
ABSTRACT
Background
The objective of this non-interventional, observational prospective cohort study (CONNECT-IBD) was to assess the use of CT-P13 (Inflectra®) in the treatment of patients with Crohn’s disease (CD) and ulcerative colitis (UC) in the context of treatment with reference infliximab (IFX; Remicade®).
Methods
Patients (recruited April 2015 to October 2018) at 150 sites across 13 European countries were followed for up to 2 years. Primary outcomes were safety, population characteristics, and drug utilization patterns. Secondary outcomes included clinical assessment of disease activity. Data were analyzed descriptively.
Results
Overall, 2543 patients (CD, n = 1676; UC, n = 867) were included. In the CT-P13 cohort (n = 1522), median disease duration was 63 (0–579) months and 30% of patients were IFX naïve; median duration of prior IFX treatment was 5 months. During the observation period, median duration of drug exposure was 14 (0–28) months. 41% of patients reported 912 all-causality treatment-emergent adverse events (TEAEs); 24% experienced treatment-related TEAEs. Most TEAEs were of mild-to-moderate severity. Treatment-emergent serious adverse events were reported by 17% of patients.
Conclusion
Safety information for CT-P13 in this large study was consistent with the known safety profile for IFX and did not alter the established benefit-risk profile of CT-P13.
1. Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) are the most common forms of inflammatory bowel disease (IBD) [Citation1]. Over the past two decades, the introduction of biologic drugs has profoundly transformed the treatment of IBD for patients with moderate-to-severe disease activity that is refractory to conventional therapies [Citation2]. Infliximab (IFX) is a chimeric human-murine immunoglobulin G1 (IgG1) monoclonal antibody (mAb). The therapeutic effect of IFX is mediated via tumor necrosis factor-alpha (TNFα) blockade. IFX neutralizes proinflammatory cytokine activity and is indicated for and used to treat CD, UC, rheumatoid arthritis (RA), ankylosing spondylitis (AS), psoriatic arthritis, and plaque psoriasis [Citation3].
A biosimilar is a biologic product that is highly similar to its licensed biologic reference product (RP) and has proven through a rigorous development program to have no clinically meaningful differences [Citation4,Citation5]. CT-P13 (Inflectra®; Pfizer Europe MA EEIG, Bruxelles, Belgium and Remsima®; Celltrion Healthcare, Hungary Kft, Budapest, Hungary) became the first registered mAb biosimilar following its approval by the Korean regulatory authority (2012) and European Medicines Agency (2013) and has subsequently been approved by regulatory agencies in approximately 110 other countries [Citation6–8]. CT-P13 (Inflectra®; infliximab-dyyb, Hospira [Pfizer], New York, NY) is approved by the US Food and Drug Administration (FDA) for all eligible indications of IFX-RP (Remicade®; Janssen Biotech, Inc., Horsham, PA and Janssen Biologics B.V., Leiden, The Netherlands) [Citation9].
The extensive development program supporting the approval of CT-P13 included two large, double-blind, randomized clinical trials in patients with AS and RA, which demonstrated the clinical equivalence of CT-P13 and IFX-RP in terms of pharmacokinetics, clinical efficacy, and safety [Citation10,Citation11]. Post licensure, CT-P13 was shown to be non-inferior to IFX-RP in patients with active CD in a randomized, double-blind, Phase III clinical trial [Citation12].
Gaining knowledge about the utilization patterns of CT-P13 in clinical practice can contribute to improving the evidence base for prescribers tasked with making informed clinical decisions that are in the best interests of their patients from a medical and an affordability standpoint. Here, we report the final data collected during a post-marketing observational cohort study (CONNECT-IBD) of patients with CD or UC in real-world clinical settings who were treated with CT-P13 or IFX-RP for up to 24 months. The primary aims of this non-interventional, prospective, observational study were to characterize the population and drug utilization patterns of patients treated with CT-P13 and to assess its long-term safety profile in the context of treatment with IFX-RP in clinical practices. A subset of interim safety data from CONNECT-IBD was previously reported as part of the pooled patient data used in the European Medicines Agency post-authorization safety studies, which added to the consortium of real-world data for CT-P13 [Citation13].
2. Materials and methods
2.1. Study design
This multinational, multi-center, observational prospective cohort study (ClinicalTrials.gov Identifier, NCT02539368; EudraCT, 2014–005192–89) was conducted at 150 academic and community sites across 13 European Union (EU) countries (Belgium, Czech Republic, Finland, France, Germany, Greece, Hungary, Italy, The Netherlands, Portugal, Slovakia, Spain, and the United Kingdom) in which CT-P13 was approved and IFX-RP or CT-P13 could be prescribed for the treatment of moderate-to-severe CD and UC according to the approved product labels (EU Summary of Product Characteristics) [Citation14,Citation15].
2.2. Patients
This was a purely prospective observational study with consecutive recruitment in the study. Patients deemed potentially eligible by their physician were recruited between 22 April 2015, and 31 October 2018, at each investigative site during usual care and invited to participate. The target study population included patients with CD or UC who were being treated or initiating treatment with CT-P13 or IFX-RP at the time of enrollment.
At the time of study invitation, consenting patients were enrolled if they were eligible for inclusion in either the CT-P13 or the IFX-RP cohort and if they met study inclusion and exclusion criteria. Patients eligible for study inclusion were ≥12 years of age at the time of initial confirmed diagnosis of CD or UC, and ≥18 years of age at enrollment. Patients with fistulizing disease, stomas, or surgery/pouch, or receiving combination therapy, were included. Surgery status was a categorical variable defined as ‘yes’ if the patient had a prior surgical procedure for the treatment of CD or UC, and ‘no’ otherwise. Patients with any reported contraindications to CT-P13 or IFX-RP, or known hypersensitivities, including severe, acute infusion reactions to IFX, its excipients, or other murine proteins, or with a prior history of failure to respond to IFX-RP or CT-P13, were excluded from study entry.
Patients were enrolled over an approximate 30-month period and study participation continued up to a 2-year follow-up period, or until the end of the last patient’s 1-year follow-up, whichever occurred first. Patients who permanently discontinued CT-P13 or IFX-RP treatment were encouraged to remain in the study until the end of the follow-up period.
2.3. Data capture and quality assurance
Data for this observational prospective study were captured using the DataTrak One database, a secure, web-based electronic data capture (EDC) system (DataTrak International, Inc.). All sites were fully trained in using the EDC system, including case report form completion guidelines. Patients’ medical information, recorded within 10 days of each treatment or clinical visit, was extracted from the EDC system at enrollment, and at least quarterly during the study period (Supplementary File 1).
2.4. Ethical considerations
Each patient completed a written informed consent form prior to enrollment. All protocol and informed consent documents were approved by the Institutional Review Boards and/or Independent Ethics Committees of each participating study site. In compliance with the observational methodology of the study, there was no study visit mandated per study protocol. Patients’ visit schedules were aligned with the local standard of care practices, which usually coincided with each patient’s individual infusion schedule; any additional visits were at the discretion of the treating physician. Sites that conduct immunogenicity analysis as part of their local clinical care practice offered their patients the option to participate in this testing, but there was no central lab option for immunogenicity testing. Immunogenicity data were described, and no attempt was made to draw an inference on similarity. The original protocol was amended twice. Details of the amendments and the final study protocol are available at ClinicalTrials.gov (https://clinicaltrials.gov/ct2/show/NCT02539368).
2.5. Study assessments
Primary endpoints included patient demographic characteristics, clinical and diagnostic characteristics (relevant medical history, including prior treatments for CD or UC), drug utilization patterns, concomitant therapies for the management of CD or UC, and long-term safety (adverse events [AEs]). For the evaluation of concomitant therapies, the World Health Organization drug dictionary enhanced B2 September 2018 coding dictionary was applied. Percentages were based on the number of patients in each treatment group. Patients were counted only once in each drug class or preferred name. Safety evaluations included the incidence of treatment-emergent AEs (TEAEs) including serious AEs (SAEs), events in a special situation (e.g. pregnancy, exposure during breast feeding, medication error, overdose, misuse), and AEs of special interest (AESIs) reported from individual investigator sites into Pfizer’s global safety database. AEs were considered treatment emergent if the event started or worsened in severity after the start of study treatment until the end of the observation period. All reported AEs corresponded with the Medical Dictionary for Regulatory Activities version 21.1, including terms and algorithms for AESI identification, as outlined in the study protocol.
Amendment 2 to the protocol in 2017 mandated non-serious AEs (NSAEs) (a subset of all AEs) to be collected prospectively from the date of the approved amendment in each represented country through the end of the study. This amendment took several months to become effective across the study locations in various regions and significantly truncated the collection period for NSAEs in some regions.
Secondary outcome measures included effectiveness based on assessments of overall disease activity (collected in patient case report forms) and as measured relative to baseline using the Harvey-Bradshaw Index (HBI; for patients with CD), which included the Global Patient Assessment, and the Partial Mayo Scoring System (PMSS; with endoscopy subscore excluded, for patients with UC and data available from ≥1 of 3 Mayo subscores) including the physician’s global assessment. Effectiveness was defined as a measure of the extent to which a specific intervention, procedure, regimen, or service in routine standard of care circumstances, performed as it was intended for the specified population. Disease activity was categorized as clinical remission (HBI score <5 or a PMSS score <2), mild (HBI score 5–7 or a PMSS score 2–4), moderate (HBI score 8–16 or a PMSS score 5–6), or severe (HBI score >16 or a PMSS score >6). Although there is no consensus for the definition of loss of response (LOR), for this analysis, LOR was defined as patients who were in remission at baseline who had a result at the specified time period, and whose disease became active at each time period.
2.6. Statistical analysis
The study planned to enroll approximately 2500 patients, including around 1900 patients in a CT-P13 cohort and the remainder anticipated being initiated or treated with IFX-RP. Descriptive statistics were used to present all data according to each treatment cohort since no statistical inferences were planned for this non-interventional study. Summary calculations for continuous variables indicate the total number (N) of observations, mean, standard deviation (±SD), median, and range (minimum–maximum). Categorical variables are reported as the population number (n) and percentage. No missing data were imputed. Primary endpoints were analyzed using the safety analysis set, defined as all patients who received ≥1 dose of study drug during the observation period. The secondary objective and exploratory endpoints were evaluated using the full analysis set, defined as all patients who received ≥1 dose of study drug and had ≥1 post-dose assessment of any effectiveness endpoints. Patient characteristics and study outcomes are reported for the CT-P13 and IFX-RP cohorts, which were defined as follows:
CT-P13 cohort: naïve to biologic therapy at enrollment and received CT-P13 continuously; or treated with CT-P13 prior to and after enrollment (i.e. continuously); or treated with CT-P13 then switched to another TNFα inhibitor (except IFX-RP) or non-biologic treatment during the study; or switched to CT-P13 from an alternative biologic therapy (not IFX-RP) due to non-responsiveness/intolerance to existing therapy.
IFX-RP cohort: naïve to biologic therapy at enrollment and received IFX-RP continuously; or treated with IFX-RP prior to and after enrollment (i.e. continuously); or treated with IFX-RP then switched to another TNFα inhibitor (except CT-P13) or non-biologic treatment during the study; or switched to IFX-RP from an alternative biologic therapy (not CT-P13) due to non-responsiveness/intolerance.
3. Results
3.1. Patient disposition
A total of 2565 patients enrolled in the study. One patient was not included in the analysis due to a missing diagnosis; overall, 22 patients did not receive treatment and were removed from the analysis set (). A total of 2543 (99.1%) patients received ≥1 dose of treatment, which comprised the safety analysis set used in the assessment of the primary outcome measures. This included 1522 patients (59.3%) in the CT-P13 cohort and 494 patients (19.2%) in the IFX-RP cohort. The remainder of the patients were those who had switched between treatments: switched IFX-RP to CT-P13 (358 [13.9%]); switched CT-P13 to IFX-RP (67 [2.6%]); and multiple switchers (102 [3.9%]). Assessment of the outcomes for patients switched between IFX products (CT-P13 and IFX-RP) was not part of the study objectives, and due to the small sample sizes and anticipated heterogeneity of results for these groups, they are not described in more detail. Overall, 2535 patients (including 1516 and 492 patients in the CT-P13 and IFX-RP cohorts, respectively) had ≥1 post-dose effectiveness assessment, which comprised the full analysis set.
Figure 1. Patient disposition. aSafety analysis set defined as all patients who received ≥1 dose of study drug during the study observation period. bMost common reasons for discontinuation (≥2% in any group). cFull analysis set defined as all patients who received ≥1 dose of study drug during the study observation period and had ≥1 post-dose assessment of any of the effectiveness endpoints.
In the CT-P13 group, 38.9% (n = 592) of patients discontinued from treatment, mainly due to AEs, including AESIs and SAEs (22.5% [n = 343]), or due to the investigator’s decision (15.8% [n = 241]). During the follow-up phase, 26.0% (n = 395) of patients discontinued from the study, predominantly for other reasons not pre-specified (10.3% [n = 157]), or they were lost to follow-up (6.4% [n = 97]). In the IFX-RP cohort, 23.7% (n = 117) of patients discontinued from treatment, primarily due to AEs (14.4% [n = 71]), whereas in the follow-up phase, 19.8% (n = 98) of patients discontinued from the study, mostly due to withdrawal of consent (7.1% [n = 35]).
3.2. Population characteristics and drug utilization patterns
The median age of patients (n = 1522) in the CT-P13 cohort was 38.0 (range, 18–87) years; 50.7% of patients were male (). Most patients in the CT-P13 cohort were White, nonsmokers, had no stoma or fistulizing disease, and did not have a history of cancer nor had they undergone surgery related to the treatment of CD or UC. The median duration of the disease was 63.0 (0–579) months. A total of 70.4% (n = 1071) of patients had received prior IFX treatment for a median duration of 5.0 (0–218.6) months. The remaining 451 (29.6%) were naïve to IFX treatment and 19.2% (n = 292) of patients were biologic naïve. In the IFX-RP cohort (n = 494), the median duration of disease was 112.5 (0–632) months and 33.4% (n = 165) of patients had undergone surgery. Overall, 93.9% (n = 464) of patients had previously received IFX treatment for a median period of 47.7 (0–256.5) months. Approximately 6% (n = 27) of patients in this cohort were biologic naïve.
Table 1. Population characteristics and drug utilization patterns (safety analysis set).
In the CT-P13 cohort, 96.9% (n = 1475) of patients received ≥1 prior medication (Supplementary File 2). Immunosuppressants (90.4% [n = 1376] of patients) and anti-diarrheal or intestinal anti-inflammatory/anti-infective agents (51.2% [n = 779]) were the most used classes of prior medication. TNFα inhibitors were the most used immunosuppressant overall (80.5% [n = 1225] of patients) in this cohort. In the IFX-RP cohort, 98.8% (n = 488) of patients received ≥1 medication previously (immunosuppressants, 97.0% [n = 479]; anti-diarrheals, intestinal anti-inflammatory/anti-infective agents, 41.5% [n = 205]).
3.3. Treatment exposure
In the CT-P13 group, most patients (52.8% [804/1522]) received treatment with a frequency of once every 8 weeks (Q8W) (). The median duration of drug exposure (from first treatment to study completion or treatment discontinuation) in the CT-P13 group was 14.0 (range, 0–27.6) months. Interruption of treatment occurred in 19.8% (n = 301) of patients, with loss of response (LOR), investigator’s decision, and AEs being indicated as the reason in 2.0%, 1.8%, and 1.1% of patients, respectively. Treatment was interrupted in three patients due to hypersensitivity. Immunosuppressants (4.8%) were the most common medication related to the treatment of IBD utilized by patients during treatment interruptions, with 13.0% of patients requiring no additional medication. Overall, 31.5% of patients (n = 479) had a change in dose, with the investigator’s decision (14.0%) and LOR (9.3%) being the most frequent reasons for the change. Dose changes due to the occurrence of AEs (including AESIs/SAEs) occurred in 1.5% of patients. Most changes resulted in a dose increase (n = 409).
In the IFX-RP cohort, the most common dosing regimen was with a frequency of Q8W (50.0% [247/494] of patients). The median duration of treatment (from first treatment to study completion or treatment discontinuation) was 17.7 (0–24.5) months. Treatment interruptions occurred in 12.8% (n = 63) of patients in the IFX-RP cohort, with LOR and AEs being indicated as the reason in 1.2% and 0.6% of patients, respectively. Immunosuppressants (1.8%) related to the treatment of IBD were the most common medication utilized by patients during treatment interruption; 10.1% of patients took no additional medication. Among the 22.3% (n = 110) of patients who had a change in dose (dose reduced, 9.1%; dose increased, 18.0%), these changes were most frequently due to the principal investigator’s decision (12.3%) and LOR (5.5%). A dose change due to the occurrence of AEs was required in 0.6% of patients.
3.4. Concomitant treatments
In the CT-P13 group, most patients (67.3% [1025/1522]) used ≥1 concomitant medication (Supplementary File 3). The most common concomitant medications used were immunosuppressants (39.4%), anti-diarrheals and intestinal anti-inflammatory/anti-infective agents (36.1%), and antineoplastic agents (6.4%). In the IFX-RP cohort, 53.0% (262/494) of patients received ≥1 concomitant medication during the study, with 24.7%, 23.5%, and 3.4% of patients receiving immunosuppressants, anti-diarrheals, and intestinal anti-inflammatory/anti-infective agents and antineoplastic agents, respectively.
3.5. Safety
In the CT-P13 cohort, 40.8% (621/1522) of patients reported 912 all-causality TEAEs (). The most reported TEAEs were drug ineffectiveness (13.0% [n = 198] of patients), maternal exposure during pregnancy (2.6% [n = 39]), hypersensitivity (2.2% [n = 34]), and CD (2.0% [n = 30]) (). A total of 24.2% (n = 369) experienced TEAEs that were considered treatment related (Supplementary File 4). Most patients experienced TEAEs that were of mild-to-moderate severity; 8.2% (n = 125) of patients reported severe TEAEs. Treatment-emergent serious AEs (TESAEs) were reported by 16.8% (n = 256) of patients (Supplementary File 5). Most TESAEs were recovered or resolved. A total of 12.4% (n = 189) of patients experienced TEAEs of special interest (TEAESIs); 4.7% (n = 72) of patients reported TEAESIs of infusion-related reactions (Supplementary File 5). The most frequently reported preferred terms (PTs) in the category of TEAESI of infusion-related reactions were hypersensitivity (2.0% [n = 30] of patients) and infusion-related reaction (1.4% [n = 22]). Two patients had a TEAESI of tuberculosis; one patient reported a serious TEAESI of disseminated tuberculosis and one patient reported a serious TEAESI of tuberculosis. TEAESIs of serious infections were reported by 3.3% (n = 50) of patients, with pneumonia (n = 9) being the most frequently reported PT. The percentage of patients who discontinued treatment due to TEAEs was 21.6% (n = 328), with 4.1% (n = 63) discontinuing from the study due to TEAEs.
Table 2. Summary of TEAEs (safety analysis set).
Table 3. Summary of all-causality TEAEs (SOC and PT ≥ 2% of patients in either cohort) (safety analysis set).a
Overall, 26.9% (133/494) of patients in the IFX-RP cohort reported 192 all-causality TEAEs (). The most frequently reported TEAEs were drug ineffectiveness (5.1% [n = 25] of patients), maternal exposure during pregnancy (3.0% [n = 15]), arthralgia (2.2% [n = 11]), and hypersensitivity (2.0% [n = 10]) (). A total of 14.2% (n = 70) of TEAEs were considered treatment related and 5.5% (n = 27) of patients reported severe TEAEs. TESAEs were reported by 8.7% (n = 43) of patients. A total of 9.9% (n = 49) of patients experienced TEAESIs. The percentage of patients who discontinued study drug due to TEAEs was 13.4% (n = 66); 2.4% (n = 12) of patients discontinued from the study due to TEAEs.
There were seven deaths overall during the treatment and follow-up phases of the study. In the CT-P13-treated cohort, sudden death occurred in one patient, and one patient died due to multiple organ dysfunction syndrome (both considered not study–drug related); one patient each died due to lung adenocarcinoma and septic shock, with both deaths considered study–drug related. In the IFX-RP treated cohort, one patient died due to a road traffic accident (not study–drug related) and one patient due to peptic ulcer perforation and septic shock (not study–drug related). One patient who switched from IFX-RP to CT-P13 died due to aortic aneurism rupture, which was not considered study–drug related.
3.6. Assessment of disease activity
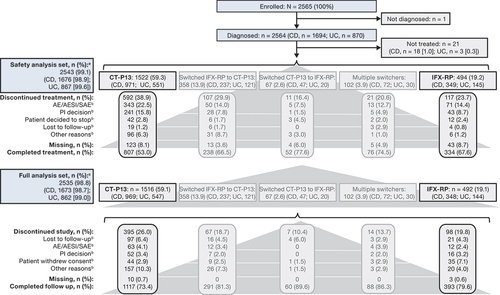
In the CT-P13 group, 57.4% (870/1516) of patients reported remission at 6 months based on physician assessment (). The rate of remission at 24 months for patients in this group was 25.5% (n = 386). In the IFX-RP group, 63.4% (312/492) of patients reported remission at 6 months () and 37.4% (n = 184) at 24 months. The proportions of patients with remission by disease type (CD and UC) for patients in the CT-P13 () and IFX-RP groups () were broadly similar to those for the overall IBD populations.
Figure 2. Rates of clinical remissiona over time following treatment with CT-P13 and IFX-RP for patients with IBD overall, (a) and (b), respectively, and by disease type (CD and UC), (c) and (d) respectively (full analysis set). aBased on physician assessment.
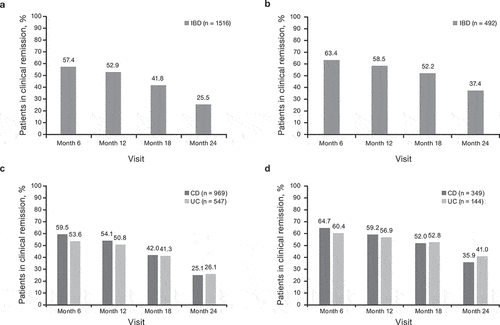
At baseline, 33.9% (514/1516) of patients in the CT-P13 group had active disease (mild, moderate, or severe). Among these patients the proportion with unchanged/worsened disease by 6 months was 38.0% (177/466) and 26.4% (101/382) from 6 months to 12 months (). At baseline, 51.1% (775/1516) of patients in the CT-P13 group were in remission. Of these patients, 14.4% (101/703) and 13.8% (86/622) had active disease (mild, moderate, or severe) by 6 months and from 6 months to 12 months, respectively.
Figure 3. Change in disease status over time for patients with IBD overall (CD and UC) treated with (a) CTP13 and (b) IFX-RP (full analysis set). aChange from baseline to analysis timepoint = (mild disease to mild disease) + (mild disease to moderate disease) + (mild disease to severe disease) + (moderate disease to moderate disease) + (moderate disease to severe disease) + (severe disease to severe disease). bChange from baseline to analysis timepoint = (clinical remission to mild disease) + (clinical remission to moderate disease) + (clinical remission to severe disease).
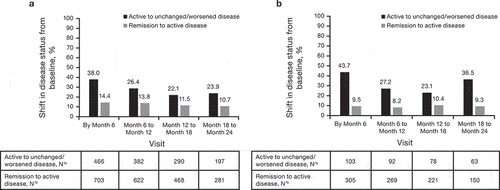
At baseline, 23.8% (117/492) of patients in the IFX-RP group had active disease. Among these patients the proportion with unchanged/worsened disease by 6 months was 43.7% (45/103) and 27.2% (25/92) from 6 months to 12 months (). At baseline, 66.9% (329/492) of patients were in remission. Of these patients, 9.5% (29/305) and 8.2% (22/269) had active disease (mild, moderate, or severe) by 6 months and from 6 months to 12 months, respectively.
4. Discussion
The primary objective of the prospective CONNECT-IBD study, the largest multinational, non-interventional prospective study of CT-P13, was to characterize the patient population receiving CT-P13 and to assess its safety in the context of standard of care (i.e. IFX-RP) in the treatment of patients with CD or UC, in real-world clinical practice. The design of this non-interventional, observational study precludes comparisons and inferences to be made between treatment cohorts, which varied markedly in size and included patients at different stages of treatment during their patient journey with CD or UC. For instance, in the CT-P13 cohort, 30% of patients had not previously received IFX and 19% were biologic naïve. Of those who had previously received IFX (70%), the median treatment duration was 5 months. In contrast, most of the patients in the IFX-RP cohort (94%) had received prior IFX treatment at the time of enrollment, with a median prior treatment duration of 48 months. The median disease duration in the CT-P13 and IFX-RP cohorts was 63 months and 113 months, respectively. During the observation period of the study, median duration of drug exposure for CT-P13 and IFX-RP groups (14 [range, 0–28] months and 18 [0–25] months, respectively) was consistent with historical reports for IFX [Citation16,Citation17].
Review of safety data for patients in this large CT-P13 cohort in CONNECT-IBD did not identify new significant safety information, and the results were consistent with the historical safety profile of IFX [Citation15,Citation18]. The safety findings were also in line with those from other observational studies of CT-P13 in patients with IBD. In the 1-year follow up from the ONWARD study, which included biologic-naive patients and those switching from IFX-RP, AEs were reported by 35% of patients with most being mild or moderate in severity [Citation19]. A lower cumulative rate of AEs (24%) was reported to week 54 in a separate prospective nationwide cohort study conducted in Hungary, with infections and infusion reactions being the most common AE [Citation20]. Based on 11 observational studies of IBD patients included in a systematic review, pooled rates of AEs were slightly higher among studies where patients switched to CT-P13 (CD 0.10 [95% CI = 0.02–0.31]; UC 0.22 [0.04–0.63]), compared with non-switching studies (CD 0.08 [0.02–0.26]; UC 0.08 [0.03–0.17]) [Citation21].
The imbalance in the numbers of patients and their characteristics between the CT-P13 and IFX-RP cohorts mandates cautious interpretation of any numerical differences in safety outcomes. Moreover, negative patient attitudes toward biosimilars or nocebo effects are known to adversely impact treatment outcomes and cannot be overlooked in the current study [Citation22]. For instance, the nocebo effect may have contributed to the greater numerical occurrence of the drug ineffectiveness PT in the CT-P13 group compared with the IFX-RP group among all-causality TEAEs (13.0% vs 5.1%) (and in treatment-related TEAEs [24.2% vs 14.2%]), which largely contributed to the overall difference between groups in the percentage of patients with TEAEs. The most frequently reported TESAEs were within the system organ class of gastrointestinal disorders (7.8% [CT-P13] and 3.2% [IFX-RP]), were largely reflective of underlying disease (PTs of UC and colitis and CD) and accounted for the overall percentages of TESAEs seen between groups (16.8% [CT-P13] and 8.7% [IFX-RP] of patients). There were no clinically meaningful differences for other TESAEs between the CT-P13 and IFX-RP treatment groups. The overall percentages of TEAESIs were similar between CT-P13 and IFX-RP treatment groups, with the rates for all observed TEAESIs consistent with those in the approved product labels [Citation14,Citation15].
Non-inferiority in effectiveness of CT-P13 and IFX-RP has been established in a clinical trial setting in patients with CD [Citation12]. A subgroup analysis of the 52-week, randomized, Phase IV NOR-SWITCH study (including a 26-week open-label extension trial) showed that there was no difference in changes in disease activity and other outcome measures between patients with stable or active CD and UC maintained on IFX-RP or switched to CT-P13 from IFX-RP at enrollment [Citation23]. A secondary objective in CONNECT-IBD was to assess the effectiveness of CT-P13 in the context of standard of care with IFX-RP in the real world. Amongst patients with active disease at baseline, 38.0% in the CT-P13 group (and 43.7% of patients in the IFX-RP group) had unchanged or worsened disease by 6 months. In the CT-P13 group, 57.4% of patients reported remission at 6 months based on physician assessment, and 63.4% of patients in the IFX-RP group were in remission at this timepoint. The remission rates observed here appear higher than those reported in randomized placebo-controlled trials of IFX-RP in biologic-naïve patients with CD [Citation24] or UC [Citation25], which most likely is due to the substantial differences in outcome definitions, study designs, and patient populations.
Of patients who were in remission at baseline, 14.4% in the CT-P13 group (and 9.5% of patients in the IFX-RP group) had active disease at 6 months. Amongst patients with active disease at baseline, the proportion with unchanged/worsened disease at 6 months determined here for CT-P13 and IFX-RP are in line with those reported for primary non-response to anti-TNFα induction in clinical trials (10–40%) and clinical case series (10–20%) [Citation26,Citation27]. Following an initial response to an anti-TNFα induction regimen, rates of LOR at 12 months of 23–46% have been reported [Citation28–31].
Interpretation of the results in CONNECT-IBD is limited by the non-interventional, observational study design, which may have introduced survivor and/or selection bias. Only patients that initiated and continued CT-P13 or IFX-RP treatment entered the study, who may differ in demographic and clinical characteristics from those who initiated but discontinued treatment prior to enrollment. The higher proportion of biologic-naïve patients in the CT-P13 cohort may have had an impact on study outcomes, with these patients potentially having less advanced disease and still establishing stable treatment doses, compared with biologic-experienced patients. The absence of statistical analyses limits comparison of the findings between the CT-P13 and IFX-RP cohorts. The study protocol did not mandate the entry of medical information into patient charts. Rather, each participating study site documented patient care and provided outcomes according to usual care, physician discretion, and local practice standards. Some study variables were unavailable for all patients at all data collection timepoints (e.g. when data were not recorded in the chart for routine medical care).
5. Conclusion
CONNECT-IBD, the largest multinational non-interventional prospective, observational cohort study of CT-P13, supports the data from previous clinical studies of CT-P13 regarding safety and effectiveness. For patients with IBD, irrespective of the duration of disease or prior treatment with IFX, treatment remission rates with CT-P13 were within expected ranges. No clinically meaningful differences were observed over 24 months in this descriptive analysis between patients receiving CT-P13 with respect to the known profile of IFX, in terms of safety, effectiveness, and other outcome measures. The results of this study add to the long-term real-world data available for the utilization of CT-P13 in clinical practice for patients with IBD and serve to further inform prescribers and clinical decision making. Although the findings from this non-comparative study were descriptive and not conclusive in nature, the safety information of this multinational, multi-center, observational cohort study was consistent with the historical safety profile of IFX [Citation15,Citation18] and did not identify new significant safety information that would change the established benefit–risk profile of CT-P13.
Declaration of interest
B Bokemeyer has received grants, research funding, consulting fees, or has served as a speaker for AbbVie, MSD, Shire, Ferring, UCB, Hospira, Takeda, Shield Therapeutics, Pfizer, Biogen, Janssen, Hexal, Celgene, Boehringer Ingelheim, Allergan, Celltrion, Merckle, Dr. Falk Pharma GmbH, HLR, Mundipharma, Given Imaging, and Galapagos. T Hlavaty has received research funding from Hospira Inc and has served as a speaker for AbbVie, Takeda Pharmaceuticals, Janssen Pharmaceuticals, Pfizer, Egis Pharmaceuticals PLC, and Amgen Inc. M Allez has served as a speaker, a consultant, and advisory member for, or has received research funding from Janssen, Roche/Genentech, Takeda, Pfizer, Celgene, Novartis, Amgen, Biogen, MSD, Ferring, and Tillotts. JP Gisbert has served as a speaker, a consultant, and advisory member for, or has received research funding from MSD, AbbVie, Hospira, Pfizer, Kern Pharma, Biogen, Takeda, Janssen, Roche, Sandoz, Celgene, Ferring, Faes Farma, Shire Pharmaceuticals, Dr. Falk Pharma, Tillotts Pharma, Chiesi, Casen Fleet, Gebro Pharma, Otsuka Pharmaceutical, and Vifor Pharma. M Mueller, S Moosavi, MJ Cadatal, and KF Liau are employees of and own stock or options in Pfizer. H Fowler has received consultancy fees from Pfizer. P Selema was an employee of Pfizer at the time the work was conducted and owns stock or options in Pfizer.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
All authors made substantial contributions to the conception or design of the work, or the acquisition, analysis, or interpretation of data. All authors contributed to the development of the first draft of the manuscript, revised it critically for important intellectual content, and read and approved the final version for publication.
Ethics statement
All protocol and informed consent documents were approved by the Institutional Review Boards and/or Independent Ethics Committees of each participating study site. Each patient completed a written informed consent form prior to enrollment.
Data sharing statement
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Trial Registration
ClinicalTrials.gov. NCT02539368
EOBT-2023-ST-0020_Supplemental_Material.docx
Download MS Word (187 KB)Acknowledgments
Medical writing support was provided by Sue Reinwald and Iain McDonald of Engage Scientific Solutions and was funded by Pfizer.
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14712598.2023.2200883.
Additional information
Funding
References
- Fakhoury M, Negrulj R, Mooranian A, et al. Inflammatory bowel disease: clinical aspects and treatments. J Inflamm Res. 2014;7:113–120.
- Cohen LB, Nanau RM, Delzor F, et al. Biologic therapies in inflammatory bowel disease. Transl Res. 2014;163(6):533–556. DOI:10.1016/j.trsl.2014.01.002
- Melsheimer R, Geldhof A, Apaolaza I, et al. Remicade(®) (infliximab): 20 years of contributions to science and medicine. Biologics. 2019;13:139–178.
- US Food and Drug Administration (FDA). Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry; 2015 [cited 2020 15 Feb]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
- US Food and Drug Administration (FDA). Patient materials, biosimilars; 2019 [cited 2020 5 Mar]. Available from: https://www.fda.gov/drugs/biosimilars/patient-materials
- Celltrion Healthcare. Remsima®; 2020 [cited 2020 19 Feb]. Available from: https://www.celltrionhealthcare.com/en-us/products/product?pkey=5
- GaBi online (Generics and Biosimilars Initiative). Biosimilars of infliximab; 2015 [cited 2020 10 Dec]. Available from: http://www.gabionline.net/Biosimilars/General/Biosimilars-of-infliximab
- Gabbani T, Deiana S, Annese V. CT-P13: design, development, and place in therapy. Drug Des Devel Ther. 2017;11:1653–1661.
- Hospira (a Pfizer Company). Inflectra (infliximab-dyyb) US prescribing information; 2016 [cited 2020 18 Oct]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125544s000lbl.pdf
- Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72(10):1605–1612. DOI:10.1136/annrheumdis-2012-203091
- Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72(10):1613–1620. DOI:10.1136/annrheumdis-2012-203090
- Ye B, Pesegova M, Alexeeva O, et al. Efficacy and safety of biosimilar CT-P13 compared with originator infliximab in patients with active Crohn’s disease: an international, randomised, double-blind, phase 3 non-inferiority study. Lancet (London, England). 2019;393:1699–1707.
- Lee SJ, Baek K, Lee S, et al. Post-marketing pooled safety analysis for CT-P13 treatment of patients with immune-mediated inflammatory diseases in observational cohort studies. BioDrugs. 2020;34(4):513–528. DOI:10.1007/s40259-020-00421-2
- European Medicines Agency. 2013; Inflectra (infliximab): summary of product characteristics [cited 2020 18 Dec]. Available from: https://www.ema.europa.eu/en/documents/product-information/inflectra-epar-product-information_en.pdf
- European Medicines Agency. Remicade (infliximab): summary of product characteristics; 2009 [cited 2020 10 Dec]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000240/WC500050888.pdf
- D’Haens G, Reinisch W, Colombel JF, et al. Five-year safety data from ENCORE, a European observational safety registry for adults with Crohn’s disease treated with infliximab [Remicade®] or conventional therapy. J Crohns Colitis. 2017;11(6):680–689. DOI:10.1093/ecco-jcc/jjw221
- Panes J, Lindsay JO, Teich N, et al. Five-year safety data from OPUS, a European observational safety registry for adults with ulcerative colitis treated with originator infliximab [Remicade®] or conventional therapy. J Crohns Colitis. 2019;13(9):1148–1157. DOI:10.1093/ecco-jcc/jjz048
- European Medicines Agency. Remsima: ePAR - risk management plan - summary; 2019 [cited 2020 10 Dec]. Available from: https://www.ema.europa.eu/en/documents/rmp-summary/remsima-epar-risk-management-plan-summary_en.pdf
- Abraham B, Eksteen B, Nedd K, et al. Impact of Infliximab-dyyb (Infliximab Biosimilar) on clinical and patient-reported outcomes: 1-year follow-up results from an observational real-world study among patients with inflammatory bowel disease in the US and Canada (the ONWARD Study). Adv Ther. 2022;39(5):2109–2127. DOI:10.1007/s12325-022-02104-6
- Gonczi L, Gecse KB, Vegh Z, et al. Long-term efficacy, safety, and immunogenicity of biosimilar infliximab after one year in a prospective nationwide cohort. Inflamm Bowel Dis. 2017;23(11):1908–1915. DOI:10.1097/MIB.0000000000001237
- Komaki Y, Yamada A, Komaki F, et al. Systematic review with meta-analysis: the efficacy and safety of CT-P13, a biosimilar of anti-tumour necrosis factor-alpha agent (infliximab), in inflammatory bowel diseases. Aliment Pharmacol Ther. 2017;45(8):1043–1057. DOI:10.1111/apt.13990
- Kim H, Alten R, Avedano L, et al. The future of biosimilars: maximizing benefits across immune-mediated inflammatory diseases. Drugs. 2020;80(2):99–113. DOI:10.1007/s40265-020-01256-5
- Jorgensen KK, Goll GL, Sexton J, et al. Efficacy and safety of CT-P13 in inflammatory bowel disease after switching from originator infliximab: exploratory analyses from the NOR-SWITCH main and extension trials. BioDrugs. 2020;34(5):681–694. DOI:10.1007/s40259-020-00438-7
- Hanauer S, Feagan B, Lichtenstein G, et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359:1541–1549.
- Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462–2476. DOI:10.1056/NEJMoa050516
- Ben-Horin S, Kopylov U, Chowers Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun Rev. 2014;13(1):24–30.
- Ding NS, Hart A, De Cruz P. Systematic review: predicting and optimising response to anti-TNF therapy in Crohn’s disease - algorithm for practical management. Aliment Pharmacol Ther. 2016;43(1):30–51.
- Gisbert JP, Panes J. Loss of response and requirement of infliximab dose intensification in Crohn’s disease: a review. Am J Gastroenterol. 2009;104(3):760–767.
- Ungar B, Ben-Shatach Z, Ben-Haim G, et al. Infliximab therapy intensification upon loss of response: is there an optimal trough level? Dig Liver Dis. 2019;51(8):1106–1111. DOI:10.1016/j.dld.2019.02.013
- Guerra Veloz MF, Arguelles-Arias F, Castro Laria L, et al. Loss of efficacy and safety of the switch from infliximab original to infliximab biosimilar (CT-P13) in patients with inflammatory bowel disease. World J Gastroenterol. 2018;24(46):5288–5296. DOI:10.3748/wjg.v24.i46.5288
- Ben-Horin S, Chowers Y. Review article: loss of response to anti-TNF treatments in Crohn’s disease. Aliment Pharmacol Ther. 2011;33(9):987–995.