
ABSTRACT
Introduction
Frontotemporal dementia (FTD) includes a group of neurodegenerative diseases characterized clinically by behavioral disturbances and by neurodegeneration of brain anterior temporal and frontal lobes, leading to atrophy. Apart from symptomatic treatments, there is, at present, no disease-modifying cure for FTD.
Areas covered
Three main mutations are known as causes of familial FTD, and large consortia have studied carriers of mutations, also in preclinical Phases. As genetic cases are the only ones in which the pathology can be predicted in life, compounds developed so far are directed toward specific proteins or mutations. Herein, recently approved clinical trials will be summarized, including molecules, mechanisms of action and pharmacological testing.
Expert opinion
These studies are paving the way for the future. They will clarify whether single mutations should be addressed rather than common proteins depositing in the brain to move from genetic to sporadic FTD.
1. Introduction
Frontotemporal dementia (FTD) includes a group of neurodegenerative diseases characterized by neurodegeneration of the anterior temporal and frontal lobes leading to atrophy. Clinically, it is characterized by progressive changes in personality and behavior, impairments in executive function, or language [Citation1]. The disease is considered the most common cause of early onset dementia after Alzheimer’s disease, with an estimated life expectancy of 6–9 years after symptom onset [Citation2].
Historically, FTD was initially described by Pick and defined later as non-Alzheimer’s dementia till the establishment of common clinical diagnostic criteria [Citation1,Citation3–6]. FTD spectrum covers different clinical presentations which have led to the identification of three principal syndromes and phenotypic variants: the behavioral variant of FTD (bvFTD), the primary progressive aphasia (PPA), which can be further subdivided into the semantic variant (svPPA) and a non-fluent variant of a primary progressive aphasia (nfvPPA) (). In addition, a new clinical phenotype has been proposed, namely the right temporal variant of FTD (rtvFTD) [Citation7–9]. Furthermore, FTD has clinical overlapping with amyotrophic lateral sclerosis (ALS), as 5–10% of FTD cases have concomitant motor neuron disease symptoms, whereas 40–50% of ALS cases demonstrate FTD symptoms [Citation10]. Together with clinical similarities, ALS and FTD share pathological ubiquitin-TDP-43 neuronal inclusions [Citation10–14].
Figure 1. Tree diagram of FTLD heterogeneity. Association of genetics and proteins to the clinical phenotypes and underlying pathology in FTLD.
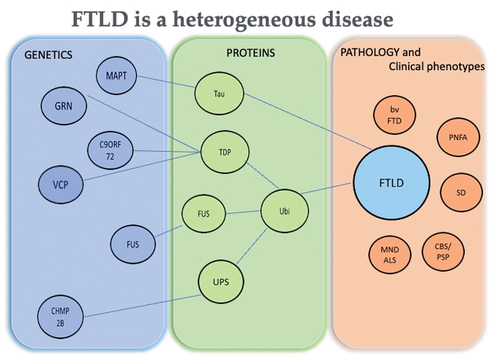
At autopsy, Frontotemporal Lobar Degeneration (FTLD) is characterized by synapse loss, gliosis, neuronal loss, and atrophy in the frontal and temporal lobes of the brain [Citation11,Citation15,Citation16]. Postmortem studies have recognized different accumulations of aggregated proteins in neurons and glial cells (). Based on the type of protein accumulated, it is possible to classify FTLD cases with neuronal intracytoplasmic inclusions composed of the microtubule-associated protein, tau, as FTLD-tau. In half of tau-positive cases, rounded bodies, known as Pick bodies, and glial tau inclusions may also be present [Citation11]. The rest of the FTLD cases present neuronal tau as neurofibrillary tangle-like structures (NFT) or more amorphous deposits (pre-tangles) [Citation17,Citation18]. The RNA- and DNA-binding protein, TDP-43, within ubiquitinated neuronal intracytoplasmic inclusions, neuritic processes or neuronal intranuclear inclusions characterize FTDL-TDP. Cases with inclusions composed of the protein Fused in Sarcoma (FUS) are termed as FTLD-FUS 9 or FTLD-FET as neuronal intracytoplasmic inclusions can also contain other members of the FET family of proteins, such as transportin-1, TAF15 and Ewing’s Sarcoma protein [Citation15,Citation16].
A recent study (FRONTIERS) estimated the incidence of FTD phenotype cases across Europe for the first time. The estimated annual incidence rate for FTD in Europe was 2.36 cases per 100,000 person-years (95% CI, 1.59–3.51 cases per 100 000 person-years). The authors reported that FTD incidence progressively increased across age, reaching its peak at the age of 71 years [Citation19]. BvFTD was the most common phenotype (40.07%), followed by PPA (28.46%) and extrapyramidal phenotypes (25.84%). FTD-ALS was the rarest phenotype (5.62%). A total of 95 patients with FTD (35.58%) had a family history of dementia. The estimated number of new FTD cases per year in Europe was 12,057 [Citation19]. Overall, the incidence was higher in males than in females [Citation19].
The results of this study suggest that FTLD-associated syndromes are more common than previously recognized, and diagnosis should be considered at any age [Citation19].
One-third to half of the FTD cases have a positive family history and are inherited in an autosomal dominant pattern. Causative mutations in three genes, each with autosomal dominant inheritance, account for most cases of familial FTD. The first is the microtubule-associated protein tau (MAPT) on chromosome 17, identified in 1998; the second is the progranulin (GRN) gene, also on chromosome 17, and the third is a hexanucleotide repeat expansion (HRE) in the gene of chromosome 9 open reading frame 72 (C9orf72) identified in 2011 [Citation17,Citation18,Citation20–22]. In addition, other rare causal genes have been identified and account for fewer than 5% of the cases [Citation22].
This review will focus on the current knowledge of FTD, starting from Pickʼs clinical description of the disease, moving toward the genetic causes and associated phenotypes, and finishing with the current therapeutic approaches to the disease, with particular emphasis on tailored treatment approaches.
2. History and clinical aspects of FTD
In 1892, Pick reported the first description of a case that would be defined today under the spectrum of FTD [Citation23]. This first case described PPA [Citation24]. The most common clinical presentation of the disease was again described by Pick 12 years later in a second patient [Citation25]. Instead, the microscopic examination of the brain of this second patient was carried out by Alzheimer, who described argyrophilic intracytoplasmic inclusions and ballooned neurons, which he called Pick bodies and Pick cells. Alzheimer soon realized that the alterations observed were distinct from changes found in the neurodegeneration later associated with his name [Citation26]. These first clinical descriptions and pathological findings were referred to later as Pick’s disease [Citation25–28]. However, no Pick pathology may be present in patients with a clinical presentation of FTD associated with motor neuron disease (MND) [Citation29,Citation30].
In the 1980s, two groups from Lund (Sweden) and Manchester (UK) tried to define this disease to establish the basis for a consensus on the diagnosis guidelines. The two groups agreed on the following: 1) FTD is more than Pick disease, 2) FTD and PPA (with two subtypes, progressive non-fluent aphasia and semantic dementia) have overlapping pathogenic bases, and 3) many cases show autosomal dominant inheritance (often with an early onset). A consensus was reached on the clinical diagnostic criteria for FTD, which were later refined to include three clinical syndromes: bvFTD, semantic dementia (SD) and progressive non-fluent aphasia (nfvPPA) [Citation1,Citation6].
In this scenario, the first descriptions of FTD symptomatology focused on the ‘frontal lobe’ clinical alterations and patients were classified as suffering from frontal lobe degeneration of non-Alzheimer type [Citation4–6]. Pathological studies in these first presentations highlighted the distribution of atrophy in the anterior temporal and frontal lobes [Citation31,Citation32].
The most common clinical presentation of FTD is bvFTD, characterized by behavioral changes and progressive deterioration of personality. Patients may show a broad spectrum of symptoms, such as behavioral alterations, including disinhibition, overeating, impulsiveness, and impairment of cognitive functions, with relative sparing of memory. In addition, socially inappropriate behaviors, loss of sympathy and empathy, and repetitive or stereotyped behaviors are early and prominent symptoms of bvFTD [Citation2,Citation33–35]. Furthermore, differential diagnosis between psychiatric patients and FTD patients is mandatory as neuropsychiatric and psychotic symptoms are described in the clinical presentation of several patients [Citation36–39].
In 2011, updated criteria for diagnosing bvFTD were proposed with a new classification of language variant presentations [Citation40,Citation41]. According to the former, the main characteristic of bvFTD is the progressive deterioration of behavior or cognition or both based on neuropsychological assessment and also on patient observation or history (provided by a knowledgeable informant). New criteria follow a hierarchic scale for diagnosis: possible, when only clinical symptoms are observed; probable, if the imaging biomarkers show frontal/temporal atrophy and definite, if a pathogenic mutation is found (or if the pathology is proven at autopsy). These new criteria are flexible in the attempt to collect the high heterogeneity of symptoms present in the patients at the first medical presentation [Citation40–42].
Other phenotypes have been described, such as progressive supranuclear palsy syndrome, corticobasal syndrome, and FTD with MND, with clinical manifestations included in the FTD spectrum [Citation43–45].
3. Neuroimaging across FTD clinical presentations
The current international FTD guidelines for the diagnosis of bvFTD rely on frontal and/or temporal abnormalities observed by magnetic resonance imaging (MRI), fluorine-18-fluorodeoxyglucose positron emission tomography (FDG-PET), or single-photon emission computed tomography for the diagnosis of ‘probable’ bv-FTD [Citation41]. The heterogeneous clinical phenotypes have been associated with typical anatomical findings in the brain [Citation46]. For example, bvFTD is typically associated with atrophy on structural MRI and hypometabolism on FDG-PET in the prefrontal cortex and anterior temporal lobes, with relative sparing of more posterior brain regions, such as the occipital lobe. The anatomical ‘frontal phenotype’ shows worse executive performance, while ‘the temporal phenotype’ shows worse episodic memory impairment [Citation46]. Furthermore, the patients with frontal dominant atrophy patterns tend to show faster clinical decline, while the subcortical subtype shows relatively slow disease progression [Citation47,Citation48]. Some bvFTD patients show mild behavioral and/or cognitive abnormalities despite frontotemporal gray matter atrophy. Further, bvFTD patients with very little atrophy on MRI show slower rates of clinical decline [Citation46,Citation49].
In conclusion, patterns of atrophy assessed on MRI demonstrate neuroanatomical signatures of the different FTD pathologies: bilateral atrophy in frontal and anterior temporal lobes with left or right predominance are observed in bvFTD, asymmetric dominant hemisphere, perisylvian atrophy in progressive non-fluent aphasia/nfvPPA and bilateral, asymmetric temporal lobe atrophy in semantic dementia/svPPA [Citation50–52].
A recent work by Ahmed et al. studied how profiles of brain atrophy might differ between clinical phenotypes using cortical and subcortical gray and white matter volume assessment on structural MRI in a large cohort of 209 participants [Citation52]. The study correlates cognitive and behavioral changes, assessed using the Addenbrooke’s Cognitive Examination and the Cambridge Behavioural Inventory, with MRI findings [Citation52]. The results of this investigation show that the motor cortex and insula can be considered as differentiating structures between clinical syndromes, with bilateral motor cortex atrophy more pronounced in ALS – FTD compared with pure-ALS, and greater left motor cortex and insula atrophy relative to bvFTD [Citation52]. The study highlighted a significant association between abnormal behavior and volume loss in a predominantly fronto-insular network involving the amygdala, striatum, and thalamus [Citation52].
Taken together, these results point to subcortical involvement in the origin of behavioral abnormalities, potentially accounting for the phenotypic variability observed across the FTD spectrum [Citation52]. Furthermore, the results presented by Ahmed et al. stressed the importance of the correlation between MRI and neuropsychological assessment in the diagnosis of bvFTD [Citation52]. Other studies have differentiated between FTD syndromes relying on MRI findings and demonstrated how behavioral alterations may correlate with specific patterns of atrophy: apathy has been linked to prominent frontal atrophy, including dorsolateral frontal cortex and medial frontal and cingulate hypoperfusion, whereas disinhibition is linked to temporal atrophy and ventromedial frontal and temporal lobe hypoperfusion [Citation53,Citation54]. Semantic dementia is often associated with the left-sided predominance of temporal lobe atrophy, and prosopagnosia has been associated with atrophy in the right temporal lobe [Citation54–56]. The clinical presentation of prosopagnosia and behavioral changes, together with right temporal atrophy, is called the ‘right temporal variant’ FTD [Citation8,Citation9]. Extensive evidence has indicated the involvement of prefrontal as well as medial/lateral temporal atrophy in memory deficits of bvFTD patients [Citation57,Citation58] in contradiction with the common view that only frontal atrophy explains memory impairment [Citation58]. Further, hippocampal atrophy has been indicated to potentially contribute to a disrupted ability to process social information [Citation59,Citation60]
4. Genetics
Although most cases of FTD are sporadic, up to 40% of the patients have a family history of FTD present or suggested to be present in at least one family member [Citation61,Citation62]. Mutations in three genes, each with autosomal dominant inheritance, account for most cases of familial FTD. The first was identified in 1998 in MAPT on chromosome 17, the second 8 years later, GRN gene, also on chromosome 17, and the third in 2011, C9orf72 [Citation17–20]. Additional causal genes that account for fewer than 5% of the cases have been identified [Citation21]. 5–20% of the cases of familial FTLD are due to MAPT mutations, 5–25% to GRN mutations and 20–30% to C9orf72 mutations [Citation63]. Sporadic cases of FTD due to mutations in MAPT are 0–2%, due to GRN or C9orf72 genes are 5% and 6%, respectively, of the total cases. Thus, the most common genetic mutation worldwide is the C9orf72 expansion [Citation21]. Geographical variations have been observed; in particular, a founder effect was described in North Wales, with more than 16 MAPT mutations in the northwest of Britain, whereas a founder effect was observed in North Italy, accounting for the high prevalence of a GRN mutation in that region [Citation64,Citation65,Citation66]. Familial and sporadic bvFTD share similar clinical characteristics, except for an earlier onset age in familial cases [Citation67].
4.1. Microtubule associated protein tau (MAPT)
MAPT was the first gene to highlight a link between chromosome 17q21.2 and an autosomal inherited form of bvFTD with Parkinsonism [Citation68]. This gene also makes FTD part of the tauopathies group, where hyperphosphorylated and insoluble tau protein aggregates inside neurons and glial cells hallmark the disease [Citation69]. So far, more than 80 mutations, such as missense mutations, deletions, and insertions, have been described and linked to different tauopathies [Citation69]. MAPT mutations lead to neurotoxic effects mainly through three different ways based on: 1) tau protein gain of function with increasing of tau aggregates that obstruct axonal transport impairing mitochondrial, lysosome, and autophagic pathways; 2) tau protein loss of function where protein aggregation and/or hyperphosphorylation cause microtubule disassembly and axonal transport deficit; 3) tau protein mislocalization into post-synaptic regions leading to synaptic impairment, due to tau hyperphosphorylation, mutations, and overexpression [Citation70,Citation71]. Interestingly, many mutations are due to alternative splicing mechanisms, primarily within MAPT exon 10, which results in a different tau protein isoform affecting their ratio (3 R/4 R) [Citation69]. The morphology and biochemistry of tau-specific isoforms determine different FTLD-tau subtypes with peculiar seeding properties and ultrastructure [Citation72,Citation73,Citation74]. Furthermore, an increased disease risk of sporadic tauopathies is associated with the common genetic variation in two major MAPT haplotypes (H1 and H2) [Citation75]. The most common clinical presentations of patients carrying MAPT mutations, even if heterogeneous within the same family, include bvFTD, PSP, and corticobasal syndrome (CBS) [Citation76].
4.2. Progranulin (GRN)
GRN, encoding the progranulin (PGRN) protein, was the second FTD causative gene discovered and genetically linked to the same chromosomal region of MAPT, 17q21 [Citation19,Citation77]. This glycoprotein, expressed in neurons and activated microglia, acts as a growth factor involved in critical processes, such as cell cycle and cellular motility when cleaved through lysosomes in smaller proteins termed granulins [Citation78]. Mutations on this gene, equally distributed within the whole coding portion and splicing sites, raise a nonfunctional allele with a premature stop codon and degradation of aberrant mRNA by nonsense-mediated decay. This produces haploinsufficiency, with neuronal cytoplasmic and evocative ‘cat eyes’ nuclear TDP-43 positive inclusions [Citation20]. Over 70 different mutations, such as frameshift, splicing, and non-sense mutations, have been reported, predominantly in heterozygous patients [Citation79]. It is worth noticing that homozygous GRN mutations cause a group of rare lysosomal disorders named neuronal ceroid lipofuscinoses characterized by seizures, cerebellar ataxia, dementia, and visual loss by the third decade of life [Citation80].
4.3. Chromosome 9 open reading frame 72 (C9orf72)
The discovery of the C9orf72 repeat expansion as the most frequent genetic cause of FTD and ALS has undoubtedly opened new horizons in the knowledge of the pathogenic mechanisms of these diseases [Citation17,Citation18]. This expansion, located in a non-coding portion of chromosome 9p21, is due to hexanucleotide repeats (GGGGCC) where a number less than 30 is physiological. In contrast, several hundreds to thousands of repeats have been reported as pathological [Citation21,Citation35]. Consequently, there is a gray area from 30 to 200 repeats where not all patients have symptoms [Citation81]. Even if the specific function of C9orf72 is still undefined, some evidence reported a role in the GTPase-interacting complex regulating autophagy, inflammation, and endosomal trafficking [Citation82]. Anyway, the C9orf72 pathogenicity is probably associated to three different mechanisms: 1) haploinsufficiency due to C9orf72 loss of function where the expanded repeats reduce the gene expression with the impairment of the synaptic vesicles recycling and the autophagy-lysosome pathway exacerbating the neuroinflammation; 2) neurotoxic gain of function of RNA foci, composed of both sense and antisense transcripts, that, via the sequestering of RNA binding proteins, affect post-transcriptional processes; and 3) toxic gain of function of the translated HRE through a non-ATG translation pathway, synthesizing the dipeptide repeat (DPR) proteins that, prone to aggregation, accumulate in intercellular inclusions [Citation82]. Each single mechanism is not able alone to induce the disease. Thus, the most likely hypothesis is that more than one mechanism is implicated, and the interplay among them could worsen the disease [Citation83]. Interestingly, a very recent work demonstrated that the excision of the HREs using the CRISP/Cas9 technology resulted in a critical reduction of RNA foci, DPRs and haploinsufficiency, the significant hallmarks of FTD in C9ALS/FTD mouse models and patient-derived iPSC paving the way toward new promising therapeutic strategies [Citation84].
The genetic similarity among FTD and ALS observed for TDP-43 is also evident for the C9orf72 gene and another cluster of genes, even if thanks to Genome-wide association studies within large projects and consortia such as Project MinE for ALS (https://www.Projectmine.com/) and Genetic Frontotemporal Dementia Initiative (GENFI)-3 for FTD (https://www.genfi.org/), more and more risk factors and new genes are emerging [Citation85]. Together, these findings have led researchers to consider ALS and FTD as a disease continuum and only sometimes easy to discern instead of two separate and different pathologies.
The C9orf72 mutation, highly penetrant, is found in 5–10% of the patients with FTD or ALS, reaching 30% in patients with both diseases, referred to as C9FTD/ALS. Age at onset is highly variable, ranging from 20 to 90 years, as well as the initial presentations and disease course [Citation86]. The most common clinical phenotype is bvFTD, with a high frequency of psychosis, delusions, and hallucinations compared to MAPT and GRN genes [Citation76].
4.4. Animal models of FTD
As in other neurodegenerative diseases, many animal models have been used to study FTD in the last decade [Citation87]. The complexity of FTD genetics and pathology and, consequently, the heterogeneity of mouse model phenotypes led to the consideration of a new holistic approach where multiple mouse models could be taken into account based on the functional alterations seen in FTD patients, such as behavioral, socioemotional, memory, language, eating, and metabolism and motor changes [Citation87,Citation88].
Therefore, based on the reported functional deficits and the genetic mutation considered, Ahmed and coworkers included available FTD mouse models in five big groups: 1) PGRN with behavior changes; 2) C9orf72 with behavioral and motor changes; 3) TDP-43 with behavioral, memory and motor changes; 4) tau with behavioral, motor and memory impairments; 5) Valosin Containing Protein (VCP) with behavioral/social alterations [Citation88]. This shows that a unique model can only contain some of the different FTD molecular and clinical presentations. Thus, it would be more appropriate to rethink a combination of multiple mouse models and other species to better define FTD’s pathological characteristics [Citation88].
Even if, in the last decades, FTD animal models have been extensively used to expand basic knowledge of molecular and cellular pathways and develop, test and refine targeted therapies, there are no animal models able to represent the whole and complex phenotypical features of FTD. Indeed, they represent rather a summary of distinctive pathological mechanisms with poor predictive power in drug efficacy [Citation89].
These models are limited and challenging for several reasons: 1) the short life span of mice is poorly adapted to aging-related diseases; 2) differences between brain development and function in mice and human; 3) genomic differences among rodents and humans regard also RNA stability and processing; 4) the use of inbred animals certainly does not reflect genetic shuffling among the population; 5) attention should be paid on the methods used for generating animal models often expressing a mild phenotype; 6) rare genetic-based animal model could be inappropriate [Citation89].
Although these limitations belong to each experimental model, animal models remain a valuable tool not only for basic research but also for applied research in the context of drug design and testing since they can target specific pathological pathways and test therapies by examining their effects.
5. Current therapeutic approaches to FTD
While recent studies are adding knowledge and shedding light on the molecular basis of FTD; currently, there are no disease-modifying treatments (DMT) for FTD. No medication or non-drug treatment has been proven to prevent, cure or slow this pathology, except symptomatic drugs for behavioral and cognitive symptoms.
Novel treatments are designed to target the underlying pathology (disease-modifying) rather than symptoms, but so far, no biomarker predicting the pathology has been identified. Moreover, the pharmacological intervention should be done very early during the pathogenesis, even in preclinical stages.
In this regard, it has been shown, in the GENFI study, that gray matter and cognitive changes can be identified 5–10 years before the expected onset of symptoms in adults at risk of genetic FTD and particularly in a cohort carrying GRN mutations [Citation90]. The development of DMTs for genetic FTD is just beginning, and clinical trials for these DMTs are ongoing; some of the results will be available in the next few years, see .
Table 1. Summary of the ongoing clinical trials for FTD-GRN and FTD-C9ORF72 (clinicaltrials.Gov).
5.1. GRN drugs
More compounds for patients with FTD-GRN are available as the frequency of the mutation is higher than MAPT. Aforementioned, more than 80 mutations in MAPT gene have been described and linked to different tauopathies without a unique mechanism to explain tau mutation effects [Citation69]. This implies different subtypes of clinical phenotypes and the complexity into new drug designing. Furthermore, decreased PGRN blood levels are good predictors of the presence of GRN null mutations in asymptomatic at-risk members of families with known GRN mutation, permitting to identify individuals at risk without carrying out the DNA sequencing [Citation91]. Moreover, most mutations consist of haploinsufficiency. Thus, restoring PGRN levels through gene replacement therapy seems easier than removing a deposited protein, such as tau.
Given these premises, restoring the levels of PGRN in FTD is currently the most promising approach in treating FTD-GRN.
5.1.1. AL001
Latozinemab (AL001), a human monoclonal immunoglobulin (IgG) G1m17,1 kappa antibody generated against human receptor sortilin, is being developed for the treatment of FTD-GRN mutation carriers to counteract neurodegeneration by increasing levels of PGRN in the blood and brain. The receptor sortilin has been identified as the primary regulator of PGRN in plasma and in the brain by guiding PGRN to lysosomal degradation.
Latozinemab has been demonstrated to elicit a long-lasting increase in PGRN in the serum and cerebrospinal fluid (CSF) of asymptomatic FTD-GRN participants and restored PGRN levels to healthy controls. Together, these findings demonstrate that the sortilin receptor is a viable target for PGRN-replacement therapy in FTD-GRN patients and supports the continued development of latozinemab [Citation92]. Clinical trials are ongoing to determine if latozinemab-induced elevations in PGRN levels can slow the progression of FTD-associated neurodegeneration. To this regard, INFRONT-3 (NCT04374136) is a Phase 3 double-blind, placebo-controlled study evaluating the efficacy and safety of AL001 administered intravenously in participants at risk for or with frontotemporal dementia due to heterozygous mutations in the GRN gene. The study was completed in October 2023, and the results are still pending. The study from Ward et al. reported the results of the first-in-human phase 1 study conducted on 8 patients with FTD-GRN. All adverse events (AEs) were mild in severity and most of treatment-related AEs were not related to the study drug [Citation93]. Only one patient experienced one treatment-related AEs (right upper quadrant pain, Murphy’s sign positive). No participants experienced a serious AE or a treatment-related AE leading to early discontinuation of the study. The reported treatment-related AEs were upper respiratory tract infection, fungal skin infection, and nasopharyngitis, and musculoskeletal disorders [Citation93]. Elevated levels of cholesterol and triglycerides have been observed, respectively, in two and three patients [Citation93]. Elevated levels of PGRN have been associated with visceral obesity and dyslipidemia as PGRN can interact with low-density lipoprotein receptor-related protein one [Citation94]
Based on data from the INFRONT-2 Phase 2 clinical trial of latozinemab in FTD-GRN participants, FDA granted Breakthrough Therapy and Fast Track designations (Alector press release: ‘FDA Grants Latozinemab Breakthrough Therapy Designation for Frontotemporal Dementia Due to a Progranulin Gene Mutation (FTD-GRN)’ 7 February 2024)
It is worth noting that a recent candidate therapy for FTD-GRN is a small molecule, called VES001that serves as a sortilin antagonist [Citation95]. VES001 selectively binds the receptor sortilin and disrupts protein interaction with progranulin at the cell membrane. In this way, VES001 acts as a progranulin uptake inhibitor. VES001 is orally available, can penetrate the central nervous system barrier, and is now in Phase 1 clinical trial (NCT06226064) [Citation95].
5.1.2. PBFT02
Preclinical data have revealed that in progranulin‐deficient mice, intraventricular injection of adeno‐associated viral (AAV) vector expressing human GRN increased PGRN levels in CSF and normalized histological and biochemical markers of GRN deficiency [Citation96]. A single vector injection into the cisterna magna of nonhuman primates achieved CSF PGRN concentrations up to 40‐fold higher than those of normal human subjects and exceeded CSF PGRN levels of successfully treated mice [Citation96].
PBFT02 is a gene-replacement candidate therapy that uses AAV1 to deliver a functional copy of the GRN gene to the human brain. PBFT02 will be delivered to the CSF by a one-time injection into the cisterna magna.
In February 2021, Passage Bio, Inc. registered UpliFT-D, a Phase 1/2 trial, to assess the safety, tolerability, and efficacy of PBFT02 in six people with early-stage symptomatic FTD-GRN mutation. The study will run at North America, Brazil, and UK centers, with primary completion scheduled for August 2024. In December 2023, Passage Bio, Inc announced interim results from the first three patients enrolled in UpliFT-D clinical trial, which show the ability of PBFT02 to elevate CSF progranulin to supraphysiologic levels at the lowest tested dose [Citation97]. Following these promising initial data, Passage Bio, Inc has also declared the willingness to extend the study of PBFT02 in other diseases, such as FTD-C9orf72, ALS, and Alzheimer’s disease [Citation97].
5.1.3. DNL593
DNL593 is a replacement therapy for FTD-GRN patients. To restore brain PGRN levels, DNL593 uses a ‘brain shuttle’ technology consisting of a protein transfer vehicle moving intravenously across the blood–brain barrier and into the central nervous system [Citation98]. DNL593 consists of progranulin protein fused to an antibody fragment that binds to the transferrin receptor. Association with transferrin receptors on brainblood barrier endothelial cells facilitates the receptor-mediated transcytosis of progranulin protein into the brain. DNL593 can prevent neurodegeneration and microglial dysfunction in PGRN-deficient mice [Citation98,Citation99]. In February 2022, Denali and Takeda began a Phase 1/2 study of the safety, tolerability, and pharmacokinetics of single and multiple doses of DNL593. The trial is running at 14 sites in Europe, Turkey, and Brazil, and will be completed by November 2025.
5.1.4. PR006 (LY3884963)
PR006 is being developed as a single-dose gene therapy for patients with FTD-GRN [Citation100]. PR006 is designed to be administered by injection into the cisterna magna, using the viral vector AAV9 with the goal to increase progranulin levels via delivery of a healthy GRN gene into the central nervous system. PR006 is currently being studied in PROCLAIM a Phase 1/2 clinical trial [Citation100]. The European Commission has granted orphan designation to PR006 for the treatment of FTD.
5.1.5. AVB-101
AVB-101 is a one-time gene replacement therapy designed to deliver a functional copy of GRN to the affected areas of the brain, restoring the PGRN levels in the brain [Citation101]. AVB-101 is delivered using a minimally invasive, stereotactic neurosurgical procedure directly in the thalamus, which has extensive connections to the other brain parts, including the frontal and temporal lobes [Citation101]. This method aims to deliver the treatment directly to the brain, reducing the dose required and the potential systemic exposure. ASPIRE-FTD is a Phase 1/2 clinical study of AVB-101 gene therapy in FTD-GRN patients. ASPIRE-FTD is an open-label, multi-center study designed to evaluate the safety and preliminary efficacy of AVB-101 in patients with FTD-GRN. Preclinical safety and biodistribution data following intrathalamic delivery to non-human primates demonstrate good tolerability and widespread progranulin expression in brain tissues (30th Annual European Society of Gene & Cell Therapy (ESGCT) meeting in Brussels, Belgium Oct. 24–27, 2023).
5.2. C9orf72 drugs
5.2.1. WVE-004
The HRE (GGGGCC) in the intron 1 of C9orf72 gene is the most common genetic cause of FTD. The pathogenicity of HRE is due to the formation of nuclear RNA granules, generated from repeat-containing spliced intron and unspliced pre-mRNA, with probable disruption of RNA processing and the unconventional translation of the GGGGCC repeats into aggregating and insoluble toxic DPR proteins [Citation102,Citation103,Citation104]. Antisense oligonucleotides (ASO) mediate the degradation of mRNAs which pathologically lead to the formation of dipeptides. WVE-004 is an ASO which selectively targets transcriptional variants containing the HRE present in the C9orf72 gene [Citation105].
On May 2023, Wave Life Sciences Ltd., announced the results from Phase 1b/2a FOCUS-C9 study evaluating WVE-004 as an investigational treatment for C9orf72-associated ALS and FTD) concluding that WVE-004 did not show clinical benefit compared with placebo; additionally, poly(GP) reductions did not correlate with clinical outcomes. Based on these results, WVE discontinued the development of WVE-004 [Citation105].
5.2.2. TPN-101
In FTD, abnormal proteins tau and TDP-43 and the consequent loss of functional nuclear TDP-43 protein initiate a cascade of events such as increased expression of long interspersed nuclear elements 1 (LINE1) and of reverse transcriptase which lead to neuron dysfunction and death [Citation106,Citation107]. LINE-1 elements are a class of retrotransposable elements that, in humans, can replicate and move to new locations within the genome. When this process becomes dysregulated, LINE-1 reverse transcriptase drives the overproduction of LINE-1 DNA, triggering an antiviral immune response and neuroinflammation [Citation108]. By inhibiting LINE1 reverse transcriptase, TPN-101 is supposed to prevent this immune response and exert neuroprotective function. TPN-101 inhibits the LINE-1 reverse transcriptase that promotes LINE-1 replication [Citation109]. Transposon will report data from three Phase 2 clinical studies of TPN-101 in four disorders, including FTD patients with C9orf72 mutations, in 2024.
6. Conclusions
The development of DMTs for genetic FTD is just beginning, and clinical trials for these DMTs are ongoing; some of the results will be available in the following years.
International consortia like the GENFI are fundamental in the knowledge of genetic FTD pathogenesis, to identify tailored treatments. Moreover, making an attempt to anticipate the diagnosis, new research criteria for prodromal bvFTD have been proposed [Citation110]. Nevertheless, we still lack deep knowledge of this phase in terms of pathological mechanisms, i.e. mutations are present since birth; it is not known how and when they start to modify the brain toward neurodegeneration. Moreover, it is plausible that other genetic and environmental factors contribute to manifest the pathological phenotype, together with potential triggering factors.
In conclusion, studies carried out so far have paved the way to personalized treatment. This concept is fundamental in diseases such as FTD, in which there are many pathogenic proteins that the clinical phenotype cannot foresee.
7. Expert opinion
Herein, we summarized the current clinical trials with disease-modifying compound ongoing. While the concept of tailored treatment for FTD is widely accepted, there are still questions and doubts on many issues. First, all compounds developed are designed to interfere with the pathogenic mechanisms caused by mutations: as regards tau, there are no trials as it is hard to identify ways to remove a deposited protein; moreover, tau exists in six isoforms, and the balance among them may play a role in homeostasis and, lastly it deposits into neurons, thus it is difficult to reach (for instance with antibodies). Concerning PGRN, mutations cause mainly haploinsufficiency; thus, it should be conceptually easier to provide (in many ways) a protein lacking rather than remove a deposited one. In addition, PGRN plasma or CSF levels have been considered surrogate measures of a clinical endpoint, although we do not actually know whether these measures reflect a benefit. Finally, it is not known whether the replacement of PGRN would be enough to limit upstream events leading to neurodegeneration. The scenario of C9orf72 is even more complicated, it still needs to be clarified whether the number of expansions matters (and which is the pathogenic threshold), which is the role of dipeptides and repeat associated non-ATG (RAN) translation and whether the latter could be a therapeutic target.
Another critical point in FTD regards the underlying pathology, i.e. to target the pathogenic protein (Tau or TDP-43) rather than address-specific mutations. This should be the ultimate goal to treat not only mutation carriers but also sporadic FTD patients. Nevertheless, even assuming to identify compounds able to target tau or TDP-43, we still do not have biomarkers to predict the underlying pathology in sporadic cases, which would be mandatory to identify the right patient for the right drug. Another consideration to be taken into account, when dealing with novel treatments, is the risk/benefit ratio, particularly with AAV, which can deliver GRN but could lead to uncontrollable aberrant mechanism activation in the brain. Last but not least, all clinical trials carried out so far addressed genetic cases only, which are a minority of the number of patients. In this regard, a question raises whether there are enough patients suffering each FTD subtype to carry out clinical trials with enough statistical power. Moreover, taking advantage of Phase II weaknesses, future Phase 3 trials should adjust inclusion and exclusion criteria.
In conclusion, looking at the future scenario, the goal will be to develop a tailored treatment for each patient. The challenge will be to predict the underlying pathology in sporadic FTD to allow all patients to have a cure. This also raises additional questions on the timing of the treatment, i.e. the preclinical, prodromal, and/or clinical phase, raising questions on the sustainability of health systems. Concerning preclinical phases of the disease, prodromal criteria have been already proposed, considering the wide heterogeneity of symptoms preceding the overt phase of the disease, ranging from behavior and memory disturbances to movement impairment or psychiatric manifestations preceding or co-occurring in patients.
Article highlights
Frontotempral dementia (FTD) includes a group of neurodegenerative diseases characterized by neurodegeneration of anterior temporal and frontal lobes leading to atrophy. Clinically, it is characterized by progressive changes in personality and behavior, impairments in executive function, or language.
One-third to half of the FTD cases have a positive family history and are inherited in an autosomal dominant pattern. Causative mutations in three genes, each with autosomal dominant inheritance, account for most cases of familial FTD: MAPT on chromosome 17, progranulin (GRN) gene, also on chromosome 17, and the third is a hexanucletide repeat expansin (HRE) in the gene of chromosome 9 open reading frame 72 (C9rf72)
Large consortia have studied carriers of mutations, also in preclinical phases. As genetic cases are the only ones in which the pathology can be predicted in life, compounds developed so far are directed toward specific proteins or mutations. Recent approved clinical trials will be summarized here, including molecules, mechanism of action and pharmacological testing.
The ongoing clinical trials will test new disease modifying drugs to clarify whether single mutations should be addressed rather than common proteins depositing in the brain to move from genetic to sporadic FTD.
Abbreviation list
| AAV | = | Adeno–Associated Viral |
| AE | = | Adverse Events |
| ALS | = | Amyotrophic Lateral Sclerosis |
| ASO | = | Antisense Oligonucleotides |
| bvFTD | = | behavioural variant of FTD |
| C9orf72 | = | Chromosome 9 Open Reading Frame 72 |
| CSF | = | cerebrospinal fluid |
| DMT | = | Disease-Modifying Treatment |
| DPR | = | Dipeptide Repeat |
| FDG-PET | = | Fluorine-18-fluorodeoxyglucose Positron Emission Tomography |
| FTD | = | Frontotemporal Dementia |
| FTLD | = | Frontotemporal Lobar Degeneration |
| FUS | = | Fused in Sarcoma |
| GENFI | = | Genetic Frontotemporal Dementia Initiative |
| GRN | = | Progranulin |
| HRE | = | Hexanucleotide Repeat Expansion |
| LINE1 | = | Long Interspersed Nuclear Elements 1 |
| MAPT | = | Microtubule Associated Protein Tau |
| MND | = | Motor Neuron Disease |
| MRI | = | Magnetic Resonance Imaging |
| NFT | = | Neurofibrillary Tangle-like Structures |
| nfvPPA | = | non-fluent variant Primary Progressive Aphasia |
| PGRN | = | progranulin |
| PPA | = | Primary Progressive Aphasia |
| RAN | = | Unconventional Translation |
| svPPA | = | semantic variant PPA |
| TDP-43 | = | TAR DNA-binding protein 43 |
| VCP | = | Valosin Containing Protein |
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Neary D, Brun A, Englund B, et al. Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester groups. J Neurol Neurosurg Psychiatry. 1994;57:416–418. doi: 10.1136/jnnp.57.4.416
- Moore KM, Nicholas J, Grossman M, et al. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 2020;19:145–156. doi: 10.1016/S1474-4422(19)30394-1
- Snowden JS. Changing perspectives on frontotemporal dementia: a review. J Neuropsychol. 2023;17:211–234. doi: 10.1111/jnp.12297
- Gustafson L. Frontal lobe degeneration of non-Alzheimer type. II. Clinical picture and differential diagnosis. Arch Gerontol Geriatr. 1987;6:209–223. doi: 10.1016/0167-4943(87)90022-7
- Miller BL, Cummings JL, Villanueva-Meyer J, et al. Frontal lobe degeneration: clinical, neuropsychological, and SPECT characteristics. Neurology. 1991;41(9):1374–1374. doi: 10.1212/wnl.41.9.1374
- Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. doi: 10.1212/wnl.51.6.1546
- Thompson SA, Patterson K, Hodges JR. Left/Right asymmetry of atrophy in semantic dementia: behavioral-cognitive implications. Neurology. 2003;61(9):1196–1203. doi: 10.1212/01.wnl.0000091868.28557.b8
- Chan D, Anderson V, Pijnenburg Y, et al. The clinical profile of right temporal lobe atrophy. Brain. 2009;132(5):1287–1298. doi: 10.1093/brain/awp037
- Erkoyun HU, Groot C, Heilbron R, et al. A clinical-radiological framework of the right temporal variant of frontotemporal dementia. Brain. 2020;143(9):2831–2843. doi: 10.1093/brain/awaa225
- Ferrari R, Kapogiannis D, Huey E, et al. FTD and ALS: a tale of two diseases. Curr Alzheimer Res. 2011;8(3):273–294. doi: 10.2174/156720511795563700
- Shi J, Shaw CL, Du Plessis D, et al. Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol. 2005;110(5):501–512. doi: 10.1007/s00401-005-1079-4
- Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536
- Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108
- Mackenzie IRA. The neuropathology of FTD associated with ALS. Alzheimer Disease & Associated Disorders. 2007;21(4):S44–S49. doi: 10.1097/WAD.0b013e31815c3486
- Mackenzie IRA, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. Journal Of Neurochemistry. 2016;138 Suppl 1(S1):54–70. doi: 10.1111/jnc.13588
- Mann DMA, Snowden JS. Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathol. 2017;27:723–736. doi: 10.1111/bpa.12486
- Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. doi: 10.1038/nature05016
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron [Internet]. 2011 [cited 2023 Dec 21];72(2):245–256. Available from. 10.1016/j.neuron.2011.09.011
- Logroscino G, Piccininni M, Graff C, et al. Incidence of syndromes associated with frontotemporal lobar degeneration in 9 European countries. JAMA Neurol. 2023 Mar 1;80(3):279–286. doi: 10.1001/jamaneurol.2022.5128
- Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. doi: 10.1038/31508
- Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.011
- Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266(8):2075–2086. doi: 10.1007/s00415-019-09363-4
- • Pick A. Uber die Beziehungen der senilen Hirnatrophie zur Aphasie. Prag Med Wchnschr. 1892;17:165–167.
- • Mesulam M‐. Slowly progressive aphasia without generalized dementia. Ann Neurol. 1982;11(6):592–598. doi: 10.1002/ana.410110607
- • Pick A. Zur Symptomatologie der linksseitigen Schläfenlappena-trophie. Monatsschr Psychiatr Neurol. 1904;16:378–388. doi: 10.1159/000219215.
- Alzheimer A. über eigenartige Krankheitsfälle des späteren Alters. Zeitschrift für die gesamte Neurologie und Psychiatrie. 1911;4:356–385. doi: 10.1007/BF02866241
- Onari K, Spatz H. Anatomische Beiträge zur Lehre von der Pickschen umschriebenen Großhirnrinden-Atrophie (“Picksche Krankheit“). Arbeiten aus der Deutschen Forschungsanstalt für Psychiatrie in München (Kaiser-Wilhelm-Institut). 1926;p. 546–587. doi: 10.1007/978-3-642-49740-7_24
- Gans A. Betrachtungen über art und ausbreitung des krankhaiten prozesses in einem fall von pickscher atrophie des stirnhirns. Zeitschrift für die gesamte Neurologie und Psychiatrie. 1923;80:10–28. doi: 10.1007/BF02866679
- Horoupian DS, Thal L, Katzman R, et al. Dementia and motor neuron disease: morphometric, biochemical, and Golgi studies. Ann Neurol. 1984;16:305–313. doi: 10.1002/ana.410160306
- Constantinjdis J, Richard J, Tissot R. Pick’s disease. Histological and clinical correlations. Eur Neurol. 1974;11(4):208–217. doi: 10.1159/000114320
- Brun A. Frontal lobe degeneration of non-Alzheimer type. I. Neuropathology. Arch Gerontol Geriatr. 1987;6(3):193–208. doi: 10.1016/0167-4943(87)90021-5
- Mann DMA, South PW, Snowden JS, et al. Dementia of frontal lobe type: neuropathology and immunohistochemistry. J Neurol Neurosurg Psychiatry. 1993;56:605–614. doi: 10.1136/jnnp.56.6.605
- Shany-Ur T, Rankin KP. Personality and social cognition in neurodegenerative disease. Curr Opin Neurol. 2011;24:550–555. doi: 10.1097/WCO.0b013e32834cd42a
- Snowden JS, Neary D, Mann DMA. Frontotemporal dementia. Br J Psychiatry. 2002;180(2):140–143. doi: 10.1192/bjp.180.2.140
- Neary D, Snowden J, Mann D. Frontotemporal dementia. Lancet Neurol. 2005;4:771–780. doi: 10.1016/S1474-4422(05)70223-4
- Fenoglio C, Scarpini E, Galimberti D. Epigenetic regulatory modifications in genetic and sporadic frontotemporal dementia. Expert Rev Neurother. 2018;18:469–475. doi: 10.1080/14737175.2018.1481389
- Snowden JS, Rollinson S, Thompson JC, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135(3):693–708. doi: 10.1093/brain/awr355
- Devenney EM, Ahmed RM, Halliday G, et al. Psychiatric disorders in C9orf72 kindreds: study of 1,414 family members. Neurology. 2018;91(16):E1498–E1507. doi: 10.1212/WNL.0000000000006344
- Assal F, Cummings JL. Neuropsychiatric symptoms in the dementias. Curr Opin Neurol. 2002;15:445–450. doi: 10.1097/00019052-200208000-00007
- Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. **: doi: 10.1212/WNL.0b013e31821103e6
- Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(9):2456–2477. doi: 10.1093/brain/awr179
- Harris JM, Gall C, Thompson JC, et al. Classification and pathology of primary progressive aphasia. Neurology. 2013;81(21):1832–1839. doi: 10.1212/01.wnl.0000436070.28137.7b
- MacKenzie IRA, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. doi: 10.1007/s00401-009-0612-2
- Mackenzie IRA, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122(1):111–113. doi: 10.1007/s00401-011-0845-8
- Kovacs GG. Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol. 2015;41(1):3–23. doi: 10.1111/nan.12208
- Whitwell JL. FTD spectrum: neuroimaging across the FTD spectrum. Prog Mol Biol Transl Sci. 2019;165:187–223. 10.1016/bs.pmbts.2019.05.009.
- Ranasinghe KG, Rankin KP, Pressman PS, et al. Distinct subtypes of behavioral variant frontotemporal dementia based on patterns of network degeneration. JAMA Neurol. 2016;73(9):1078–1088. doi: 10.1001/jamaneurol.2016.2016
- Josephs KA, Whitwell JL, Weigand SD, et al. Predicting functional decline in behavioural variant frontotemporal dementia. Brain. 2011;134(2):432–448. doi: 10.1093/brain/awq348
- Borroni B, Cosseddu M, Pilotto A, et al. Early stage of behavioral variant frontotemporal dementia: clinical and neuroimaging correlates. Neurobiol Aging. 2015;36:3108–3115. doi: 10.1016/j.neurobiolaging.2015.07.019
- Rohrer JD, Warren JD, Modat M, et al. Patterns of cortical thinning in the language variants of frontotemporal lobar degeneration. Neurology. 2009 [cited 2023 Dec 28];72(18):1562–1569. doi: 10.1212/WNL.0b013e3181a4124e
- Rohrer JD. Structural brain imaging in frontotemporal dementia. Biochim Biophys Acta. 2012;1822:325–332. doi: 10.1016/j.bbadis.2011.07.014
- Ahmed RM, Bocchetta M, Todd EG, et al. Tackling clinical heterogeneity across the amyotrophic lateral sclerosis-frontotemporal dementia spectrum using a transdiagnostic approach. Brain Commun. 2021 Oct 23;3(4):fcab257. doi: 10.1093/braincomms/fcab257
- Zamboni G, Huey ED, Krueger F, et al. Apathy and disinhibition in frontotemporal dementia: insights into their neural correlates. Neurology. 2008;71(10):736–742. doi: 10.1212/01.wnl.0000324920.96835.95
- Le Ber I, Guedj E, Gabelle A, et al. Demographic, neurological and behavioural characteristics and brain perfusion SPECT in frontal variant of frontotemporal dementia. Brain. 2006;129(11):3051–3065. doi: 10.1093/brain/awl288
- Whitwell JL, Josephs KA. Neuroimaging in frontotemporal lobar degeneration—predicting molecular pathology. Nat Rev Neurol. 2012;8(3):131–142. doi: 10.1038/nrneurol.2012.7
- Josephs KA, Whitwell JL, Knopman DS, et al. Two distinct subtypes of right temporal variant frontotemporal dementia. Neurology. 2009;73:1443–1450. 10.1212/WNL.0b013e3181bf9945.
- Bertoux M, Flanagan EC, Hobbs M, et al. Structural anatomical investigation of long-term memory deficit in behavioral frontotemporal dementia. J Alzheimers Dis. 2018;62(4):1887–1900. doi: 10.3233/JAD-170771
- Hornberger M, Piguet O. Episodic memory in frontotemporal dementia: a critical review. Brain. 2012 Mar;135(Pt 3):678–692. doi: 10.1093/brain/aws011. PMID: 22366790.
- Wilson NA, Ramanan S, Roquet D, et al. Scene construction impairments in frontotemporal dementia: evidence for a primary hippocampal contribution. Neuropsychologia. 2020 Feb 3;137:107327.10.1016/j.neuropsychologia.2019.107327
- Wilson NA, Ahmed RM, Hodges JR, et al. Constructing the social world: impaired capacity for social simulation in dementia. Cognition. 2020 Sep;202:104321. doi: 10.1016/j.cognition.2020.104321
- Ratnavalli E, Brayne C, Dawson K, et al. The prevalence of frontotemporal dementia. Neurology. 2002;58(11):1615–1621. doi: 10.1212/wnl.58.11.1615
- Bird T, Knopman D, VanSwieten J, et al. Epidemiology and genetics of frontotemporal dementia/Pick’s disease. Ann Neurol. 2003;Suppl 54(S5):S29–S31. doi: 10.1002/ana.10572
- Grossman M, Seeley WW, Boxer AL, et al. Frontotemporal lobar degeneration. Nat Rev Dis Primers. 2023 Aug 10;9(1):40. doi: 10.1038/s41572-023-00447-0
- Goldman JS, Farmer JM, Wood EM, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology. 2005;65(11):1817–1819. doi: 10.1212/01.wnl.0000187068.92184.63
- Borroni B, Bonvicini C, Galimberti D, et al. Founder effect and estimation of the age of the progranulin Thr272fs mutation in 14 Italian pedigrees with frontotemporal lobar degeneration. Neurobiology Of Aging. 2011;32(3):.e555.1–.e555.8. doi: 10.1016/j.neurobiolaging.2010.08.009
- Pickering-Brown SM, Baker M, Nonaka T, et al. Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain. 2004;127(6):1415–1426. doi: 10.1093/brain/awh147
- Heuer HW, Wang P, Rascovsky K, et al. Comparison of sporadic and familial behavioral variant frontotemporal dementia (FTD) in a North American cohort. Alzheimers Dement. 2020;16:60–70. doi: 10.1002/alz.12046
- Lynch T, Sano M, Marder KS, et al. Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia-parkinsonism-amyotrophy complex. Neurology. 1994;44(10):1878–1878. doi: 10.1212/wnl.44.10.1878
- Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):22–35. doi: 10.1038/nrn.2015.1
- Lines G, Casey JM, Preza E, et al. Modelling frontotemporal dementia using patient-derived induced pluripotent stem cells. Mol Cell Neurosci. 2020;109. doi: 10.1016/j.mcn.2020.103553
- Gomez-Suaga P, Mórotz GM, Markovinovic A, et al. Disruption of ER-mitochondria tethering and signalling in C9orf72-associated amyotrophic lateral sclerosis and frontotemporal dementia. Aging Cell. 2022;21. doi: 10.1111/acel.13549
- Sanders DW, Kaufman SK, DeVos SL, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82(6):1271–1288. doi: 10.1016/j.neuron.2014.04.047
- Shi Y, Zhang W, Yang Y, et al. Structure-based classification of tauopathies. Nature. 2021;598(7880):359–363. doi: 10.1038/s41586-021-03911-7
- Narasimhan S, Guo JL, Changolkar L, et al. Pathological tau strains from human brains recapitulate the Diversity of Tauopathies in Nontransgenic Mouse Brain. J Neurosci. 2017;37(47):11406–11423. doi: 10.1523/JNEUROSCI.1230-17.2017
- Yokoyama JS, Karch CM, Fan CC, et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol. 2017;133(5):825–837. doi: 10.1007/s00401-017-1693-y
- Snowden JS, Adams J, Harris J, et al. Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:497–505. doi: 10.3109/21678421.2015.1074700
- Cruts M, Gijselinck I, Van Der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–924. doi: 10.1038/nature05017
- Paushter DH, Du H, Feng T, et al. The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 2018;136(1):1–17. doi: 10.1007/s00401-018-1861-8
- Yu CE, Bird TD, Bekris LM, et al. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol. 2010;67(2):161–170. doi: 10.1001/archneurol.2009.328
- Mole SE. The genetic spectrum of human neuronal ceroid-lipofuscinoses. Brain Pathol. 2004;14:70–76. doi: 10.1111/j.1750-3639.2004.tb00500.x
- McGoldrick P, Zhang M, van Blitterswijk M, et al. Unaffected mosaic C9orf72 case: RNA foci, dipeptide proteins, but upregulated C9orf72 expression. Neurology. 2018;90(4):e323–e331. doi: 10.1212/WNL.0000000000004865
- Smeyers J, Banchi EG, Latouche M. C9ORF72: what it is, what it does, and why it matters. Front Cell Neurosci. 2021;15. doi: 10.3389/fncel.2021.661447
- Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. 2018;14(9):544–558. doi: 10.1038/s41582-018-0047-2
- Meijboom KE, Abdallah A, Fordham NP, et al. CRISPR/Cas9-mediated excision of ALS/FTD-causing hexanucleotide repeat expansion in C9ORF72 rescues major disease mechanisms in vivo and in vitro. Nat Commun. 2022;13(1):1–17. doi: 10.1038/s41467-022-33332-7
- Kirola L, Mukherjee A, Mutsuddi M. Recent updates on the genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Mol Neurobiol. 2022;59(9):5673–5694. doi: 10.1007/s12035-022-02934-z
- Van Der Ende EL, Jackson JL, White A, et al. Unravelling the clinical spectrum and the role of repeat length in C9ORF72 repeat expansions. J Neurol Neurosurg Psychiatry. 2021;92(5):502–509. doi: 10.1136/jnnp-2020-325377
- Roberson ED. Mouse models of frontotemporal dementia. Ann Neurol. 2012 Dec;72(6):837–849. doi: 10.1002/ana.23722
- Ahmed RM, Irish M, van Eersel J, et al. Mouse models of frontotemporal dementia: a comparison of phenotypes with clinical symptomatology. Neurosci Biobehav Rev. 2017 Mar;74:(Pt A):126–138.
- Dawson TM, Golde TE, Lagier-Tourenne C. Animal models of neurodegenerative diseases. Nat Neurosci. 2018 Oct;21(10):1370–1379. doi: 10.1038/s41593-018-0236-8
- Rohrer JD, Nicholas JM, Cash DM, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the genetic frontotemporal dementia initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. 2015;14:253–262. doi: 10.1016/S1474-4422(14)70324-2
- Galimberti D, Fumagalli GG, Fenoglio C, et al. Genetic FTD initiative (GENFI). Progranulin plasma levels predict the presence of GRN mutations in asymptomatic subjects and do not correlate with brain atrophy: results from the GENFI study. Neurobiol Aging. 2018;62:.e245.9–.e245.12. doi: 10.1016/j.neurobiolaging.2017.10.016
- Kurnellas M, Mitra A, Schwabe T, et al. Latozinemab, a novel progranulin-elevating therapy for frontotemporal dementia. J Transl Med. 2023;21(1):1–18. doi: 10.1186/s12967-023-04251-y
- Ward M, Carter LP, Huang JY, et al. Phase 1 study of latozinemab in progranulin-associated frontotemporal dementia. Alzheimers Dement (N Y). 2024;10(1):e12452. doi: 10.1002/trc2.12452
- Youn BS, Bang SI, Klöting N, et al. Serum progranulin concentrations may be associated with macrophage infiltration into omental adipose tissue. Diabetes. 2009 Mar;58(3):627–636. doi: 10.2337/db08-1147
- Vesper Bio. Available from: https://www.vesperbio.com/
- Hinderer C, Miller R, Dyer C, et al. Adeno-associated virus serotype 1-based gene therapy for FTD caused by GRN mutations. Ann Clin Transl Neurol. 2020;7(10):1843–1853. doi: 10.1002/acn3.51165
- Kariolis MS, Wells RC, Getz JA, et al. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci Transl Med. 2020;12. doi: 10.1126/scitranslmed.aay1359
- Passage Bio, Inc. - Passage bio announces promising initial data from phase 1/2 clinical trial of PBFT02 in FTD-GRN and updated strategic priorities. Available from: https://www.passagebio.com/investors-and-news/press-releases-and-statements/news-details/2023/Passage-Bio-Announces-Promising-Initial-Data-From-Phase-12-Clinical-Trial-of-PBFT02-in-FTD-GRN-and-Updated-Strategic-Priorities/default.aspx
- Logan T, Simon MJ, Rana A, et al. Rescue of a lysosomal storage disorder caused by grn loss of function with a brain penetrant progranulin biologic. Cell. 2021;184(18):4651–4668.e25. doi: 10.1016/j.cell.2021.08.002
- Prevail. AAV-based gene therapy. Available from: https://www.prevailtherapeutics.com/
- AviadoBio. Available from: https://aviadobio.com/en/
- Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 2016 Jun;17(6):383–395. doi: 10.1038/nrn.2016.38
- Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013 Mar 15;339(6125):1335–1338. doi: 10.1126/science.1232927
- Gao FB, Richter JD, Cleveland DW. Rethinking unconventional translation in neurodegeneration. Cell. 2017 Nov 16;171(5):994–1000. doi: 10.1016/j.cell.2017.10.042
- Wave life Sciences.Available from: https://ir.wavelifesciences.com/news-releases/news-release-details/wave-life-sciences-announces-topline-results-Phase-1b2a-focus-c9
- Li W, Jin Y, Prazak L, et al. Transposable elements in TDP-43-mediated neurodegenerative disorders. PLoS One. 2012;7(9):e44099. doi: 10.1371/journal.pone.0044099
- Liu EY, Russ J, Cali CP, et al. Loss of Nuclear TDP-43 Is Associated with decondensation of LINE retrotransposons. Cell Reports. 2019;27(5):1409–1421.e6. doi: 10.1016/j.celrep.2019.04.003
- Rodriguez-Martin B, Alvarez EG, Baez-Ortega A, et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat Genet. 2020;52(3):306–319. doi: 10.1038/s41588-019-0562-0
- Transposon Therapeutics, Inc. Available from: https://www.transposonrx.com/
- Barker MS, Gottesman RT, Manoochehri M, et al. Proposed research criteria for prodromal behavioural variant frontotemporal dementia. Brain. 2022;145(3):1079–1097. doi: 10.1093/brain/awab365