacologic Inhibition of Hypoxia-Inducible Factor (HIF) Hydroxylases. Methods Mol Biol. 2018;1742:1–14. doi:10.1007/978-1-4939-7665-2_1.
- Stiehl DP, Wirthner R, Köditz J, Spielmann P, Camenisch G, Wenger RH. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen-sensing system. J Biol Chem. 2006;281:23482–23491. doi:10.1074/jbc.M601719200.
- Wenger RH, Kurtcuoglu V, Scholz CC, Marti HH, Hoogewijs D. Frequently asked questions in hypoxia research. Hypoxia. 2015;3:35–43. doi:10.2147/HP.S92198.
- Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods. 2022;19:679–682. doi:10.1038/s41592-022-01488-1.
- Perez-Riverol Y, Bai J, Bandla C, García-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, Eisenacher M, et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi:10.1093/nar/gkab1038.