
ABSTRACT
A 23-year-old man presented with behavioral disinhibition, stereotypies, motor apathy, flattened affect, and inappropriate laughter. CT demonstrated generalized cerebral atrophy. He was admitted with a diagnosis of unspecified psychosis and discharged on antipsychotic medication. He was readmitted 3 months later, was diagnosed with schizophrenia, and antipsychotic medication was continued. Owing to symptom progression and aggressive behavior, he was readmitted 2 months later. CT again demonstrated moderate central and cortical cerebral atrophy. MRI showed severe, stable atrophy with frontotemporal predominance, and he was diagnosed with probable behavioral variant frontotemporal dementia (bvFTD). Over the next year he rapidly deteriorated, with loss of cognitive abilities. Genetic testing revealed several variants, none of which are clearly disease-causing.
Case presentation
Because this patient was under a permanent conservatorship through Ventura County, the Ventura County Public Guardian’s Office gave permission for his genetic testing and presentation of this case report.
In September 2019, a 23-year-old Hispanic man was seen in the emergency department owing to personality changes and altered mental status. He was well until 3 months prior, when he was observed speaking to himself on multiple occasions, including when dropping off his young daughter at school. He began obsessively counting the doors of cars while driving, took an excessive number of daily showers, and ritualistically repeated the same daily routine. He insisted on watching cartoons all day, and his diet consisted mainly of the same fast foods he purchased daily. Formerly social, he became more reserved and spent his time aimlessly standing while speaking and laughing to himself. According to his family, he exhibited odd behaviors such as standing on the sidewalk for hours on end, staring into the windows of neighbors’ homes, and responding to internal stimuli. He continuously repeated the same phrase and frequently inserted his thumbs into his nostrils, which he stated was to check if he was still breathing. Despite his legal resident status, he had a persistent fear of being deported.
His symptoms progressed considerably within the first month of onset, and his wife felt it was no longer safe for her and their young daughter to live with him. He seemed distraught over his wife’s decision to divorce him and repetitively stated, “she divorced me.” The patient moved in with his parents and then with his sister. He was fired from his job in sanitation, owing to his bizarre behaviors of speaking to himself and taking trash to his home.
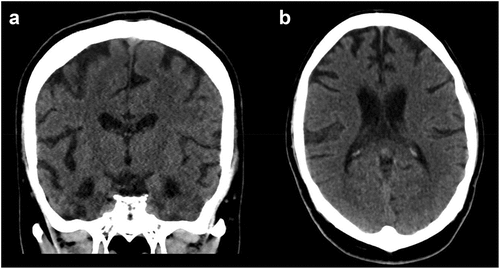
During his first visit to the emergency department, the patient’s sister reported his withdrawn, compulsive, and odd behaviors. He had no family history of psychiatric or neurological disorders. His childhood was uneventful, except for nocturnal enuresis ages 7–15; otherwise, he was described as being very bright. He and his family denied current drug and alcohol use; however, he had a history of intermittent marijuana use from 2013 until a few months prior, and alcohol use until a citation 2-years prior for driving while intoxicated. He denied any history of head trauma or loss of consciousness, although he reported having been assaulted 1 month prior and did not think to defend himself at that time. On examination, he had fluent speech and denied all reported behaviors, but he was repeatedly inserting his thumbs into his nostrils. His drug screen was negative. CT of the head () demonstrated generalized cerebral atrophy, which initially was attributed to a psychotic process, given these changes have been reported in young adult patients with early schizophrenia (Vita et al., Citation2012).
Figure 1. a. Coronal and b. Axial sections of head CT at initial Emergency Department visit in September 2019, 3 months after onset of symptoms.) .
The patient was admitted to a psychiatric facility. Multiple psychiatric evaluations during this hospitalization revealed the patient to have blunted affect, illogical thought process, responses to internal stimuli, and repeated movements of inserting his thumbs into his nostrils. A presumptive diagnosis of unspecified psychosis was made, and he was prescribed an antipsychotic medication. He was discharged 12 days later, with some resolution of his psychotic symptoms, including diminished paranoid thoughts and lessened worry about his breathing. He maintained a blunted affect.
Three months after discharge and six months after symptom onset, in December 2019, he was questioned by the police while standing in front of a house that he claimed was his own. The owner of the house stated the patient did not reside there, and the patient was re-admitted to the same psychiatric facility. He continued to exhibit bizarre behavior, and he lacked insight into his symptoms and the reason for hospitalization. He continued to respond to internal stimuli and repeatedly placed his thumbs into his nostrils. He displayed intermittent auto-echolalia, displayed by his persistent repetition of the psychiatrist’s name throughout the interview, although he was still able to communicate. His drug and alcohol screens were negative. Schizophrenia was diagnosed, and he was discharged on orders to continue antipsychotics.
In February 2020, 8 months after symptom onset, the patient again was admitted to the same psychiatric hospital on an involuntary hold for aggressive behavior toward his sister’s young son. His sister reported the family was being evicted, owing to the patient’s intrusive and aggressive behaviors. The patient had unprovoked screaming, talked excessively, trespassed on neighboring property, and wandered from home. In the hospital, he denied such behavior and showed paranoid thinking, stating that neighbors had stolen his property, and he was trying to get it back. He remained with poor insight into his behaviors and condition. His speech was monotone, with little spontaneity, increased repetition, and echolalia. He continued to loudly respond to internal stimuli. During his stay, his echolalia worsened, and he would urinate on the floor. He became progressively more constricted in his behavior and toward the end of hospitalization was minimally interactive with peers, often pacing around the unit, mumbling to himself. At this time, his family stated they were unable to continue his care.
Physical examinations (except neurological examinations) during his admissions were within normal limits. Neurological examination in May 2020 indicated he was alert, had mumbling speech, intermittently followed simple commands, had intact naming, and had impaired repetition. Cranial nerves were within normal limits: Pupils were equal, regular, and reacted to light; there was no papilledema, visual fields were full, extraocular muscles were intact, his face was symmetrical, tongue was midline and palate was symmetrical, and hearing was intact bilaterally. His gait was intact, reflexes were 5/5 bilaterally, and muscle tone was normal. Sensory examination could not be accomplished reliably.
Temperature, pulse, respiratory rate, and blood pressure were normal. Between September 2019 and July 2020, the patient had fluctuating, low-to-normal red blood cell count (range: 3.84–4.93 10^6/uL), hemoglobin concentration (range: 12.3–15.0 gm/dL), and hematocrit (range: 35.3–44.8%); other hematological measures were normal. CBCs with differentials during this time were normal, except for two incidents of increased monocytes and one incident in July 2020 of neutropenia (43.4%) and lymphocytosis (41.9%). Routine blood chemistry panels were within normal limits, except for consistently elevated serum lipids (triglycerides 111–185 mg/dL, LDL 167–180 mg/dL, total cholesterol 202–239 mg/dL), and fluctuating normal-to-increased bicarbonate concentrations (25–34 mEq/L) with a decreased anion gap. Other laboratory values included increased vitamin B12 (1304 pg/mL), normal folate and copper, and decreased ammonia (<10 µmol/L). Urine toxicology screening was negative for benzodiazepines, cocaine, phencyclidine, amphetamine, cannabis, opiates, and barbiturates. He tested negative for the HIV 1–2 antibody/antigen, syphilis-treponemal antibody, and HSV-1 and HSV-2 DNA.
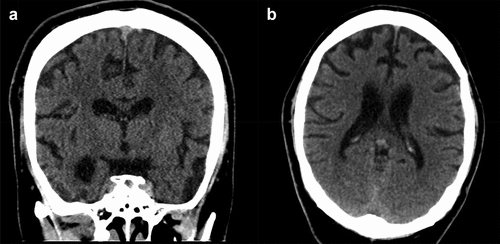
In March 2020, 9 months after symptom onset, a CT of the brain was ordered, which indicated progressive central and cortical cerebral atrophy, without evidence of white matter change, atherosclerosis, intracranial hemorrhage, mass, or acute infarction (). Based on the patient’s clinical presentation and brain imaging results, he met the revised diagnostic criteria for probable behavioral variant frontotemporal dementia (bvFTD) (Rascovsky et al., Citation2011), and this diagnosis was made.
Figure 2. a. coronal and b. axial sections of head CT during hospitalization in March 2020, 6 months after the scan shown in and 9 months after initial symptoms.
During the following month in the psychiatric facility, the patient’s symptoms progressed rapidly. He was mostly mute, with bursts of echolalia and loud, odd pitched tones. For hours, he repeated phrases he heard from the TV. He urinated on himself and the floor, spread his feces on the wall, grabbed food off other patients’ trays, and walked through any open door. His thought process was disorganized and incoherent, with progressive psychomotor retardation; however, he showed no deficit in reading staff name tags. He had little insight into his condition and was emotionally blunted.
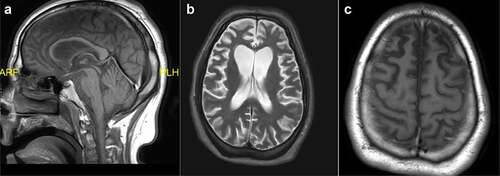
The neurology service was consulted, and an MRI of the brain with and without contrast was ordered to rule out a rapidly progressive dementia. The MRI, performed 11 months after initial symptom onset, showed severe, stable atrophy with frontotemporal predominance, consistent with frontotemporal lobar degeneration (). No acute infarct, hemorrhage, mass lesion, or abnormal enhancement were noted. More specifically, the frontal atrophy was particularly seen to involve the superior frontal and cingulate gyri. There was also the presence of ex-vacuo ventriculomegaly asymmetrically affecting the frontal horns of the lateral ventricles and widening of the medial longitudinal fissure and superior sulcus more pronounced on the right. White matter was unremarkable, without evidence of edema. The diffusion sequence was normal, without evidence of infarct.
Figure 3. Head MRI during hospitalization in May 2020, 11 months after initial symptom onset. a. Sagittal T1-weighted midline image. b. Axial T2-weighted image. c. Axial T1-weighted image.
Further neurological examinations in April and July 2022 indicated he was alert but almost completely nonverbal, and he did not follow simple commands. He was incontinent and had to be spoon-fed. Pupils were equal, regular, and reacted to light; extraocular muscles were intact, his face was symmetrical, and his tongue was midline. He moved all four extremities spontaneously, muscle tone was normal, reflexes were 2+ bilaterally, and he had positive Babinski, palmomental, Hoffman’s, grasp, and snout reflexes. He was discharged from a chronic care facility, continued to deteriorate, and died in early November 2022. His family denied permission for a postmortem examination. summarizes his course over the 41 months from initial behavioral changes to his demise.
Table 1. Patient’s course over 41 months from initial behavioral changes to demise.
Family history
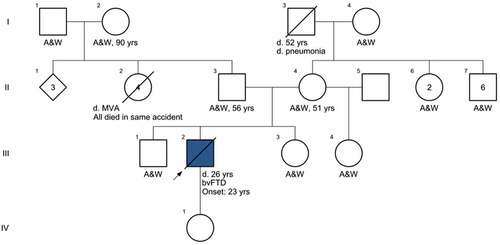
presents the genetic pedigree of the patient as fully as it could be determined from his mother and sister. Both the patient’s paternal and maternal ancestors were from Mexico, and his father and living grandparents reside in Mexico. There is no history of psychiatric illness or dementia in any of his relatives. Consanguinity was denied.
Figure 4. Four-generation family pedigree. A&W, alive and well. d., died. MVA, motor vehicle accident. bvFTD, behavioral variant frontotemporal dementia.
Discussion
This previously healthy 23-year-old man initially presented with personality changes. He developed debilitating dementia with rapidly progressive loss of cognitive abilities in just under 1 year. Given his unique presentation, several diagnoses were considered, as follows:
Psychiatric disorders
Schizophrenia spectrum and psychotic disorders are characterized by severe impairment in five key domains: delusions, hallucinations, thought process, negative symptoms, and psychomotor abnormalities (American Psychiatric Association, Citation2022). Negative symptoms include diminished affect, anhedonia, avolition, alogia, and asociality (Marder & Galderisi, Citation2017).
At initial evaluation, our patient expressed persecutory and somatic delusional beliefs, e.g., he endorsed a fear of deportation despite his legal resident status. He also demonstrated concern and fear that he would stop breathing, and he stereotypically inserted his fingers into his nostrils to check. He displayed grossly disorganized motor behavior marked by both prolonged lingering and inappropriate laughter and child-like noises. Changes in cognition and behavior underscored his negative symptoms, including decreased social interactions, decreased motivation, and a flattened affect. He also exhibited echolalia, which at that point was not severe enough to characterize disorganization of speech, though his mental status exam indicated an illogical thought process.
These initial symptoms were suggestive of an underlying psychiatric disorder. Our patient’s initial working diagnosis, therefore, was unspecified psychosis. His condition, however, was concerning for first-break psychosis. Although his age at symptom manifestation was consistent with the peak age of schizophrenia onset in males (Charlson et al., Citation2018; American Psychiatric Association, Citation2022), there was no history of prodromata such as childhood developmental disorder or previous psychiatric illness. The initial CT scan revealed a reduction of cerebral volume marked by dilatation of the sulci and ventricles. Brain volume reduction with accompanying ventricle enlargement, however, is not infrequently seen in schizophrenic individuals compared to healthy individuals of similar age, as noted above (Olabi et al., Citation2012; Vita et al., Citation2012). As well, our patient showed notable improvement in behavior and mental state after initial treatment with risperidone. Upon his return 3 months later, his continuing symptoms were marked predominantly by negative symptomatology in the setting of possible non-adherence to medication, so that the working diagnosis at this point was schizophrenia.
Neurological disorders
In May 2020, 11 months after the onset of the patient’s symptoms, a brain MRI revealed bilateral generalized cortical atrophy predominantly in the frontotemporal cortices and the hippocampal region. This finding, in conjunction with the patient’s clinical presentation, suggested possible frontotemporal dementia (FTD), although FTD in an individual of such a young age (23) is quite rare (Olabi et al., Citation2012). As well, his mother and sister denied any history of psychiatric or neurological conditions in their family, including dementia.
FTD is a disorder involving deficits in cognition, behavior, language, and movement, with a neuropathology of frontotemporal lobar degeneration (Olney et al., Citation2017; Rabinovici & Miller, Citation2010). It is the second leading cause of dementia and commonly presents in the fifth or sixth decade of life, though occurrences at earlier adult ages have been reported (Cooper & Ovsiew, Citation2013; Olney et al., Citation2017; Rabinovici & Miller, Citation2010). Behavioral variant FTD (bvFTD) is a subtype of FTD with psychotic symptomatology. It is often misclassified as schizophrenia in patients presenting at an early age (Hugo & Ganguli, Citation2014; Landqvist Waldö et al., Citation2015; Rabinovici & Miller, Citation2010). Domains of dysfunction in bvFTD include cognitive dysfunction, disinhibition, apathy, lack of empathy, stereotypical behavior, and hyperorality. Possible bvFTD is diagnosed when a patient demonstrates at least three symptoms (Cooper & Ovsiew, Citation2013; Olney et al., Citation2017); probable bvFTD is diagnosed if the prior criteria are met, along with congruent neuroimaging changes and prominent functional decline. A diagnosis of definite bvFTD additionally requires histopathological confirmation or a pathogenic mutation (Rascovsky et al., Citation2011).
Our patient’s initial behavioral changes included disinhibition and stereotypical and compulsive behaviors such as tapping, echolalia, prolonged showers, and collecting trash from work. Apathy and lack of empathy manifested as decreased affect, inappropriate laughter, and striking out at a young child. Motor apathy was prominent, as he was often seen lingering in fast food restaurants or standing in front of neighbors’ houses. As his illness progressed, his disinhibition and hyperorality increased. He would urinate on the floor, inappropriately touch others, take food from other patients’ trays, and display coprophagia. His symptoms progressed rapidly over the 6 months following his initial presentation, and within nine to 10 months from symptom onset, he demonstrated profound cognitive dysfunction, as he was unable to perform daily activities including feeding, grooming, and toileting.
CT imaging at initial ER presentation in September 2019, 3 months after the onset of symptoms, demonstrated generalized cerebral atrophy with no intracranial hemorrhage, white matter disease, or atherosclerosis. Repeat CT imaging 6 months after onset demonstrated consistent moderate central and cortical cerebral atrophy. The absence of vascular pathology on imaging makes vascular dementia a highly unlikely explanation for the patient’s cognitive decline.
Similar changes in behavior and cognition may occur in several other neurological conditions such as Huntington's disease and leukoencephalopathies like CADASIL. Our patient did not demonstrate any derangements in gross motor function including gait disturbances, muscle weakness, dyskinesia, spasticity, dystonia, or any sensory abnormalities or white matter changes on MRI, making these other conditions less likely. Other neuroanatomical etiologies that can be discounted are chronic traumatic encephalopathy, as the patient has no history of head trauma or brain injury, and malignancy or space occupying lesions, which were not present on neuroimaging.
Other medical conditions
Other conditions that may present with psychotic-like symptoms and mental status abnormalities but have been excluded based on history, chronicity, physical examination, and laboratory evaluation include infections such as viral encephalitis, syphilis, and HIV; metabolic derangements such as thyroid disease, Wilson’s disease, and electrolyte abnormalities; nutritional deficiencies; effects of toxins; and substance use, all of which were ruled out during the serial medical workups.
Genetic disorders
Many cases with early onset bvFTD with prominent psychosis show FUS-aggregates without a known genetic etiology, but genetic causes have been described (Miller et al., Citation2015). Genetic testing was done sequentially by looking for genetic causes of both neurodegenerative diseases and neuropsychiatric disorders. First, Invitae’s ALS/FTD/Alzheimer’s disease panel was completed in order to rule out genetic causes of FTD and other known high-penetrance genetic causes of dementia. This included the most common genetic causes of FTD (C9orf72, GRN, and MAPT), as well as much less prevalent genetic causes. No pathogenic variants (PV), likely pathogenic (LP) variants, or variants of unknown significance (VUS) were identified. Because chromosomal copy number variants (CNV) are enriched in patient populations with schizophrenia (Marshall et al., Citation2017; Raznahan et al., Citation2022), an SNP chromosomal microarray then was performed by GeneDx. Again, no pathogenic, LP or VUS copy number variants nor any regions of homozygosity were identified.
Lysosomal dysfunction has been implicated as a mechanism for FTD both in cases with clear genetic causes (e.g., GRN mutations (Mohan et al., Citation2021) and in cases without a known monogenetic cause that have decreased levels of lysosome gene expression (Sawyer et al., Citation2022). We therefore tested for variants in lysosomal genes via Invitae’s Comprehensive Lysosomal Storage Disorders Panel. The patient was found to be heterozygous for a pathogenic variant in NPC1: c.2974 G>C (p.Gly992Arg) which is consistent with being an unaffected carrier of autosomal recessive Niemann-Pick disease type C. We then tested for genetic causes for leukoencephalopathy. While no white matter disease was found on MRI, it is known that leukoencephalopathies can present with an FTD-like clinical syndrome (Wong et al., Citation2011). Testing identified five additional variants: ATP7A c.2626 + 16 G>A (hemizygous, likely benign), LAMA2 c.6236C>T (p.Thr2079Met) [heterozygous, variant of unknown significance (VUS)], RPS6KC1 c.1729 G>T (p.Asp577Tyr) [heterozygous, VUS], SHPK c.1352C>T (p.Pro451Leu) [heterozygous, VUS], SYNE1 c.613A>G and (p.Ile205Val) [heterozygous, VUS]. See supplementary materials for methods used in each genetic test.
The patient’s lack of motor symptoms made the variants in LAMA2 and SYNE1 unlikely to be significant contributors to his disease. RPS6KC1 has been proposed to be an autosomal recessive cause of brain malformations. Compound heterozygous variants have been identified in a patient with developmental delay, white matter volume loss, periventricular leukomalacia, delayed myelination, cortical dysgenesis, corpus callosum abnormality, and axial hypotonia (Charng et al., Citation2016). Both parents of the patient were heterozygous for their respective variants and neither was reported to have any neurologic disease (Charng et al., Citation2016). Because our patient’s variant is heterozygous, and no second variant was identified on sequencing or microarray, he is likely to be an unaffected carrier, even if this variant was re-classified as pathogenic. Similarly, SHPK variants are proposed to be an autosomal recessive cause of sedoheptulokinase deficiency. The heterozygous parents of the patients in the literature were apparently healthy (Wamelink et al., Citation2015), also pointing to a high likelihood that if this variant was reclassified as pathogenic, our patient would be an unaffected carrier.
The ATP7A gene is located on the X chromosome, so our male patient is hemizygous for the ATP7A c.2626 + 16 G>A variant. If this variant is pathogenic, he would be at risk for ATP7A-related disorders. Pathogenic variants in ATP7A are known to cause malabsorption of copper by the intestine and affect copper transport/homeostasis, thus causing lowered serum copper concentration (Kaler, Citation2011). Serum copper was determined in our patient as a part of Wilson Disease screening and was normal.
The clinical presentations associated with ATP7A currently include Menkes disease, occipital horn syndrome (OHS), and distal motor neuropathy (DMN) (Kaler, Citation2011). Our patient’s normal serum copper does not rule out the possibility that this variant is pathogenic, because copper levels in ATP7A-related DMN are typically normal. This may be because the variants that have been associated with DMN mislocalize copper rather than reduce copper concentrations (Yi & Kaler, Citation2014). This leaves open the possibility that this variant caused our patient’s neurologic disease. Variants associated with DMN also expose a UBX domain in ATP7A, which then interacts with valosin-containing protein (VCP) (Yi & Kaler, Citation2014).
Pathogenic variants in VCP are pleiotropic and can cause one or any combination of the following diseases: Paget’s disease of the bone, inclusion body myopathy, FTD, ALS, or peripheral neuropathy (Charcot-Marie-Tooth disease, type 2Y). The c.2626 + 16 G>A splice variant that was identified could change the structure of the ATP7A protein in multiple ways. While this is a novel, rare variant, it is classified as likely benign, owing to Illumina’s Splice AI in silico model predicting that this variant does not interfere with splicing. In previous work, we found a mutation in the VCP-binding co-factor (SVIP) that specifically recruits VCP to lysosomes that caused bvFTD syndrome in a man in his 50s (Johnson et al., Citation2021). More individuals with this ATP7A variant will need to be identified in order to know if this is a novel lysosomal link to FTD or simply a chance finding.
Functional studies to investigate the effect of this variant on ATP7A-VCP interactions were not able to be done owing to logistical issues; however, RNA sequencing is underway to attempt to better understand the effect of this variant on splicing. Because this variant is novel, the extent to which it impacts the interaction with VCP and/or its relationship to FTD is currently unknown. Whole exome sequencing was completed, but analysis of the data did not lead to any additional variants being identified.
It is possible that some of the variants that were identified contributed to a multigenic cause of disease in our patient. Further analyses and understanding of multigenic disease will be needed before we can ascertain what caused our patient to have bvFTD at such a young age. Parental testing may uncover a de novo cause of our patient’s disease and lead to better understanding of the variants identified so far. Unfortunately, this has not been able to be accomplished to date. Because no pathogenic variants were identified, genetic testing thus was unable to contribute to the diagnostic criteria for bvFTD in our patient.
In summary, we report a very rare presentation of bvFTD in a young man (age 23). Owing to the rapid progression of his symptoms, additional testing could include lumbar puncture to identify infectious, inflammatory, and autoimmune etiologies, including proteins associated with Creutzfeldt-Jakob disease, although his symptoms were not consistent with the latter. Because of the patient’s difficulty in remaining still without sedation, a lumbar puncture was not attempted. To date, no clear genetic cause of his disease has been identified. Whole genome sequencing, RNA studies, and familial testing all could be next steps in better understanding this very early-onset case.
Acknowledgements
Yuri Brito, MD provided early clinical information on the patient. Eric Wallace, MD provided helpful comments on the interpretation of the CT and MRI images. Adit Friedberg, MD provided helpful comments on the preparation of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Because this is a case report, there are no associated research data to be shared.
Additional information
Funding
References
- American Psychiatric Association. (2022). Schizophrenia spectrum and other psychotic disorders. In diagnostic and statistical manual of mental disorders (Fifth Edition, Text Revision ed.). American Psychiatric Association Publishing. https://doi.org/10.1176/appi.books.9780890425787
- Charlson, F. J., Ferrari, A. J., Santomauro, D. F., Diminic, S., Stockings, E., Scott, J. G., McGrath, J. J., & Whiteford, H. A. (2018). Global epidemiology and burden of schizophrenia: Findings from the global burden of disease study 2016. Schizophrenia Bulletin, 44, 1195–1203. https://doi.org/10.1093/schbul/sby058
- Charng, W. L., Karaca, E., Coban Akdemir, Z., Gambin, T., Atik, M. M., Gu, S., Posey, J. E., Jhangiani, S. N., Muzny, D. M., Doddapaneni, H., Hu, J., Boerwinkle, E., Gibbs, R. A., Rosenfeld, J. A., Cui, H., Xia, F., Manickam, K., Yang, Y., Faqeih, E. A. … Lupski, J. R. (2016). Exome sequencing in mostly consanguineous Arab families with neurologic disease provides a high potential molecular diagnosis rate. BMC Medical Genomics, 9, 42. https://doi.org/10.1186/s12920-016-0208-3
- Cooper, J. J., & Ovsiew, F. (2013). The relationship between schizophrenia and frontotemporal dementia. Journal of Geriatric Psychiatry and Neurology, 26, 131–137. https://doi.org/10.1177/0891988713490992
- Hugo, J., & Ganguli, M. (2014). Dementia and cognitive impairment: Epidemiology, diagnosis, and treatment. Clinical and Geriatric Medicine, 30, 421–442. https://doi.org/10.1016/j.cger.2014.04.001
- Johnson, A. E., Orr, B. O., Fetter, R. D., Moughamian, A. J., Primeaux, L. A., Geier, E. G., Yokoyama, J. S., Miller, B. L., & Davis, G. W. (2021). SVIP is a molecular determinant of lysosomal dynamic stability, neurodegeneration and lifespan. Nature Communications, 12, 513. https://doi.org/10.1038/s41467-020-20796-8
- Kaler, S. G. (2011). ATP7A-related copper transport diseases —emerging concepts and future trends. Nature Reviews Neurology, 7, 15–29. https://doi.org/10.1038/nrneurol.2010.180
- Landqvist Waldö, M., Gustafson, L., Passant, U., & Englund, E. (2015). Psychotic symptoms in frontotemporal dementia: A diagnostic dilemma? International Psychogeriatrics, 27, 531–539. https://doi.org/10.1017/S1041610214002580
- Marder, S. R., & Galderisi, S. (2017). The current conceptualization of negative symptoms in schizophrenia. World Psychiatry, 16, 14–24. https://doi.org/10.1002/wps.20385
- Marshall, C. R., Howrigan, D. P., Merico, D., Thiruvahindrapuram, B., Wu, W., Greer, D. S., Antaki, D., Shetty, A., Holmans, P. A., Pinto, D., Gujral, M., Brandler, W. M., Malhotra, D., Wang, Z., Fajarado, K., Maile, M. S., Ripke, S., Agartz, I., Albus, M. … Sebat, J. (2017). Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nature Genetics, 49, 27–35. https://doi.org/10.1038/ng.3725
- Miller, B. L., Dickerson, B. C., Lucente, D. E., Larvie, M., Frosch, M. P., Cabot, R. C., Rosenberg, E. S., Harris, N. L., Shepard, J. A. O., Cort, A. M., Ebeling, S. H., & McDonald, E. K. (2015). Case records of the Massachusetts General Hospital. case 9-2015: A 31-year-old man with personality changes and progressive neurologic decline. The New England Journal of Medicine, 372, 1151–1162. https://doi.org/10.1056/NEJMc1505794
- Mohan, S., Sampognaro, P. J., Argouarch, A. R., Maynard, J. C., Welch, M., Patwardhan, A., Courtney, E. C., Zhang, J., Mason, A., Li, K. H., Huang, E. J., Seeley, W. W., Miller, B. L., Burlingame, A., Jacobson, M. P., & Kao, A. W. (2021). Processing of progranulin into granulins involves multiple lysosomal proteases and is affected in frontotemporal lobar degeneration. Molecular Neurodegeneration, 16, 51. https://doi.org/10.1186/s13024-021-00472-1
- Olabi, B., Ellison-Wright, I., Bullmore, E., & Lawrie, S. M. (2012). Structural brain changes in first episode schizophrenia compared with fronto-temporal lobar degeneration: A meta-analysis. BMC Psychiatry, 12, 104. https://doi.org/10.1186/1471-244X-12-104
- Olney, N. T., Spina, S., & Miller, B. L. (2017). Frontotemporal dementia. Neurology Clinics, 35, 339–374. https://doi.org/10.1016/j.ncl.2017.01.008
- Rabinovici, G. D., & Miller, B. L. (2010). Frontotemporal lobar degeneration: Epidemiology, pathophysiology, diagnosis and management. CNS Drugs, 24, 375–398. https://doi.org/10.2165/11533100-000000000-00000
- Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., Neuhaus, J. Van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain: A Journal of Neurology, 134, 2456–2477. https://doi.org/10.1093/brain/awr179
- Raznahan, A., Won, H., Glahn, D. C., & Jacquemont, S. (2022). Convergence and divergence of rare genetic disorders on brain phenotypes: A review. JAMA Psychiatry, 79, 818–828. https://doi.org/10.1001/jamapsychiatry.2022.1450
- Sawyer, R. P., Hill, E. J., Yokoyama, J., Medvedovic, M., Ren, Y., Zhang, X., Choubey, D., Shatz, R. S., Miller, B., & Woo, D. (2022). Differences in peripheral immune system gene expression in frontotemporal degeneration. Medicine, 101, e28645. https://doi.org/10.1097/MD.0000000000028645
- Vita, A., De Peri, L., Deste, G., & Sachetti, E. (2012). Progressive loss of cortical gray matter in schizophrenia: A meta-analysis and meta-regression of longitudinal MRI studies. Translational Psychiatry, 2, e190. https://doi.org/10.1038/tp.2012.116. Correction at Translational Psychiatry, 3, e275 doi:10.1038/tp.2013.52
- Wamelink, M. M. C., Ramos, R. J. J. F., van den Elzen, A. P. M., Ruijter, G. J. G., Bonte, R., Diogo, L., Garcia, P., Neves, N., Nota, B., Haschemi, A., Tavares de Almeida, I., & Salomons, G. S. (2015). First two unrelated cases of isolated sedoheptulokinase deficiency: A benign disorder? Journal of Inherited Metabolic Disease, 38, 889–894. https://doi.org/10.1007/s10545-014-9809-1
- Wong, J. C., Chow, T. W., & Hazrati, L. -N. (2011). Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia can present as frontotemporal dementia syndrome. Dementia and Geriatric Cognitive Disorders, 32, 150–158. https://doi.org/10.1159/000331422
- Yi, L., & Kaler, S. (2014). ATP7A trafficking and mechanisms underlying the distal motor neuropathy induced by mutations in ATP7A. Annals of the New York Academy of Sciences, 1314, 49–54. https://doi.org/10.1111/nyas.12427
Appendix
Invitae Hereditary Amyotrophic Lateral Sclerosis, Frontotemporal Dementia and Alzheimer Disease Panel: This analysis included next-generation sequencing and deletion/duplication analysis of the following genes: ALS2, ANG, ANXA11, APP, CHCHD10, CHMP2B, DCTN1, ERBB4, FUS, GRN, HEXA, HNRNPA2B1, ITM2B, KIF5A, MAPT, OPTN, PFN1, PRNP, PSEN1, PSEN2, SETX, SNCA, SOD1, SORL1, SPG11, SQSTM1, TARDBP, TBK1, TFG, TREM2, UBQLN2, VAPB, VCP. This testing used next-generation sequencing via a hybridization-based protocol on an Illumina platform with at least 50× read depth. Analysis focused on the coding regions and the 20 base pairs flanking the intron/exon borders. Reads were to the GRCh37 reference sequence. The octapeptide repeat variants in PRNP are not analyzed or reported.
GeneDx SNP microarray: This was performed using the Affymetrix CytoScan HD microarray system, containing 2.67 million probes (1.9 million for CNV detection and 750,000 for SNP detection). The probes were more concentrated in gene-rich regions than gene-sparse regions: on average 880 bases apart vs. 1,700 bases apart, respectively. This microarray is able to identify deletions of ≥25 kb and duplications of ≥50 kb as long as there are enough consecutive probes in the region (25 for deletions and 50 for duplications). Regions of homozygosity are reported if there is one region ≥10 Mb or two ≥8 Mb. The platform is designed to map the human genome build GRCh37.
Invitae Lysosomal Storage Diseases Panel: This analysis included next-generation sequencing and deletion/duplication analysis of the following genes: AGA, ARSA, ARSB, ASAH1, CLN3, CLN5, CLN6, CLN8, CTNS, CTSA, CTSD, CTSK, FUCA1, GAA, GALC*, GALNS, GLA, GLB1, GM2A, GNPTAB, GNPTG, GNS, GUSB, HEXA, HEXB, HGSNAT, HYAL1, IDS*, IDUA, KCTD7, LAMP2, LIPA, MAN2B1, MANBA, MCOLN1, MFSD8, NAGA, NAGLU, NEU1, NPC1, NPC2, PPT1, PSAP, SGSH, SLC17A5, SMPD1, SUMF1, and TPP1. Due to technical limitations, this testing cannot analyze copy number variant of exon 6 of GALC nor complex re-arrangements of IDS.
Invitae Leukodystrophy and Genetic Leukoencephalopathy Panel: This analysis included next generation sequencing and deletion/duplication analysis of the following genes: AARS, AARS2, ABAT, ABCA1, ABCD1, ACADS, ACER3, ACO2, ACOX1, ACP5, ACY1, ADAR, ADGRG1, ADK, ADSL, AGA, AHDC1, AIFM1, AIMP1, AIMP2, ALDH3A2, ALDH5A1, ALDH6A1, ALG2, AMACR, AMPD2, ANK3, AP1S2, AP4B1, AP4E1, AP4M1, AP4S1, APOPT1, APP, ARCN1, ARHGAP31, ARNT2, ARSA, ARX*, ASNS, ASPA, ASXL1, ASXL2, ATP13A2, ATP7A, ATP7B, ATP8A2, ATPAF2, ATRN, AUH, B3GALNT2, BCAP31, BCL11B, BCS1L, BMP4, BOLA3, BRAT1, C12orf65, C19orf12, CACNA1A*, CARS2, CCDC88A, CHMP2B, CLCN2, CLCN7, CLN6, CLP1, CLPP, CNTNAP1, COASY, COL4A1, COL4A2*, COQ2, COQ7, COQ8A, COQ9, COX10*, COX14, COX15, COX20, COX6B1, COX7B, COX8A, CP, CPLX1, CPS1, CRAT, CSF1R, CTBP1, CTC1, CTDP1*, CTNS, CTSA, CYP27A1, CYP2U1, CYP7B1, D2HGDH, DAG1, DARS, DARS2, DBT, DCAF17, DDC*, DDHD2, DDOST, DEAF1, DEGS1, DGUOK, DHFR, DLL4, DNM1L, DNM2, DOCK6, DPYS, DYRK1A, EARS2, EDNRB, EGR2, EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5, ELOVL4, ENTPD1, EPG5, EPRS, ERCC2, ERCC3, ERCC6, ERCC8, ETFA, ETFB, ETFDH, ETHE1, FA2H, FAM126A, FARS2, FARSB, FASTKD2, FBXL4, FDX2, FGD4, FGFRL1, FH*, , FKRP, FKTN, FOLR1, FOXC1, FOXG1, FOXRED1, FTL, FUCA1, GAA, GALC*, GALT, GAN, GBE1, GCDH, GDAP1, GFAP, GFM1, GFM2, GJA1, GJB1, GJC2, GLA, GLB1, GLDC, GLRX5, GLUL, GLYCTK, GNAO1, GRM7, GRN, GTF2H5, GTPBP2, HEPACAM, HEXA, HIBCH, HIKESHI, HK1, HMGCL, HSD17B4, HSPD1, HTRA1, IBA57, IDH2, IDS*, IDUA, IER3IP1, IFIH1, ISCA1, ISCA2, ITPA, JAM3, KARS, KCNJ10, KCNT1, KIAA1161, KIF1A, KIF5A, L2HGDH, LAMA1, LAMA2, LAMB1, LARGE1, LETM1, LIAS, LIPT1, LIPT2, LMNB1, LONP1, LRPPRC, LYRM7, MAG, MAN2B1, MAPT, MARS2, MAT1A, MCOLN1, MEF2C, MGP, MLC1, MLYCD, MOCS1, MOCS2A, MOCS2B, MPLKIP, MPV17, MPZ, MRPL44, MRPS16, MRPS22, MTFMT, MTHFR*, MTR, MTRR, MUT, NADK2, NAXD, NAXE, NDRG1, NDUFA1, NDUFA10, NDUFA11, NDUFA12, NDUFA2, NDUFA9, NDUFAF1, NDUFAF2, NDUFAF3, NDUFAF4, NDUFAF5, NDUFAF6, NDUFB3, NDUFB8, NDUFB9, NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS6, NDUFS7, NDUFS8, NDUFV1, NDUFV2, NFU1, NGLY1, NKX6-2, NOTCH1, NOTCH3, NPC1, NPC2, NRXN1, NUBPL, NUP62, OCRL, OSGEP, OTC, PAFAH1B1, PAH, PANK2, PARS2, PC, PCDH12, PDYN, PET100, PEX1, PEX10, PEX11B, PEX12, PEX13, PEX14, PEX16, PEX19, PEX2, PEX26, PEX3, PEX5, PEX6, PEX7, PGAP1, PGK1, PHGDH, PHYH, PIGA, PINK1, PLA2G6, PLEKHG2, PLP1, PMP22, PNPT1, POLG, POLG2, POLR1A, POLR1C, POLR3A, POLR3B, POMGNT1, POMK, POMT1, POMT2, PPP2R1A, PPT1, PRF1, PRKDC, PRNP*, PRPS1, PSAP, PSAT1, PSEN1, PTEN*, PURA, PUS3, PYCR2, QARS, RARS*, RBPJ, REPS1, RMND1, RNASEH2A, RNASEH2B, RNASEH2C, RNASET2, RNF216*, RPIA, RPS6KC1, RRM2B, SAMHD1, SCO1, SCO2, SCP2, SDHA*, SDHAF1, SDHB, SDHD, SEPSECS, SERAC1, SH3TC2, SHPK, SLC13A3, SLC16A2, SLC17A5, SLC19A3, SLC1A2, SLC1A4, SLC20A2, SLC25A1, SLC25A12, SLC25A22, SLC25A4, SLC25A42, SLC33A1, SLC35A2, SLC46A1, SLC6A8, SLC6A9, SNIP1, SNORD118, SNRPB, SON, SOX10, SPART, SPAST, SPATA5, SPG11, SPG7, SPTAN1, SQSTM1, SSR4, STAMBP, STAT1, STN1, STX11, STXBP1, STXBP2, SUCLA2, SUMF1, SURF1, SYNE1, TACO1, TAF2*, TARS2, TBC1D24, TBCD, TBCK, TIMM50, TM4SF20*, TMEM106B, TMEM126B, TMEM165, TMEM70, TMTC3, TPI1, TRAPPC11, TRAPPC9, TREM2, TREX1, TRMT10A, TRMT5, TSC1*, TSEN54, TTC19, TUBB2A*, TUBB4A, TUFM, TWNK, TYMP, TYROBP, UBE2A, UFM1, UGT1A1, UNC13D, UPB1, VARS2, VCP, VPS11, VPS33A, WARS2, WDR45, WHSC1, WWOX, ZEB2, ZFYVE26 and ZNF335. For technical reasons, this test cannot analyze exon 2 of TUBB2A, exons 2 or 6 of RNF216, exon 6 of COX10, variants in CTDP1 other than c.863 + 389C>T, more than 10 cds around exon 21 of TSC1, the octapeptide repeats of PRNP, copy number variants of exon 6 of GALC, copy number variants of exons 10–11 of DDC, FH sequencing for more than 10 cds of exon 9, complex rearrangements of IDS, analysis of exon 2 of TAF2, CNV analysis of exon 14 of RARS, analysis of econ 21 of COL4A2, CNV analysis of exon 1 of TM4SF20, more than 10 cds of exon 8 of PTEN, polyaniline expansions of ARX, CNV analysis of SDHA, analysis of exon 14 of SDHA, more than 10 cds of exons 6–8 of SDHA, or trinucleotide repeat expansions of CACNA1A. MTHFR c.665C>T (p.Ala222Val) (aka 677C>T) and c.1286A>C (p.Glu429Ala) (aka 1298A>C) variants are not reported unless specifically requested.
Variants identified in our patient: