
Abstract
Background
Longitudinal changes in neurofilament light chain (NfL) levels were evaluated alongside prespecified clinical assessments 24 months into the patisiran Global open-label extension (OLE) study in patients with ATTRv amyloidosis with polyneuropathy.
Methods
All patients enrolled in the Global OLE, from phase III APOLLO and phase II OLE parent studies, received patisiran. Assessments included measures of polyneuropathy (modified Neuropathy Impairment Score+7 (mNIS+7)), quality of life (QOL; Norfolk QOL-Diabetic Neuropathy questionnaire (Norfolk QOL-DN)), and plasma NfL.
Results
Patients receiving patisiran in the parent study (APOLLO-patisiran, n = 137; phase II OLE-patisiran, n = 25) demonstrated sustained improvements in mNIS+7 (mean change from parent study baseline (95% confidence interval): APOLLO-patisiran −4.8 (−8.9, −0.6); phase II OLE-patisiran −5.8 (−10.5, −1.2)) and Norfolk QOL-DN (APOLLO-patisiran −2.4 (−7.2, 2.3)), and maintained reduced NfL levels at Global OLE 24 months. After initiating patisiran in the Global OLE, APOLLO-placebo patients (n = 49) demonstrated stabilized mNIS+7, improved Norfolk QOL-DN, and significantly reduced NfL levels. Patisiran continued to demonstrate an acceptable safety profile. Earlier patisiran initiation was associated with a lower exposure-adjusted mortality rate.
Conclusions
Long-term patisiran treatment led to sustained improvements in neuropathy and QOL, with NfL demonstrating potential as a biomarker for disease progression and treatment response in ATTRv amyloidosis with polyneuropathy.
Introduction
Hereditary transthyretin-mediated amyloidosis, also known as ATTRv (hereditary transthyretin (v for variant)) amyloidosis, is a rapidly progressive and fatal disease caused by variants in the transthyretin (TTR) gene [Citation1–3]. Misfolded variant and wild-type TTR accumulate as amyloid deposits in multiple tissues, including the nerves, heart, and gastrointestinal tract [Citation1,Citation3], and the majority of patients develop a mixed phenotype of polyneuropathy and cardiomyopathy [Citation4–7].
ATTRv amyloidosis is associated with substantial morbidity and mortality, yet diagnosis is difficult and often delayed [Citation8,Citation9]. As approved therapies appear to be more effective when given early [Citation10,Citation11], the need for earlier diagnosis is apparent. Further to the diagnostic challenges, monitoring disease progression and treatment response is also difficult, due to the multisystem nature and heterogeneous clinical presentation [Citation12–16]. Hence, identification of reliable biomarkers for ATTRv amyloidosis would be of benefit.
Elevated plasma neurofilament light chain (NfL) levels in diseases presenting with neuropathy are presumed to result from NfL release following neuroaxonal injury [Citation17–24], making NfL a biomarker for nerve damage [Citation25,Citation26]. NfL levels are elevated in patients with ATTRv amyloidosis [Citation27,Citation28], most likely due to neuronal injury or degeneration resulting from deposition of TTR amyloid fibrils [Citation29].
In the phase III APOLLO study in patients with ATTRv amyloidosis with polyneuropathy, the RNA interference (RNAi) therapeutic patisiran demonstrated improvements in the primary endpoint of neuropathy, measured by modified Neuropathy Impairment Score + 7 (mNIS+7) [Citation30], and all secondary endpoints, including quality of life (QOL; Norfolk Quality of Life-Diabetic Neuropathy questionnaire (Norfolk QOL-DN)), vs. placebo [Citation6]. During APOLLO, patisiran treatment also led to a decrease in NfL levels from baseline over 18 months, whereas an increase was seen with placebo [Citation28]. Notably, changes in mNIS+7 in APOLLO were significantly correlated with changes in NfL levels, suggesting that NfL may act as a biomarker in ATTRv amyloidosis [Citation28].
The ongoing patisiran Global open-label extension (OLE) study [Citation10], which enrolled eligible patients from APOLLO [Citation6] and the patisiran phase II OLE study [Citation31], is evaluating the long-term safety and efficacy of patisiran. The first interim data analysis at 12 months has been presented previously [Citation10]. Here we present the second interim analysis at 24 months, together with an evaluation of longitudinal changes in NfL levels during patisiran treatment.
Materials and methods
The primary objective of the current analysis was to present updated efficacy and safety results at 24 months in the patisiran Global OLE study. In addition, an analysis of NfL levels was undertaken to: confirm the previously observed decrease in NfL levels upon patisiran treatment; describe longitudinal changes in NfL, alongside clinical assessments, with long-term patisiran treatment; and better understand the temporal relationship between NfL lowering and patisiran treatment.
Trial design
The patisiran Global OLE (NCT02510261) is an ongoing, international, multicenter trial [Citation10], which enrolled patients with ATTRv amyloidosis with polyneuropathy who had completed the phase III APOLLO (NCT01960348) [Citation6] or phase II OLE (NCT01961921) [Citation31] parent studies, and, in the opinion of the investigator, tolerated the study drug. Methods for the Global OLE study have been reported previously [Citation10] and are summarized below. In the Global OLE study, patients will receive patisiran 0.3 mg/kg intravenously every 3 weeks for up to 5 years, although patients had not completed the full duration of the study at the time of this 24-month interim analysis.
Measures
In the Global OLE study, mNIS+7, Norfolk QOL-DN, and polyneuropathy disability (PND) scores were assessed at Global OLE baseline, 12 months, and 24 months. mNIS+7 is a composite clinical measure of motor, sensory, and autonomic neuropathy, which uses standardized assessments to quantify nerve conduction, muscle weakness, muscle stretch reflexes, sensory loss, and autonomic impairment (scored 0 − 304; higher scores indicate greater impairment) [Citation32]. Norfolk QOL-DN is a 35-item, patient-reported questionnaire with five domains comprizing activities of daily living, physical functioning/large-fiber neuropathy, small-fiber neuropathy, autonomic neuropathy, and symptoms (scored −4 to 136; higher scores indicate lower QOL) [Citation33]. PND score, which is based on ambulatory function, is also used to assess neuropathy in patients with ATTRv amyloidosis (scored I, II, IIIa, IIIb, and IV; higher scores indicate more limited ambulatory function).
NfL levels were measured at 12 and 24 months from Global OLE baseline. NfL plasma measurements were done in duplicate at a central location (PPD Bioanalytical Lab, Richmond, VA, USA) using the Quanterix Simoa platform. NfL plasma level assessment is a post hoc exploratory analysis in the Global OLE.
Patients returned to the clinical site for safety evaluations, including laboratory assessments, at 12, 26, and 52 weeks after the first patisiran dose in the Global OLE study and yearly thereafter. Patients were continuously assessed for adverse events (AEs).
Statistical analysis
The sample size of this study was not prespecified [Citation10]. Patients who enrolled in the Global OLE are presented in groups based on their participation in the parent studies: the APOLLO-patisiran group comprizes patients who received patisiran for 18 months in APOLLO and continue receiving patisiran in the Global OLE, the APOLLO-placebo group comprizes patients who started receiving patisiran for the first time in the Global OLE, and the phase II OLE-patisiran group comprizes patients who received patisiran for 24 months in the phase II OLE and continue receiving patisiran in the Global OLE.
mNIS+7 and Norfolk QOL-DN efficacy data are reported as an integrated analysis including all patients who had ≥1 dose of study treatment in the parent studies and had an efficacy assessment at a given timepoint. Data for the Global OLE 24-month timepoint are given for patients who had completed the 24-month efficacy assessment by the 24-month interim cut-off for the Global OLE on 7 October 2019. mNIS+7 and Norfolk QOL-DN are described as change from parent study or Global OLE baseline.
NfL levels were analysed in a subset of consenting patients who had received ≥ 1 dose of patisiran and had available samples [Citation28]. NfL data are described as plasma levels or change in plasma levels from parent study or Global OLE baseline.
Improvement in mNIS+7 score, Norfolk QOL-DN score, and NfL levels was defined as a negative mean change from the baseline value. Unless otherwise stated, baseline was defined as the initiation of patisiran treatment.
A safety analysis for the total Global OLE population is presented. A post hoc analysis assessed integrated exposure-adjusted mortality rate from the onset of patisiran treatment in each group, using pooled data from the parent and Global OLE studies. The integrated exposure-adjusted mortality rate was calculated overall and by treatment group as the total number of deaths per total patient-years of exposure multiplied by 100. Deaths occurring between the first patisiran dose and up to 90 days after the last dose were included in the calculation.
Number and percentage of patients were reported for categoric variables. Mean (standard deviation (SD)) or median (interquartile range) were calculated for continuous variables. Comparisons of NfL levels between groups were tested for significance using linear regression, controlling for age, using R (R Foundation, Vienna, Austria). Sex was not found to affect NfL levels and was not included in the regression model. Comparisons of NfL levels in a given group at different timepoints were tested for significance using a Wilcoxon signed-rank test. Statistical analyses of efficacy data were made using SAS, version 9.4 (SAS Institute Inc, Cary, NC, USA).
The study protocol is publicly available [Citation34].
Results
Patients
The study population for this analysis comprized a subset of patients from the Global OLE and its parent studies; details of enrolment have been described previously [Citation10]. Of the 148 APOLLO patients treated with patisiran, 138 (93.2%) completed the parent study and 137 enrolled in the Global OLE (APOLLO-patisiran group). Of the 77 patients treated with placebo in APOLLO, 55 (71.4%) completed the parent study and 49 enrolled in the Global OLE (APOLLO-placebo group) (). There were 27 patients treated with patisiran in the phase II OLE study, 25 of whom completed the study and enrolled in the Global OLE (). At the 24-month data cut-off (7 October 2019), 169 of 211 (80.1%) patients remained on patisiran treatment in the Global OLE study. Across the parent and Global OLE studies, 224 patients had received patisiran for a mean of 43.3 months and had a cumulative drug exposure of 808.5 patient-years. The reasons for study withdrawal up to the 24-month data cut-off in the Global OLE were death (26/211 (12.3%)), AEs (10/211 (4.7%)), patient decision (3/211 (1.4%)), and physician decision (3/211 (1.4%)).
Figure 1. Patient disposition. AE: adverse event; OLE: open-label extension.
At parent study baseline, patients included in the NfL analysis in each patient group () had similar characteristics to the total population of the respective patient group [Citation31,Citation35].
Table 1. Demographics and disease characteristics at parent study baseline of patients with NfL measurements.
Efficacy endpoints
The Global OLE 24-month efficacy analyses were prespecified. Assessments for mNIS+7 and Norfolk QOL-DN were available for at least 100/137 (73.0%) patients from the APOLLO-patisiran group and 24/49 (49.0%) patients from the APOLLO-placebo group; only mNIS+7 was assessed and available for 16/25 (64.0%) patients from the phase II OLE-patisiran group.
At Global OLE 24 months, APOLLO-patisiran and phase II OLE-patisiran groups demonstrated durable improvement in neuropathy vs. parent study baselines, indicated by negative mean changes (95% confidence interval (CI)) from parent study baseline in mNIS+7 of −4.8 (−8.9, −0.6) and −5.8 (−10.5, −1.2), respectively ().
Figure 2. Integrated changes in mNIS+7 during the parent studies and the Global OLE. (A) APOLLO. (B) Phase II OLE. Data are integrated change from parent study baseline in mNIS+7. (C) Integrated change in Norfolk QOL-DN score during the parent studies and the Global OLE from the APOLLO study. aFor APOLLO patients initiating alternative ATTRv amyloidosis treatment, mNIS+7 assessments after alternative treatment are treated as missing. The APOLLO-placebo arm began patisiran treatment at the start of the Global OLE. APOLLO mNIS+7 parent study baseline (mean (SD)): APOLLO-placebo = 74.6 (37.0); APOLLO-patisiran = 80.9 (41.5) [Citation6]. bPhase II OLE mNIS+7 parent study baseline (mean (SD)): 53.0 (35.6). cData are integrated change from parent study baseline in Norfolk QOL-DN. The APOLLO-placebo arm began patisiran treatment at the start of the Global OLE. APOLLO Norfolk QOL-DN parent study baseline (mean (SD)): APOLLO-placebo = 55.5 (24.3); APOLLO-patisiran = 59.6 (28.2) [Citation6]. ATTRv: hereditary transthyretin (v for variant); BL: baseline; CI: confidence interval; mNIS+7: modified Neuropathy Impairment Score + 7; Norfolk QOL-DN: Norfolk Quality of Life-Diabetic Neuropathy questionnaire; OLE: open-label extension; SD: standard deviation.
![Figure 2. Integrated changes in mNIS+7 during the parent studies and the Global OLE. (A) APOLLO. (B) Phase II OLE. Data are integrated change from parent study baseline in mNIS+7. (C) Integrated change in Norfolk QOL-DN score during the parent studies and the Global OLE from the APOLLO study. aFor APOLLO patients initiating alternative ATTRv amyloidosis treatment, mNIS+7 assessments after alternative treatment are treated as missing. The APOLLO-placebo arm began patisiran treatment at the start of the Global OLE. APOLLO mNIS+7 parent study baseline (mean (SD)): APOLLO-placebo = 74.6 (37.0); APOLLO-patisiran = 80.9 (41.5) [Citation6]. bPhase II OLE mNIS+7 parent study baseline (mean (SD)): 53.0 (35.6). cData are integrated change from parent study baseline in Norfolk QOL-DN. The APOLLO-placebo arm began patisiran treatment at the start of the Global OLE. APOLLO Norfolk QOL-DN parent study baseline (mean (SD)): APOLLO-placebo = 55.5 (24.3); APOLLO-patisiran = 59.6 (28.2) [Citation6]. ATTRv: hereditary transthyretin (v for variant); BL: baseline; CI: confidence interval; mNIS+7: modified Neuropathy Impairment Score + 7; Norfolk QOL-DN: Norfolk Quality of Life-Diabetic Neuropathy questionnaire; OLE: open-label extension; SD: standard deviation.](/cms/asset/b4e102da-4738-41b2-a9af-270046e602c7/iamy_a_2232520_f0002_c.jpg)
Rapid polyneuropathy progression in the APOLLO-placebo group was observed during placebo treatment in the parent study and at Global OLE baseline. However, polyneuropathy progression in the APOLLO-placebo group was stabilized upon initiation of patisiran treatment in the Global OLE. This stabilization of polyneuropathy progression was sustained over 24 months of patisiran treatment in the Global OLE (mean change (95% CI) from Global OLE baseline 1.2 (–5.6, 8.0)). However, the mNIS+7 values for APOLLO-placebo patients remained worse than their parent study baseline at Global OLE 24 months, likely due to polyneuropathy progression while on placebo in APOLLO (mean change (95% CI) from APOLLO baseline 27.0 (17.0, 37.0)).
The majority of patients in the APOLLO-patisiran (53.4%) and phase II OLE-patisiran (66.7%) groups had an improved or stable PND score at Global OLE 24 months compared with parent study baselines (). In the APOLLO-placebo group, more patients with evaluable data had a worsened PND score (29.9%) than a stable score (14.3%) at Global OLE 24 months compared with APOLLO baseline, and no patients with evaluable data in this group had an improved PND score compared with APOLLO baseline. However, when compared with Global OLE baseline, the majority of the APOLLO-placebo patients had a stable or improved PND score (57.1%) at Global OLE 24 months, reflecting the effect of patisiran initiation on entry to the Global OLE ().
Table 2. Change in PND score from parent study baseline and Global OLE enrolment to Global OLE 24 months.
After 24 months of additional patisiran treatment in the Global OLE, durable improvement in Norfolk QOL-DN was observed in the APOLLO-patisiran group compared with the parent study baseline (mean change (95% CI) from APOLLO parent study baseline −2.4 (−7.2, 2.3); ). For patients in the APOLLO-placebo group, switching to patisiran treatment led to an improvement in Norfolk QOL-DN over 24 months in the Global OLE (mean change (95% CI) from Global OLE baseline −4.1 (–10.8, 2.6)). Similar to the mNIS+7 results, Norfolk QOL-DN scores in APOLLO-placebo patients did not return to their parent study baseline by Global OLE 24 months, likely due to progression on placebo during APOLLO (mean change (95% CI) from APOLLO baseline 15.9 (6.7, 25.0)).
NfL levels
The Global OLE NfL analyses were post hoc. At parent study baseline, phase II OLE-patisiran patients had significantly lower mean NfL levels than APOLLO patients (32.9 vs. 69.4 pg/mL, respectively; estimated difference (95% CI) −38.0 pg/mL (−54.5, −21.6)) (Supplementary Figure 1).
At APOLLO baseline, mean (SD) plasma NfL levels were comparable between the patisiran (72.0 (45.7) pg/mL) and placebo (63.2 (32.2) pg/mL) groups (estimated difference (95% CI) −7.4 pg/mL (−21.8, 6.9)). Similarly, assessment at Day 21 of the APOLLO study did not show a significant difference in NfL levels between the two groups (estimated difference (95% CI) 0.2 pg/mL (−6.9, 7.3)) (, ). A significant difference between mean (SD) NfL levels in the patisiran (51.6 (29.4) pg/mL) and placebo groups (82.2 (34.0) pg/mL; estimated difference (95% CI) 30.6 pg/mL (21.8, 42.0)) was first apparent at Day 126 of the APOLLO study (, ). In addition, there was also a significant reduction in NfL levels in the patisiran group compared with APOLLO baseline by Day 126 (mean change (95% CI) from APOLLO baseline −20.0 (–25.7, −14.4) pg/mL; p<.001). In contrast, there was a significant increase in NfL levels from baseline in the placebo group at the same timepoint (mean change (95% CI) from APOLLO baseline 19.0 (12.2, 25.8) pg/mL; p<.001).
Figure 3. Integrated change in NfL levels during the parent studies and the Global OLE. (A) APOLLO. (B) Phase II OLE. Data are integrated change from parent study baseline in NfL level. The APOLLO-placebo arm began patisiran treatment at the start of the Global OLE. BL: baseline; CI: confidence interval; NfL: neurofilament light chain; OLE: open-label extension.
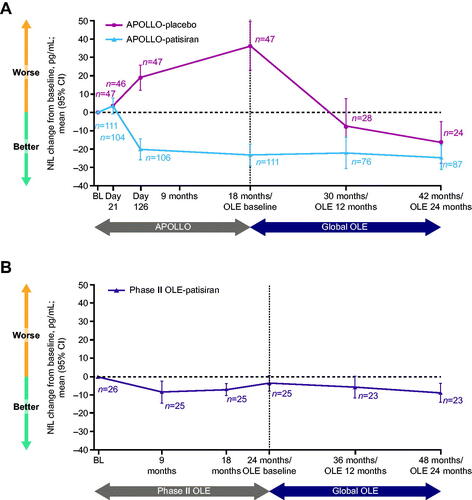
Table 3. Mean NfL levels (SD) in the APOLLO and Global OLE studies.
The significant decrease in NfL levels from baseline to Day 126 in the patisiran group of the APOLLO study was sustained at 18 months (mean change (95% CI) from APOLLO baseline −23.2 (–29.2, −17.2) pg/mL; p<.001), and there was a further increase in NfL levels from baseline in the placebo group at this timepoint (mean change (95% CI) from APOLLO baseline 36.3 (23.1, 49.5) pg/mL; p < .001). In the phase II OLE, NfL levels also decreased from baseline to 24 months with patisiran treatment (mean change (95% CI) from phase II OLE baseline −3.6 (–7.9, 0.6) pg/mL; , ).
Table 4. Mean NfL levels (SD) in the phase II OLE and Global OLE studies.
In the Global OLE, significant reductions in NfL levels from parent study baseline were maintained following an additional 24 months of patisiran treatment in both the APOLLO-patisiran (, ) (mean change (95% CI) from APOLLO baseline −24.7 (–31.4, −18.0) pg/mL; p<.001) and phase II OLE-patisiran (, ) groups (mean change (95% CI) from phase II OLE baseline −8.9 (–14.1, −3.7) pg/mL; p<.001). In the APOLLO-placebo group, following 12 months of patisiran treatment in the Global OLE, NfL levels were reduced from APOLLO baseline (mean change (95% CI) −7.6 (–22.6, 7.5) pg/mL) and significantly reduced from Global OLE baseline (mean change (95% CI) −37.5 (–22.0, −52.9) pg/mL; p<.001). There was continued significant reduction of NfL levels in the APOLLO-placebo group at 24 months in the Global OLE (mean change (95% CI) from APOLLO baseline −16.5 (–27.8, −5.3) pg/mL; p<.001) to reach a mean of 42.8 pg/mL, similar to the mean level in the APOLLO-patisiran group at 24 months in the Global OLE (44.1 pg/mL; ).
Global OLE safety
Safety of, and total exposure to, patisiran at 24 months in the Global OLE are summarized in . AEs were reported in 209/211 (99.1%) patients in the Global OLE, with the majority of AEs in the APOLLO-patisiran and phase II OLE-patisiran groups being mild or moderate in severity. Serious AEs were more common in the APOLLO-placebo group (34/49 (69.4%) patients with ≥ 1 event) compared with the APOLLO-patisiran group (59/137 (43.1%)) or phase II OLE-patisiran group (11/25 (44.0%)). The most common treatment-related AEs were mild or moderate infusion-related reactions (IRRs). The frequency of IRRs decreased over time, consistent with observations in APOLLO [Citation6], and there were no serious IRRs or discontinuations due to IRRs between the Global OLE baseline and 24-month data cut-off. Over the first 24 months of the Global OLE, the APOLLO-patisiran and APOLLO-placebo groups reported alanine aminotransferase elevation AEs in one (0.7%) patient and three (6.1%) patients, respectively, and aspartate aminotransferase elevation AEs in one (0.7%) patient and two (4.1%) patients, respectively. Neither AE was reported in the phase II OLE-patisiran group.
Table 5. Exposure and overall safety in the Global OLE.
Deaths were reported in 29 (13.7%) patients in the Global OLE, and all were considered unrelated to patisiran by the investigators; causes of death were consistent with the natural history of ATTRv amyloidosis. The proportion of deaths in the APOLLO-placebo group (16 (32.7%)) was higher than in the APOLLO-patisiran (13 (9.5%)) and phase II OLE-patisiran (zero) groups.
Throughout the parent studies and Global OLE, the exposure-adjusted mortality rate for patients who received ≥1 dose of patisiran was 4.3 per 100 patient-years (95% CI 3.1, 5.9), based on 35 deaths and 808.5 patient-years of cumulative exposure (), which is below the estimated range for patients with ATTRv amyloidosis (6.8 − 29 per 100 patient-years) [Citation36–40]. The exposure-adjusted mortality was lowest for patients in the phase II OLE, who were treated with patisiran for the longest duration and from the earliest stage of disease, and lower in the APOLLO-patisiran group compared with the APOLLO-placebo group.
Table 6. Integrated exposure-adjusted mortality rates in the parent and Global OLE studies.
Discussion
The second interim analysis of the Global OLE study presents the results at 24 months, including data from up to 48 months of patisiran treatment, which represents the longest treatment period reported to date for an RNAi therapeutic. These data indicate that the improvements in mNIS+7 and Norfolk QOL-DN scores seen with patisiran in parent studies were durable over 24 months of additional patisiran treatment. Furthermore, the overall safety of patisiran during this additional treatment period in the Global OLE was consistent with that previously reported up to 12 months [Citation10], and patisiran continued to demonstrate an acceptable safety profile at Global OLE 24 months.
Of interest for this post hoc assessment was the significant and sustained reduction in NfL levels observed with patisiran treatment in patients with ATTRv amyloidosis with polyneuropathy, supporting the potential utility of NfL as a biomarker for this disease.
In the APOLLO and phase II OLE studies, patisiran treatment lowered levels of NfL and led to improvements in neuropathy measured by mNIS+7 compared with their parent study baselines. Additionally, patisiran also improved QOL vs. parent study baseline, with changes in Norfolk QOL-DN score mirroring the pattern seen in the mNIS+7 results. The lowering of NfL levels, as well as the improvements in the clinical efficacy assessments of mNIS+7 and Norfolk QOL-DN, were sustained over an additional 24 months of treatment in the Global OLE study, demonstrating that the effect of patisiran on NfL levels corresponds with the clinical effectiveness of patisiran. In the Global OLE, initiation of patisiran in the APOLLO-placebo group also led to significant reduction in NfL levels and prevented further worsening of polyneuropathy and QOL, which had been seen during APOLLO [Citation6], further supporting an association between NfL levels and clinical outcomes with patisiran.
At parent study baseline, patients from both APOLLO groups had more severe disease and higher NfL levels compared with the phase II OLE-patisiran group. This observation supports the potential relationship between increasing disease severity (as measured by, for example, worse PND score) and higher NfL that was observed by Kapoor et al. and Maia et al. in ATTRv amyloidosis [Citation27,Citation41,Citation42], and suggests the possibility that NfL levels, in addition to being a marker of disease progression or treatment response, could also serve as an indicator of disease severity. An association between increased disease severity and higher NfL levels has also been observed in other neurologic diseases, such as Huntington’s disease [Citation20], chronic inflammatory demyelinating polyneuropathy [Citation22], and Charcot–Marie–Tooth disease [Citation24]. Interestingly, however, a prior analysis of the APOLLO data showed that there was no correlation between an individual’s NfL levels and mNIS+7 scores at study baseline, regardless of age adjustment [Citation28]. This lack of correlation may relate to differences in patient populations and/or outcomes assessed in previous studies on NfL in ATTRv amyloidosis compared with APOLLO, which included a broader range of TTR variants and patients at different disease stages [Citation6,Citation27,Citation41–43]. These different study groups may have included patients with involvement of varying nerve fibres, which could differentially affect NfL levels, as NfL is not equally expressed in all peripheral nerve types affected by ATTRv amyloidosis [Citation42]. In addition, correlations between NfL and PND score were assessed in two of the other studies that investigated NfL, rather than between NfL and mNIS+7 [Citation27,Citation42].
It is of interest that the pattern of change in NfL and the scores measuring polyneuropathy (mNIS+7) and QOL (Norfolk QOL-DN) demonstrate certain differences. For example, in the Global OLE, while initiation of patisiran treatment led to a clear reduction in NfL in the APOLLO-placebo group, there was a relatively small improvement in Norfolk QOL-DN, and mNIS+7 remained stable. These differences may be because NfL is a biologic indicator of active nerve damage, while scores such as mNIS+7 reflect the burden of cumulative nerve damage on the patient’s overall clinical picture. Thus, NfL may have the potential to serve as an ‘acute-phase’ marker of nerve damage in ATTRv amyloidosis. Indeed, it has been referred to as ‘the neurologist’s troponin’ and ‘the neurologist’s C-reactive protein’ in the literature [Citation44,Citation45]. Notably, the differences in the behaviours of different assessment methods to demonstrate treatment response indicate the value of using multiple measures to monitor a multisystem disease like ATTRv amyloidosis.
The decrease in NfL levels in APOLLO with patisiran treatment became significant between Day 21 and Day 126. Although this reduction in NfL levels occurred later than the reduction in TTR levels in the APOLLO study, in which the first documented TTR reduction took place around Week 3 of treatment in the patisiran group, eventually levels of both biomarkers demonstrated a similar trend, namely a significant decrease from baseline [Citation6]. Although RNAi has been shown to lead to a reduction in TTR levels within days, it takes several months for the beneficial effects of treatment to become clinically measurable [Citation46].
Following the relatively steep reduction in NfL levels in the early stage of treatment in the APOLLO-patisiran group, the subsequent decrease in NfL levels with continuing treatment became more gradual. Once NfL levels had decreased to a certain point, they appeared to remain stable over an additional treatment period, and during this follow-up period they did not decrease to levels seen in healthy controls [Citation28]. The NfL levels of patients in the APOLLO-placebo group decreased to a similar value to the APOLLO-patisiran group at Global OLE 24 months, but the overall clinical burden of the APOLLO-placebo group remained higher, as demonstrated by higher mNIS+7 and Norfolk QOL-DN scores. Of the three treatment groups, the NfL level at Global OLE 24 months was lowest in phase II OLE-patisiran patients, although they did not decrease to levels seen in healthy controls either [Citation28]. This suggests that the lowest level of NfL reached in patients with ATTRv amyloidosis receiving patisiran treatment may depend on pre-treatment levels of NfL and/or disease severity. It is unclear whether this lowest base level of NfL in patients with ATTRv amyloidosis is due to an ongoing, low level of axon injury or axon injury accumulated prior to treatment; for some patients, this may also represent central nervous system involvement. However, it is also possible that with longer-term continued patisiran treatment, NfL levels may decrease further towards those seen in healthy controls [Citation28].
The stabilization or improvement of neuropathy and QOL during long-term patisiran treatment indicates the value of early intervention. Importantly, the onset of treatment stabilized the deterioration in ambulation, which is part of the natural history of the disease [Citation47,Citation48]. These results are further supported by the post hoc analysis of mortality, in which the lowest exposure-adjusted mortality rates were observed in phase II OLE-patisiran patients, who were treated for the longest time and from the earliest stage of disease.
The results presented here collectively support the value of monitoring NfL levels in ATTRv amyloidosis, although some limitations of the study should be recognized. The healthy control group referenced was not age-, sex-, and race-matched to the phase II OLE-patisiran patients and no variables other than age were adjusted for when making between-group comparisons, which limits the comparisons made between both groups. Furthermore, although NfL is a promising potential biomarker for monitoring disease progression and treatment response, it is not specific to ATTRv amyloidosis. However, it may still have potential utility for clinicians treating the disease as either a marker of disease severity, a measure of disease progression, or a method of assessment of response to treatment. NfL testing could be especially valuable since existing assessment methods for polyneuropathy, such as mNIS+7 [Citation30], can be cumbersome to perform, require training, and some are not routinely used in clinical practice.
The results of this study suggest several promising areas for further investigation. An evaluation of potential correlations between NfL levels and baseline measures of disease (such as the mNIS+7 or PND score) may also be of interest. However, interpretation of such an analysis would be challenging given that NfL levels may reflect active or continuing neuronal damage at a single time point and mNIS+7 or PND scores would reflect the clinical impact of the cumulative nerve damage over time. It may also be useful to conduct studies to examine whether NfL can be used as a biomarker for early detection of disease onset in patients carrying TTR variants, enabling earlier intervention and potentially preventing polyneuropathy progression. Further studies to elucidate NfL levels in patients with ATTR amyloidosis with predominant cardiomyopathy could also be conducted. In addition, measuring NfL levels over time in patients who receive treatments for ATTR amyloidosis other than patisiran could provide insights into treatment response/non-response for these medications.
| Abbreviations | ||
| AE | = | adverse event |
| ATTRv | = | hereditary transthyretin-mediated (v for variant) |
| CI | = | confidence interval |
| IQR | = | interquartile range |
| IRR | = | infusion-related reaction |
| mNIS+7 | = | modified Neuropathy Impairment Score + 7 |
| NfL | = | neurofilament light chain |
| Norfolk QOL-DN | = | Norfolk Quality of Life-Diabetic Neuropathy questionnaire |
| OLE | = | open-label extension |
| PND | = | polyneuropathy disability |
| QOL | = | quality of life |
| RNAi | = | RNA interference |
| SAE | = | serious adverse event |
| SD | = | standard deviation |
| TTR | = | transthyretin |
Supplemental Material
Download MS Word (243.8 KB)Acknowledgments
The authors thank the patients and their families for their valued contribution to this study and the members of the patisiran Global OLE collaborators for their work on the study (patisiran Global OLE collaborators list is available here). This study was funded by Alnylam Pharmaceuticals (Cambridge, MA, USA). Medical writing support was provided by Ed Childs of Adelphi Communications (Macclesfield, UK), in accordance with Good Publication Practice guidelines, funded by Alnylam Pharmaceuticals.
Disclosure statement
ST, EA, JV, and PN are all employed by Alnylam Pharmaceuticals and report ownership of shares in Alnylam Pharmaceuticals. CH was employed by Alnylam Pharmaceuticals at the time of this analysis and reports ownership of shares in Alnylam Pharmaceuticals. MP has participated in clinical trials sponsored by Akcea, Alnylam Pharmaceuticals, and Pfizer, and has received consulting fees from Akcea, Alnylam Pharmaceuticals, Biogen-Idec, Pfizer, and Vertex Pharmaceutical. DA has participated in clinical trials sponsored by Akcea and Alnylam Pharmaceuticals, and has received consulting fees advisory from Alnylam Pharmaceuticals, Bridgebio, Pfizer, and AstraZeneca. TC has participated in clinical trials sponsored by Akcea, Alnylam Pharmaceuticals, Eidos, Ionis, Prothena, and Pfizer. MU reports grants, personal fees, and non-financial support from Alnylam Pharmaceuticals and Pfizer; grants from Prothena; and personal fees from Janssen Pharmaceutical K.K., outside the submitted work.
Data availability statement
Anonymized individual participant data that support these results would be made available in a secure-access environment 12 months after study completion and when the product and indication have been approved for no less than 12 months in the US and/or the EU.
Access will be provided contingent upon the approval of a research proposal and the execution of a data sharing agreement. Requests for access to data can be submitted via the website www.vivli.org.
Correction Statement
This article was originally published with errors, which have now been corrected in the online version. Please see Correction (http://dx.doi.org/10.1080/13506129.2023.2246797)
Additional information
Funding
References
- Adams D, Koike H, Slama M, et al. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15(7):387–404. doi: 10.1038/s41582-019-0210-4.
- Damy T, Judge DP, Kristen AV, et al. Cardiac findings and events observed in an open-label clinical trial of tafamidis in patients with non-Val30Met and non-Val122Ile hereditary transthyretin amyloidosis. J Cardiovasc Transl Res. 2015;8(2):117–127. doi: 10.1007/s12265-015-9613-9.
- Hawkins PN, Ando Y, Dispenzeri A, et al. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625–638. doi: 10.3109/07853890.2015.1068949.
- Rapezzi C, Quarta CC, Obici L, et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J. 2013;34(7):520–528. doi: 10.1093/eurheartj/ehs123.
- Coelho T, Maurer MS, Suhr OB. THAOS – The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63–76. doi: 10.1185/03007995.2012.754348.
- Adams D, Gonzalez-Duarte A, O'Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi: 10.1056/NEJMoa1716153.
- Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. doi: 10.1056/NEJMoa1716793.
- Adams D, Suhr OB, Hund E, et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. 2016;29(Supplement 1):S14–S26. doi: 10.1097/WCO.0000000000000289.
- Obici L, Kuks JB, Buades J, et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016;29(Supplement 1):S27–S35. doi: 10.1097/WCO.0000000000000290.
- Adams D, Polydefkis M, González-Duarte A, et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. 2021;20(1):49–59. doi: 10.1016/S1474-4422(20)30368-9.
- Gundapaneni BK, Sultan MB, Keohane DJ, et al. Tafamidis delays neurological progression comparably across Val30Met and non-Val30Met genotypes in transthyretin familial amyloid polyneuropathy. Eur J Neurol. 2018;25(3):464–468. doi: 10.1111/ene.13510.
- Adams D, Lozeron P, Lacroix C. Amyloid neuropathies. Curr Opin Neurol. 2012;25(5):564–572. doi: 10.1097/WCO.0b013e328357bdf6.
- Adams D, Lozeron P, Theaudin M, et al. Regional difference and similarity of familial amyloidosis with polyneuropathy in France. Amyloid. 2012;19(Supplement 1):61–64. doi: 10.3109/13506129.2012.685665.
- Mazzeo A, Russo M, Di Bella G, et al. Transthyretin-related familial amyloid polyneuropathy (TTR-FAP): a single-center experience in Sicily, an Italian endemic area. J Neuromuscul Dis. 2015;2(s2):S39–S48. doi: 10.3233/JND-150091.
- Alves-Ferreira M, Coelho T, Santos D, et al. A trans-acting factor may modify age at onset in familial amyloid polyneuropathy ATTRV30M in Portugal. Mol Neurobiol. 2018;55:3676–3683. doi: 10.1007/s12035-017-0593-4.
- Plante-Bordeneuve V, Carayol J, Ferreira A, et al. Genetic study of transthyretin amyloid neuropathies: carrier risks among French and Portuguese families. J Med Genet. 2003;40:e120. doi: 10.1136/jmg.40.11.e120.
- Gunnarsson M, Malmeström C, Axelsson M, et al. Axonal damage in relapsing multiple sclerosis is markedly reduced by natalizumab. Ann Neurol. 2011;69(1):83–89. doi: 10.1002/ana.22247.
- Lewczuk P, Ermann N, Andreasson U, et al. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimers Res Ther. 2018;10:71. doi: 10.1186/s13195-018-0404-9.
- Lin YS, Lee WJ, Wang SJ, et al. Levels of plasma neurofilament light chain and cognitive function in patients with Alzheimer or Parkinson disease. Sci Rep. 2018;8(1):17368. doi: 10.1038/s41598-018-35766-w.
- Byrne LM, Rodrigues FB, Blennow K, et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington’s disease: a retrospective cohort analysis. Lancet Neurol. 2017;16(8):601–609. doi: 10.1016/S1474-4422(17)30124-2.
- Bischof A, Manigold T, Barro C, et al. Serum neurofilament light chain: a biomarker of neuronal injury in vasculitic neuropathy. Ann Rheum Dis. 2018;77(7):1093–1094. doi: 10.1136/annrheumdis-2017-212045.
- van Lieverloo GGA, Wieske L, Verhamme C, et al. Serum neurofilament light chain in chronic inflammatory demyelinating polyneuropathy. J Peripher Nerv Syst. 2019;24(2):187–194. doi: 10.1111/jns.12319.
- Mariotto S, Farinazzo A, Magliozzi R, et al. Serum and cerebrospinal neurofilament light chain levels in patients with acquired peripheral neuropathies. J Peripher Nerv Syst. 2018;23(3):174–177. doi: 10.1111/jns.12279.
- Sandelius Å, Zetterberg H, Blennow K, et al. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology. 2018;90(6):e518–e524. doi: 10.1212/WNL.0000000000004932.
- Lycke JN, Karlsson JE, Andersen O, et al. Neurofilament protein in cerebrospinal fluid: a potential marker of activity in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1998;64(3):402–404. doi: 10.1136/jnnp.64.3.402.
- Preische O, Schultz SA, Apel A, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat Med. 2019;25(2):277–283. doi: 10.1038/s41591-018-0304-3.
- Louwsma J, Brunger AF, Bijzet J, et al. Neurofilament light chain, a biomarker for polyneuropathy in systemic amyloidosis. Amyloid. 2021;28(1):50–55. doi: 10.1080/13506129.2020.1815696.
- Ticau S, Sridharan GV, Tsour S, et al. Neurofilament light chain as a biomarker of hereditary transthyretin-mediated amyloidosis. Neurology. 2021;96(3):e412–e422. doi: 10.1212/WNL.0000000000011090.
- Shin SC, Robinson-Papp J. Amyloid neuropathies. Mt Sinai J Med. 2012;79(6):733–748. doi: 10.1002/msj.21352.
- Suanprasert N, Berk JL, Benson MD, et al. Retrospective study of a TTR FAP cohort to modify NIS + 7 for therapeutic trials. J Neurol Sci. 2014;344(1–2):121–128. doi: 10.1016/j.jns.2014.06.041.
- Coelho T, Adams D, Conceição I, et al. A phase II, open-label, extension study of long-term patisiran treatment in patients with hereditary transthyretin-mediated (hATTR) amyloidosis. Orphanet J Rare Dis. 2020;15(1):179. doi: 10.1186/s13023-020-01399-4.
- Dyck PJB, González-Duarte A, Obici L, et al. Development of measures of polyneuropathy impairment in hATTR amyloidosis: from NIS to mNIS+7. J Neurol Sci. 2019;405:116424. doi: 10.1016/j.jns.2019.116424.
- Vinik EJ, Vinik AI, Paulson JF, et al. Norfolk QOL-DN: validation of a patient reported outcome measure in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2014;19(2):104–114. doi: 10.1111/jns5.12059.
- Alnylam Pharmaceuticals Inc. Patisiran global open-label long-term extension study protocol; 2021 [cited 2023 Mar 27]. Available from: https://www.alnylam.com/OLE-Study-Protocol-Amendment.pdf.
- Adams D, Coelho T, Conceição I, et al. Phase 2 open-label extension (OLE) study of patisiran, an investigational RNAi therapeutic for the treatment of polyneuropathy due to hereditary ATTR (hATTR) amyloidosis: final 24-month data. Neurology. 2017;88(16 Supplement):S24.004.
- Sattianayagam PT, Hahn AF, Whelan CJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J. 2012;33(9):1120–1127. doi: 10.1093/eurheartj/ehr383.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi: 10.1056/NEJMoa1805689.
- Ruberg FL, Maurer MS, Judge DP, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the transthyretin amyloidosis cardiac study (TRACS). Am Heart J. 2012;164(2):222–228 e1. doi: 10.1016/j.ahj.2012.04.015.
- Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658–2667. doi: 10.1001/jama.2013.283815.
- Arruda-Olson AM, Zeldenrust SR, Dispenzieri A, et al. Genotype, echocardiography, and survival in familial transthyretin amyloidosis. Amyloid. 2013;20(4):263–268. doi: 10.3109/13506129.2013.845745.
- Kapoor M, Foiani M, Heslegrave A, et al. Plasma neurofilament light chain concentration is increased and correlates with the severity of neuropathy in hereditary transthyretin amyloidosis. J Peripher Nerv Syst. 2019;24(4):314–319. doi: 10.1111/jns.12350.
- Maia LF, Maceski A, Conceição I, et al. Plasma neurofilament light chain: an early biomarker for hereditary ATTR amyloid polyneuropathy. Amyloid. 2020;27(2):97–102. doi: 10.1080/13506129.2019.1708716.
- Luigetti M, Di Paolantonio A, Guglielmino V, et al. Neurofilament light chain as a disease severity biomarker in ATTRv: data from a single-centre experience. Neurol Sci. 2022;43(4):2845–2848. doi: 10.1007/s10072-021-05850-7.
- Thebault S, Booth RA, Freedman MS. Blood neurofilament light chain: the neurologist’s troponin? Biomedicines. 2020;8(11):523. doi: 10.3390/biomedicines8110523.
- Giovannoni G. Peripheral blood neurofilament light chain levels: the neurologist’s C-reactive protein? Brain. 2018;141(8):2235–2237. doi: 10.1093/brain/awy200.
- Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–829. doi: 10.1056/NEJMoa1208760.
- Mariani LL, Lozeron P, Theaudin M, et al. Genotype-phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Ann Neurol. 2015;78(6):901–916. doi: 10.1002/ana.24519.
- Ericzon BG, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847–1854. doi: 10.1097/TP.0000000000000574.