
ABSTRACT
Introduction
Autoimmune hemolytic anemia (AIHA) treatment has been revolutionized by the introduction of target therapies, mainly monoclonal antibodies (MoAbs).
Areas covered
The anti-CD20 rituximab, which targets Ab production by B-cells, induces 80% of response in warm-type AIHA (wAIHA) and 50–60% in cold agglutinin disease (CAD). Other B-cell targeting MoAbs including ianalumab, povetacicept, and obexelimab are under active study. The anti-CD38 MoAb daratumumab has been used in several reports to target long-lived plasma-cells responsible for AIHA relapse, being effective even in multi-refractory cases. Anti-complement MoAbs will soon change the treatment paradigm in CAD; the anti-C1s sutimlimab rapidly increased Hb in more than 80% of the cases. Finally, MoAbs inhibiting the neonatal Fc receptor (FcRn), such as nipocalimab, can reduce the half-life of the pathogenic autoAbs, representing a promising treatment for wAIHA.
Expert opinion
MoAbs offer the potential to improve efficacy by reducing toxicity. However, there is a huge need for clinical trials exploring response duration rather than short-term efficacy. Complement inhibitors and anti-FcRns do not abrogate autoAb production and are being developed as long-term therapies. Thus, the combination of B-cell/plasma cell targeting drugs deserves to be explored. On the other hand, their rapid efficacy should be exploited for the acute AIHA phase.
1. Introduction
Autoimmune hemolytic anemias (AIHAs) are a heterogeneous group of diseases involving the immune destruction of erythrocytes due to autoantibodies and complement, either within the circulation (intravascular hemolysis, IVH) or in the extravascular space of the spleen and liver (extravascular hemolysis, EVH) [Citation1,Citation2]. After several decades of steroids and splenectomy as the main treatment strategies, the advent of targeting therapies revolutionized the approach by improving long-term efficacy and reducing toxicities. Monoclonal antibodies (MoAbs), particularly rituximab, borrowed from the field of lymphoproliferative disorders, were the protagonists of such change in the late 2000s [Citation1,Citation3]. Since then, several other MoAbs have been investigated in the management of AIHAs with the aim of targeting specific pathogenic mechanisms including B-cell activation and proliferation, long-lived plasma cells, complement cascade, and persistence of the pathogenic autoantibodies [Citation1]. In this review, we will briefly recapitulate AIHAs classification and management and then focus on the various MoAbs, discussing how they may target AIHA pathogenesis and be positioned in the patient journey.
2. Disease overview
2.1. Epidemiology and classification
AIHAs have an incidence rate of about 0.8–3/100.000/year and are diagnosed through the direct antiglobulin test (DAT), which is able to demonstrate anti-erythrocyte autoantibodies on autologous red blood cells (RBCs) [Citation1]. AIHAs are generally classified according to the thermal characteristics and isotypes of the autoantibody into ‘warm’ (wAIHA), “cold agglutinin diseases’’ (CAD), and mixed forms. summarizes the classification of AIHA. WAIHA is sustained by an IgG reactive at 37°C (monospecific DAT positive for IgG with or without complement), whilst CAD is generally due to a pentameric cold reactive IgM (<37°C, cold agglutinins) able to activate the classical complement cascade (DAT positive for complement fractions and cold agglutinin titer >64). If cold AIHA is secondary to an infection (i.e. Mycoplasma pneumonia) or lymphoproliferative disease (i.e. chronic lymphocytic leukemia and non-Hodgkin lymphomas), it is named ‘cold agglutinin syndrome’ (CAS). It is worth mentioning the rare paroxysmal cold hemoglobinuria (PCH), usually secondary to infections in children, which is caused by a biphasic hemolysin that binds to erythrocytes at 4°C and reacts at 37°C. Mixed forms include AIHAs with both a warm IgG and cold IgM and DAT positive for IgG and complement with high titer cold agglutinin. Atypical AIHAs include those with negative DAT, IgA mediated, and the so-called ‘warm IgM AIHAs’ characterized by diagnostic delay, marked intravascular hemolysis, and high mortality [Citation2].
Table 1. Epidemiology and classification of autoimmune hemolytic anemia.
2.2. Physiopathology
From a physiopathology point of view, in wAIHA, IgG mediates antibody-dependent cell-mediated cytotoxicity by macrophages and activated lymphocytes, as well as phagocytosis by the reticuloendothelial system particularly in the spleen (EVH) [Citation3]. Complement activation, which is minor as compared to CAD, is an increasingly recognized event, observed in about one-third of patients [Citation4]. It contributes to the severity of anemia and refractoriness to available treatments [Citation4–6]. Hemolysis is generally acute since it happens in vivo at 37°C and the patient often requires hospital admission and transfusion. In CAD, patients often display an IgM monoclonal component in the peripheral blood and a B-cell clonal lymphoproliferative disorder in the bone marrow without clinical or radiological evidence of malignancy. This led to the inclusion of CAD in the WHO 2022 classification of lymphoproliferative disorders [Citation7]. Cold agglutinins are usually IgM kappa with anti-I specificity and may induce erythrocyte agglutination and complement-mediated hemolysis by activation of the classical pathway. Hemolysis is mainly extravascular in the liver, through opsonization by C3b, but terminal complement activation with IVH may also occur to a lesser extent. Anemia in CAD is often chronic with worsening of fatigue and peripheral symptoms (acrocyanosis and Raynaud’s phenomenon) after exposure to low temperature; the frequency and severity of clinical manifestations also depend on the season and on the thermal range of the autoantibody [Citation1]. Both wAIHA and CAD are at risk of AIHA-related complications, particularly thromboses and infections, which may complicate 10–20% of cases and be related to disease morbidity and mortality [Citation4].
3. Current therapy
It has become clear that therapy should be differentiated in wAIHA and CAD, whilst mixed and atypical forms are often treated like wAIHA [Citation1]. For wAIHA steroids (i.e. prednisone 1 mg/kg day) are used as frontline, followed by rituximab and splenectomy as further lines [Citation3]. All these therapies show high rates of response (generally ~80%), which are, however, durable only in one-third of cases and carry the risk of side effects [Citation3,Citation4,Citation8–10]. Available literature supporting the efficacy and safety of rituximab use as a second line will be discussed in the following paragraph. Splenectomy is currently recommended in patients unresponsive to or relapsing after rituximab, with responses of about 80%. The risk of severe infection and thrombosis should be known and prevented [Citation11]. For patients who are refractory to or relapsing after rituximab and not candidates for splenectomy, few options exist, including cytotoxic immunosuppressants [Citation3,Citation4,Citation8,Citation12] with limited efficacy and several toxicities.
Regarding CAD, a proportion of patients with mild anemia avoidance of low temperatures and folic acid may be sufficient [Citation8,Citation13,Citation14]. In those with symptomatic anemias or disabling peripheral symptoms, rituximab has become the preferred first-line therapy with response rates of 45–60%, and mainly partial. In fact, steroids are effective only at high doses, and splenectomy is contraindicated (since complement mediated EVH mainly occurs in the liver). The advent of complement inhibitors, discussed in a dedicated paragraph, will likely revolutionize treatment for rituximab relapsing/refractory patients and possibly for those experiencing hemolytic exacerbations [Citation15–19].
Mixed AIHA is an uncommon condition, often exhibiting a more aggressive course. Treatment options are limited and primarily involve the use of steroids, rituximab, and splenectomy, with only limited supporting evidence available. Regarding DAT, negative AIHA is diagnosed after the exclusion of other causes of hemolysis (paroxysmal nocturnal hemoglobinuria, congenital hemolytic anemias, thrombotic microangiopathies, intravascular devices, etc.) and on the basis of the response to steroid therapy. Extensive diagnostic work up at referral centers is advised before further therapy lines [Citation1,Citation2].
In all AIHAs, bone marrow compensatory response has emerged as an important determinant of clinical severity. It has been shown that reticulocytosis may be inadequate in more than one-third of patients as did the levels of endogenous erythropoietin [Citation20,Citation21]. In these cases, the use of recombinant erythropoietin may significantly improve the outcome, by increasing Hb levels, while immunosuppressive agents start to work [Citation5]. Finally, for patients with very severe and acute presentation, transfusion support, plasma exchange, and intravenous Ig infusions are available options.
3.1. B-cell targeting Ab based therapies
Rituximab is a B-cell depleting, anti-CD20 monoclonal antibody that has proven effective in a wide range of antibody-mediated autoimmune disorders, including AIHA. Within the PubMed library, over 600 results were generated when utilizing the keywords ‘rituximab’ and ‘autoimmune hemolytic anemia.’ In this context, rituximab acts not only by the elimination of B-lymphocytes responsible for anti-red blood cells auto-antibodies production but also by hampering the efficacy of lymphocytes as antigen-presenting cells and by reducing the production of cytokines [Citation22,Citation23]. Initially utilized in the setting of multi-refractory AIHA, rituximab (375 mg/sm/week for 4 weeks) is nowadays utilized as second-line therapy for wAIHA and as first line for CAD [Citation8]. Of note, although widely available, the drug is not formally licensed for wAIHA or CAD in many countries. Overall, rituximab is well tolerated with only mild infusion reactions, although a proportion of patients may experience prolonged lymphopenia/hypogammaglobulinemia, reduced response to vaccinations, and 3–27% of the patients, mainly with lymphoproliferative disorders, may develop late onset neutropenia. summarizes the most relevant prospective and retrospective studies, together with the most relevant meta-analysis and reviews regarding the use of rituximab in AIHA. The first report dates back to 1998, when Lee and Colleagues described the case of a 75-years old woman with CAD secondary to non-Hodgkin lymphoma treated with the anti-CD20 MoAb [Citation43]. In the following years, an increasing amount of evidence, mostly case reports or series, highlighted the efficacy of rituximab both in wAIHA and CAD, mainly in the refractory setting. With respect to wAIHA, the overall response rate (ORR) ranges from ~70% to ~90% across different studies, with the complete response (CR) ranging from ~40% to ~75%. The timing of response is usually slow, requiring on average 1 month to manifest, with some patients achieving a response even after 3–6 months. In the context of newly diagnosed wAIHA, the superiority of steroids plus rituximab versus steroids alone was demonstrated by two clinical trials [Citation27,Citation29]. Birgens and colleagues led a phase 3 trial comparing rituximab plus steroids versus steroids alone; CR at 1 year was 75% in rituximab-treated patients versus 39% in the steroids alone group. At 36 months, 70% of the patients treated with steroids plus rituximab were still in remission [Citation27]. The results of a subsequent phase 3 clinical trial were published in 2017 by Michael et al. [Citation29]; 32 patients with newly diagnosed wAIHA were randomized to receive steroids plus rituximab or steroids alone. At 1 year of follow-up, an ORR of 75% was observed in the rituximab-treated group, versus 31% in patients treated with steroids alone.
Table 2. Rituximab in autoimmune hemolytic anemia.
In CAD, rituximab has proven effective both alone and in combination with other drugs, such as fludarabine and bendamustine, resembling therapy schemes utilized in lymphomas. In 2004, Berentsen et al. first reported the results of a phase 2 clinical trial exploring the rituximab role in CAD. In this trial, 14/27 patients obtained a response after the first course of treatment and 6/10 to re-treatment, with a median hemoglobin increase of 4 g/dL in responders [Citation31]. A study published by Schöllkopf and Colleagues displayed similar outcomes, with an ORR of 45% [Citation32]. However, responses were rarely complete and protracted (median duration of response ~11 months). Two distinct phase 2 clinical trials demonstrated the better efficacy of rituximab in combination with chemotherapy. In the first, the combination of fludarabine and rituximab was able to induce an ORR of 76% (21% CR and 55% PR) [Citation33]. Similar outcomes were obtained with bendamustine and rituximab [Citation34], with an ORR of 71%; of note, the CR rate was higher, with 40% of patients obtaining complete response. Both treatments were considered safe, being the mortality rate (2/29 with fludarabine and 3/45 with bendamustine) not unexpected in a cohort of elderly patients; however, the relatively high rate of febrile neutropenia in both trial warns against the use of this combination in unfit CAD patients and moves this option to the second-line setting [Citation8]. Additionally, the use of these associations in real life has not been systematically reported yet.
Of note, in a phase 2 clinical trial, Barcellini et al. demonstrated that lower doses of rituximab (100 mg/sm/week for 4 weeks) induced sustained response in AIHA, with better response rate in warm than in cold forms [Citation40]. This is based on the above-mentioned rationale that rituximab not only reduces the B-cell burden but mainly exerts immunomodulatory properties. Finally, evidence of the use of anti-CD20 other than rituximab has been reported in the literature: ofatumumab was successfully used in treating AIHA secondary to LES or CLL and obinutuzumab in treating AIHA secondary to CLL.
Among the novel drugs under-development, it is worth mentioning a clinical trial investigating the anti-B-cell activating factor (BAFF) MoAb ianalumab in wAIHA [Citation44]. BAFF belongs to the superfamily of tumor necrosis factor (TNF) and is a major cytokine that regulates B-cell survival, maturation, and differentiation. Its levels have been found increased in AIHA [Citation45], and its use may exert an immunomodulatory effect in these patients, also reducing relapse occurrence. Similarly, povetacicept, an engineered double inhibitor of BAFF and APRIL, is under study in autoimmune cytopenias [Citation46].
Finally, the bispeciphic Moab obexelimab, which simultaneously binds CD19 and FcγRIIb, resulting in the down regulation of B cell activity, is under active investigation in a phase 2/3 clinical trial [Citation47].
Although they are not MoAbs, it is worth mentioning the B-cell targeting agents ibrutinib that was shown effective in a recent case series of 15 CAD patients (primary or secondary) [Citation48], and parsaclisib and rilzabrutinib under investigation for wAIHA [Citation49,Citation50]. These agents are orally available and target the B-cell receptor signaling. Finally, the spleen tyrosine kinase inhibitor fostamatinib reduces ADCC and B-cell activation and induced around 50% response in wAIHA patients in a phase 2 study [Citation51], but the phase 3 trial failed to meet the primary endpoint.
3.2. Plasma cells targeting Ab based therapies
The plasma cell compartment is not appropriately targeted by conventional therapies for AIHA (e.g. steroids, immunoglobulins, cytotoxic agents, or rituximab). The residual long-lived CD38+ plasma cells in the spleen and bone marrow may perpetuate the production of autoantibodies and explain disease refractoriness/relapse. The use of plasma cells targeting agents may therefore have a rationale in treating AIHA, as highlighted by the efficacy of the proteasome inhibitor bortezomib (one cycle of 1.3 mg/m2 iv for total four biweekly doses), backbone of different myeloma therapies, in several reports [Citation52] and in a phase 2 clinical trial in CAD (ORR of 30%) [Citation53].
Regarding MoAbs, daratumumab is an IgGk monoclonal antibody (MoAb) targeting CD38, a transmembrane receptor highly expressed in malignant but also nonmalignant plasma cells. summarizes the main publications of the use of daratumumab in AIHA, the majority being case reports or case series [Citation61]. Of note, daratumumab was administered as per myeloma-based schedules (16 mg/kg weekly for the first cycle and then fortnightly) and the time to respond was generally short (about 2 weeks). No major adverse events to daratumumab were reported. In detail, in 2018, Schuetz and Colleagues [Citation54] first reported the therapeutic effect of daratumumab in two pediatric patients with multi-refractory post-stem cell transplantation (SCT) wAIHA; one of the patients showed an initial response to daratumumab but eventually relapsed and died. The efficacy of daratumumab in post-SCT AIHA was also demonstrated by different case reports. Even-Or et al. reported the case of a 2-year-old boy with post-SCT refractory AIHA who was successfully treated with daratumumab [Citation57]. Blennerhasset et al. published the case of a 35-year-old woman who developed Evans syndrome after SCT for severe aplastic anemia and showed no response to different treatment lines, including bortezomib, and eventually responded to daratumumab [Citation56]. Driouk and colleagues [Citation62] described the case of a 6-year-old boy with post-SCT multi-refractory mixed-AIHA who did not either respond to salvage daratumumab and died. Further six patients with refractory wAIHA responding to daratumumab were later published [Citation55,Citation58,Citation59]. Only few data are available for CAD. In 2020, Tomkins and colleagues [Citation60] reported the first success of daratumumab in a 48-year-old man with CAD, who was refractory to rituximab, bortezomib, and lenalidomide. Zaninoni et al. reported a similar case of a 59-year-old man diagnosed with CAD unresponsive to rituximab and bortezomib, who eventually achieved a partial response with daratumumab [Citation61]. All these reports suggest that daratumumab is safe and effective for refractory AIHAs, although with the caveat of being mostly case reports/series often reporting successful treatments only.
Table 3. Daratumumab in autoimmune hemolytic anemia.
More recently, the use of isatuximab, another anti-CD38 MoAb that was developed in the multiple myeloma setting, was investigated in a phase 1 clinical trial as a subcutaneous infusion in adult patients with warm AIHA but results have not been published yet [Citation63].
3.3. Complement inhibitors
In AIHA, complement plays a crucial role in erythrocyte destruction; this is especially true for CADs, where IgM autoantibodies bind to erythrocytes and fix complement proteins via a classical pathway, eventually leading to the formation of membrane attack complex on the cell surface with consequent hemolysis. C3 fragments also deposit on the erythrocyte surface as opsonins and are recognized by specific receptors in the reticuloendothelial system, particularly in the liver (i.e. complement mediated EVH). Thus, complement inhibitors have a rationale for treating AIHAs, particularly CAD.
Eculizumab is a first-in-class, humanized MoAb targeting the terminal complement protein C5, preventing the generation of C5 and C5b-9. This molecule was first developed for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and dramatically changed the natural history of this disease and is now FDA-approved for PNH and atypical hemolytic uremic syndrome. It is the first anti-complement MoAb to be used in AIHAs, and the main experiences are listed in . In 2009, Roth and Colleagues [Citation15] reported a CAD patient refractory to various therapies (steroids, immunoglobulins, and rituximab) who achieved transfusion independence with eculizumab. Subsequent case reports supported the efficacy of eculizumab and the rapid onset of response in the CAD setting [Citation64–68]. In a phase 2 clinical trial [Citation16], eculizumab was able to induce a significant reduction of hemolysis in CAD patients: mean Hb increased from 9.35 g/dL to 10.15 g/dL, three patients maintained and eight acquired transfusion independence, and one patient showed reduced transfusion requirement. However, the drug was not effective on peripheral symptoms. This is likely linked to the absence of an effect on erythrocyte agglutination, since the drug does not eliminate the production of the pathogenic autoantibody. Classical pathway inhibition was first explored in murine models, where the anti-C1s MoAb TNT003 efficiently blocked opsonization of erythrocytes with C3b [Citation74]. These experiments led to the development of sutimlimab (TNT009 or BIVV009). summarizes main evidence of the use of sutimlimab in CAD. In 2019, Jager and colleagues published the results of a first-in-human phase 1 trial of sutimlimab in 10 patients with CAD [Citation75]. Of them, seven subjects achieved a response, with an increase in Hb > 2 g/dl. Based on this experience, two phase 3 clinical trials were conducted.
Table 4. Eculizumab in autoimmune hemolytic anemia.
Table 5. Sutimlimab in autoimmune hemolytic anemia.
CARDINAL was a phase 3 multicenter, open-label, single-group study to assess the efficacy of sutimlimab in patients with a history of recent transfusion (<6 months) [Citation76]; 24 patients received a fixed, biweekly dose of 6.5 or 7.5 g according to body weight <75 or ≥75 kg. A mean increase in Hb level of 2.6 g/dl was reported. Bilirubin and C4 levels normalized by week 3 and fatigue markedly improved within 1 week. Participants were vaccinated against N. meningitidis, S. pneumoniae, and H. influenzae but did not receive prophylactic antibiotics. There were no meningococcal infections or other serious AEs related to the study drug. In the 2-years extension, normalization of Hb level was reported in half of the patients and almost the totality of patients maintained transfusion independence (19/22) [Citation18]. CADENZA was a 26-week placebo-controlled phase 3 trial investigating the use of sutimlimab in patients without a recent history of transfusion support [Citation19]. Sutimlimab significantly increased Hb levels and reduced bilirubin values within 1 week of treatment, showing clearcut superiority versus placebo. Sustained response to sutimlimab has been recently reported, with ~77% of the patients treated with sutimlimab maintaining Hb levels >11 g/dl at 79 weeks [Citation78]. Importantly, in both studies, sutimlimab was shown to be able to improve patients' reported outcomes, particularly fatigue [Citation79]. The drug has been recently FDA and EMA approved.
Since complement inhibitors do not eliminate antibody production, they have been proposed as a continuous therapy. In the post-trial experience of sutimlimab, a large proportion of CAD patients relapsed during the 8-week interruption period and regained response by restarting sutimlimab [Citation80].
Although it is not a MoAb, it is worth mentioning the C3 inhibitor pegcetacoplan (APL-2), also used in PNH. This is a pegylated compstatin that inhibits both the classical and alternative complement pathways and showed good activity in CAD and wAIHA with IgG + C DAT positivity, and a phase 3 trial is ongoing [Citation81]. Finally, the orally available factor B inhibitor iptacopan is currently being evaluated in a trial including CAD and immune thrombocytopenia [Citation82].
In wAIHA, complement activation may occur in about one-third to half of patients, but it is usually not the pivotal pathogenic mechanism [Citation83]. These IgG + C+ wAIHAs usually display more severe anemia, higher hemolysis, and increased relapses after rituximab comparing with IgG+ wAIHA [Citation4]. Eculizumab was effective also in a few case reports of relapsed/refractory wAIHA patients with quick onset of efficacy [Citation17,Citation69–71,Citation73,Citation84]. Interestingly, only one case report was published of the use of sutimlimab as a successful salvage therapy in a 30-years old, critically ill woman with wAIHA secondary to systemic lupus erythematosus refractory to steroids, rituximab, hydroxychloroquine, intravenous immunoglobulins, cyclophosphamide, and plasmapheresis [Citation72]. A phase 2 study with the C3 inhibitor pegcetacoplan and a phase 2 trial with the novel C1q inhibitor ANX00580 are currently ongoing [Citation85,Citation86].
3.4. Neonatal Fc receptor inhibitors
The neonatal Fc receptor (also FcRn, IgG receptor FcRn large subunit p51, or Brambell receptor) is an IgG Fc receptor with a structure similar to the MHC class I molecule and also associates with beta-2-microglobulin. It was originally discovered in rodents as the receptor that transports maternal immunoglobulin IgG from mother to neonatal offspring via milk, leading to its name. In humans, FcRn has the same function in the placenta, but also plays a role in regulating IgG and serum albumin turnover. In fact, its binding to the Fc portion of the IgGs prevents their degradation through the endosome during pinocytosis. This results in the long half-life of human immunoglobulins (2 to 3 weeks). FcRn expression is up-regulated by pro-inflammatory cytokines, thus being particularly active in chronic inflammatory conditions such as AIHAs. In these forms, the blocking of FcRn by MoAbs is able to dampen IgG recycling, thus increasing the clearance of pathogenic autoantibodies. Intravenous nipocalimab [Citation87] and subcutaneous RVT-1401 [Citation88] are being evaluated in phase 2 trials.
4. Conclusions
In conclusion, AIHAs management did significantly benefit from the introduction of MoAbs in the therapeutic armamentarium (). The anti-CD20 rituximab was the very first targeted therapy to be used in AIHA. It is administered at 375 mg/sm/week for 4 weeks and is the preferred second-line choice for wAIHA, leading to an ORR greater than 70% with a median duration of 18–24 months. In CAD responses are less pronounced, with mainly partial short-lasting ones, and the combination with purine analogues may be effective, albeit at the price of increased toxicity. Rituximab itself has a very good safety profile; however, a proportion of patients may experience prolonged hypogammaglobulinemia, reduced response to vaccines, and late onset neutropenia, suggesting monitoring. Novel B-cell targeting MoAbs, such as ianalumab, povetacicept, and obexelimab, are under active investigation in clinical trials with the aim of establishing long-term immunomodulation and prolonging relapse-free survival in wAIHA.
Figure 1. Monoclonal antibody-based therapies for autoimmune hemolytic anemia (AIHA).
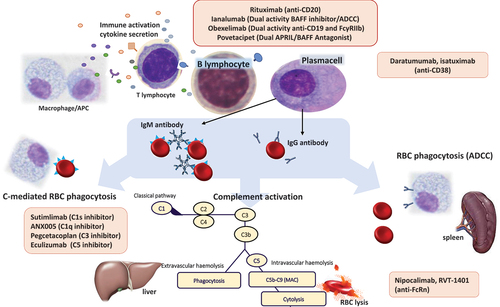
The anti-plasma cell agents used in AIHAs include bortezomib and the anti-CD38 MoAb daratumumab, for which several case reports/series are available. With the rationale of targeting long-lived plasma cells resistant to rituximab, these compounds have shown efficacy in very severe orphan settings such as multi-refractory post-transplant AIHAs and should, therefore, be taken into account. The anti-CD38 isatuximab is under investigation for immune cytopenias, including wAIHA.
Anti-complement MoAbs are revolutionizing the treatment of CAD by targeting the key pathogenic mechanism of the disease. The anti-C5 eculizumab was the first to be used but held limited efficacy since hemolysis is mainly extravascular in CAD. Sutimlimab induced quick Hb recovery and normalization in a significant proportion of CAD patients and was recently approved by the FDA and EMA. Its use is also supported by a favorable safety profile in such an elderly and comorbid patient population, and by long-term data showing improvement in fatigue and patient reported outcomes. Other complement inhibitors such as anti-C3 pegcetacoplan, anti-C1r ANX05, and the anti-factor B iptacopan are under investigation in CAD and wAIHA with complement activation. These drugs carry the caveat of not eliminating the pathogenic autoantibody and are therefore not effective in CAD-associated peripheral symptoms and require them to be administered long term. Finally, anti-FcRn MoAbs by reducing IgG half-life represent an intriguing treatment strategy for wAIHA. The harmful reduction of total IgGs appears to be overcome by the quick reconstitution after therapy discontinuation.
5. Expert opinion
The first modern advance in the field of AIHAs treatment was the introduction of rituximab in the late 2000s. This was the first targeted therapy in this setting and showed efficacy in real-life even before entering clinical trials. This is typical of therapies borrowed from other diseases such as lymphoproliferative disorders and multiple myeloma. The same occurred for anti-plasma cell agents that have been mainly used off-label in multi-refractory patients. Even complement inhibitors were first used in small independent experiments based on the rationale of complement activation in AIHAs and on the efficacy matured for PNH. However, single-patient experiences, abundant in the literature, carry several biases since the reasons for treating, disease type and severity, and outcome measures are heterogeneous by definition. In the end, we remain with more questions than answers, underlining the need to conduct prospective clinical trials even in these rare diseases. The latter requires an extraordinary effort, with the need for including several centers worldwide and harmonizing diagnosis, disease phase, and concomitant treatments. For instance, the CARDINAL trial required years to enroll 23 patients, but the value of the evidence collected provided a reliable basis to establish a new treatment era for CAD.
A second consideration regards the use of ‘relapse free survival’ or ‘duration of response’ as endpoints for evaluating treatment efficacy in modifying disease course. Rituximab remains the drug with the longest response duration in both wAIHA and CAD experiences, and novel drugs struggle to compare it. The notion that rituximab can be repeated at 1–2-year intervals in case of relapse, usually reestablishing a response, further stresses its importance in AIHA therapeutic armamentarium. Although CAD complement inhibitors establish durable responses, they need to be administered indefinitely since they do not abrogate the production of the cold agglutinin. In the near future, a combination of sutimlimab and rituximab will possibly be the answer to obtain quick disease control along with a possible durable remission off-therapy. Similarly, in wAIHA, FcRn Moabs target the final step of the hemolytic process and do not shut down autoimmunity. Although no published experience is available, it is likely that a treatment combination with drugs targeting autoantibody source will be needed.
Both complement inhibitors in CAD and anti-FcRn in wAIHA carry the potential for rapid control of hemolysis and might be useful ‘on demand’ tools in the acute phase, rather than in relapsed/refractory cases only. The acute AIHA phase represents in fact an unmet clinical need, since it is marked by the highest rate of complications, need of ICU admission, and mortality, and rituximab is a relatively slow-acting agent.
Other specific subsets include post-transplant AIHAs, as well as those forms arising after treatment with novel anti-cancer drugs. The latter include checkpoint inhibitors that are aimed at restoring T-cell aggression against cancer cells, as well as chimeric antigen receptor T-cells. By reactivating the host immune system against cancer, they may induce autoimmunity manifestations including AIHAs. These forms are often DAT negative, with consistent diagnostic delay, and multi-refractory. Additionally, there is the fear of excessive immunosuppression possibly leading to cancer relapse. In this setting, the use of targeting agents, such as anti-plasma cell ones, showed efficacy without necessarily impacting immune surveillance.
A reflection should also be made regarding physiopathology, in fact, with the above-mentioned drugs we mainly target B-cells, plasma-cells, complement, and IgG-mediated hemolysis but not the T-cell compartment. The latter is largely implied in antigen presentation, cytokine production, and establishment of a pro-inflammatory, pro-apoptotic milieu likely contributing to disease transition to the chronic phase. In the near future, studies addressing these key pathogenic players will possibly enrich therapeutic options in AIHAs.
Finally, several trials on novel targeting agents ongoing will allow us to further retail treatment on specific disease and patient characteristics, while also accounting for routes and schedules of administration. This precision medicine approach will likely include combination strategies with the new endpoint for obtaining quick but long-lasting remissions of therapy along with short- and long-term safety.
Article highlights
Treatment of autoimmune hemolytic anemias (AIHAs) traditionally relies on immunosuppression with steroids, cytotoxic agents, and splenectomy.
The advent of monoclonal antibodies (MoAbs) has revolutionized AIHA therapy by improving efficacy and limiting toxicity.
Rituximab, an anti-CD20 MoAb, B-cell depleting agent, has become the preferred second line for wAIHAs and frontline for CAD.
Other B-cell targeting agents including ianalumab and obexelimab are under study to improve response duration and reduce toxicity.
Plasma cell-targeting agents like bortezomib and daratumumab have shown efficacy in refractory and severe cases of AIHAs.
Complement inhibitors such as sutimlimab are changing the paradigm of CAD treatment.
Declaration of interest
B Fattizzo received consultancy for Amgen, Alexion, Annexon, Apellis, Momenta, Novartis, and Sobi. W Barcellini received consultancy for Agios, Alexion, Annexon, Sanofi, Novartis, and Sobi. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Berentsen S, Barcellini W. Autoimmune Hemolytic Anemias. N Engl J Med. 2021 Oct 7;385(15):1407–1419. doi: 10.1056/NEJMra2033982.
- Barcellini W, Fattizzo B. Strategies to overcome the diagnostic challenges of autoimmune hemolytic anemias. Expert Rev Hematol. 2023 Jul;16(7):515–524. doi: 10.1080/17474086.2023.2216930
- Barcellini W, Fattizzo B. How I treat warm autoimmune hemolytic anemia. Blood. 2021 Mar 11;137(10):1283–1294. Erratum in: Blood. 2023 Jan 26;141(4):438-439. doi: 10.1182/blood.2019003808
- Barcellini W, Fattizzo B, Zaninoni A, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. 2014 Nov 6;124(19):2930–2936. doi: 10.1182/blood-2014-06-583021.
- Barcellini W, Zaninoni A, Fattizzo B, et al. Predictors of refractoriness to therapy and healthcare resource utilization in 378 patients with primary autoimmune hemolytic anemia from eight Italian reference centers. Am J Hematol. 2018 Sep;93(9):E243–E246. doi: 10.1002/ajh.25212
- Wang Z, Bo L, Xu Y, et al. Features of serum complement C3 and C4 levels in autoimmune hemolytic anemia patients. Int J Lab Hematol. 2021;43(5):1154–1158. doi: 10.1111/ijlh.13469
- Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022 Jul;36(7):1703–1719. doi: 10.1038/s41375-022-01613-1
- Jäger U, Barcellini W, Broome CM, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: recommendations from the first international consensus meeting. Blood Rev. May 2020;41:100648. doi: 10.1016/j.blre.2019.100648.
- Petz LD, Garratty G. Drug-induced haemolytic anemia. Clin Haematol. 1975 Feb;4(1):181–197. doi: 10.1016/S0308-2261(21)00631-7
- Reynaud Q, Durieu I, Dutertre M, et al. Efficacy and safety of rituximab in auto-immune hemolytic anemia: a meta-analysis of 21 studies. Autoimmun Rev. 2015 Apr;14(4):304–313. doi: 10.1016/j.autrev.2014.11.014
- Ho G, Brunson A, Keegan THM, et al. Splenectomy and the incidence of venous thromboembolism and sepsis in patients with autoimmune hemolytic anemia. Blood Cells Mol Dis. 2020 Mar;81:102388.
- Fattizzo B, Cantoni S, Giannotta JA, et al. Efficacy and safety of cyclosporine a treatment in autoimmune cytopenias: the experience of two Italian reference centers. Ther Adv Hematol. 2022 May 14;13:20406207221097780. doi: 10.1177/20406207221097780
- Berentsen S, Barcellini W, Longo DL. Autoimmune Hemolytic Anemias. N Engl J Med. 2021 Oct 7;385(15):1407–1419. doi: 10.1056/NEJMra2033982
- Berentsen S. How I treat cold agglutinin disease. Blood. 2021 Mar 11;137(10):1295–1303. doi: 10.1182/blood.2019003809
- Röth A, Hüttmann A, Rother RP, et al. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009 Apr 16; 113(16):3885–3886. doi: 10.1182/blood-2009-01-196329
- Röth A, Bommer M, Hüttmann A, et al. Eculizumab in cold agglutinin disease (DECADE): an open-label, prospective, bicentric, nonrandomized phase 2 trial. Blood Adv. 2018 Oct 9; 2(19):2543–2549. doi: 10.1182/bloodadvances.2018024190
- Ma K, Caplan S. Refractory IgG warm autoimmune hemolytic anemia treated with eculizumab: a novel application of anticomplement therapy. Case Rep Hematol. 2016;2016:9181698. doi: 10.1155/2016/9181698
- Roth A, Barcellini W, D’Sa S, et al. Complement C1s inhibition with sutimlimab results in durable response in cold agglutinin disease: CARDINAL study 1-year interim follow-up results. Haematologica. 2022 Jul 1; 107(7):1698–1702. doi: 10.3324/haematol.2021.279812
- Röth A, Berentsen S, Barcellini W, et al. Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo-controlled phase 3 CADENZA trial. Blood. 2022 Sep 1; 140(9):980–991. doi: 10.1182/blood.2021014955
- Fattizzo B, Zaninoni A, Gianelli U, et al. Prognostic impact of bone marrow fibrosis and dyserythropoiesis in autoimmune hemolytic anemia. Am J Hematol. 2018 Aug;93(4):E88–E91. doi: 10.1002/ajh.25020
- Fattizzo B, Pasquale R, Carpenedo M, et al. Bone marrow characteristics predict outcome in a multicenter cohort of primary immune thrombocytopenia patients treated with thrombopoietin analogs. Haematologica. 2019 Oct;104(10):e470–e473. doi: 10.3324/haematol.2019.216804
- Reff ME, Carner K, Chambers KS, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994 Jan 15;83(2):435–445. doi: 10.1182/blood.V83.2.435.435
- Stasi R. Rituximab in autoimmune hematologic diseases: not just a matter of B cells. Semin Hematol. 2010 Apr;47(2):170–179. doi: 10.1053/j.seminhematol.2010.01.010
- Motto DG, Williams JA, Boxer LA. Rituximab for refractory childhood autoimmune hemolytic anemia. Isr Med Assoc J. 2002 Nov;4(11):1006–1008.
- Narat S, Gandla J, Hoffbrand AV, et al. Rituximab in the treatment of refractory autoimmune cytopenias in adults. Haematologica. 2005 Sep;90(9):1273–1274.
- Bussone G, Ribeiro E, Dechartres A, et al. Efficacy and safety of rituximab in adults’ warm antibody autoimmune haemolytic anemia: retrospective analysis of 27 cases. Am J Hematol. 2009 Mar;84(3):153–157. doi: 10.1002/ajh.21341
- Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013 Nov;163(3):393–399. doi: 10.1111/bjh.12541
- Maung SW, Leahy M, O’Leary HM, et al. A multi-centre retrospective study of rituximab use in the treatment of relapsed or resistant warm autoimmune haemolytic anaemia. Br J Haematol. 2013 Oct;163(1):118–122. doi: 10.1111/bjh.12486
- Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017 Jan;92(1):23–27. doi: 10.1002/ajh.24570
- Gutiérrez Jomarrón I, López Rubio M, Arias MM, et al. Autoimmune haemolytic anaemias: a retrospective study of 93 patients. Med Clin. 2020 May 8;154(9):331–337. English, Spanish. doi: 10.1016/j.medcli.2019.06.024
- Berentsen S, Ulvestad E, Gjertsen BT, et al. Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood. 2004 Apr 15; 103(8):2925–2928. doi: 10.1182/blood-2003-10-3597
- Schöllkopf C, Kjeldsen L, Bjerrum OW, et al. Rituximab in chronic cold agglutinin disease: a prospective study of 20 patients. Leuk Lymphoma. 2006 Feb;47(2):253–260. doi: 10.1080/10428190500286481
- Berentsen S, Randen U, Vågan AM, et al. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood. 2010 Oct 28; 116(17):3180–3184. doi: 10.1182/blood-2010-06-288647
- Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017 Jul 27; 130(4):537–541. doi: 10.1182/blood-2017-04-778175
- Berentsen S, Barcellini W, D’Sa S, et al. Cold agglutinin disease revisited: a multinational, observational study of 232 patients. Blood. 2020 Jul 23; 136(4):480–488. doi: 10.1182/blood.2020005674
- Zaja F, Iacona I, Masolini P, et al. B-cell depletion with rituximab as treatment for immune hemolytic anemia and chronic thrombocytopenia. Haematologica. 2002 Feb;87(2):189–195. Erratum in: Haematologica 2002 Mar;87(3):336.
- Zecca M, Nobili B, Ramenghi U, et al. Rituximab for the treatment of refractory autoimmune hemolytic anemia in children. Blood. 2003 May 15; 101(10):3857–3861. doi: 10.1182/blood-2002-11-3547
- Dierickx D, Verhoef G, Van Hoof A, et al. Rituximab in auto-immune haemolytic anaemia and immune thrombocytopenic purpura: a Belgian retrospective multicentric study. J Intern Med. 2009 Nov;266(5):484–491. doi: 10.1111/j.1365-2796.2009.02126.x
- Peñalver FJ, Alvarez-Larrán A, Díez-Martin JL, et al. Multi-institutional retrospective study on the use of rituximab in refractory AIHA. Rituximab is an effective and safe therapeutic alternative in adults with refractory and severe autoimmune hemolytic anemia. Ann Hematol. 2010 Nov;89(11):1073–1080. doi: 10.1007/s00277-010-0997-y
- Barcellini W, Zaja F, Zaninoni A, et al. Sustained response to low-dose rituximab in idiopathic autoimmune hemolytic anemia. Eur J Haematol. 2013 Dec;91(6):546–551. doi: 10.1111/ejh.12199
- Ducassou S, Leverger G, Fernandes H, et al. Benefits of rituximab as a second-line treatment for autoimmune haemolytic anaemia in children: a prospective French cohort study. Br J Haematol. 2017 Jun;177(5):751–758. doi: 10.1111/bjh.14627
- Chao SH, Chang YL, Yen JC, et al. Efficacy and safety of rituximab in autoimmune and microangiopathic hemolytic anemia: a systematic review and meta-analysis. Exp Hematol Oncol. 2020 Apr 15;9(1):6. doi: 10.1186/s40164-020-00163-5
- Lee EJ, Kueck B. Rituxan in the treatment of cold agglutinin disease. Blood. 1998;92(9):3490–3491. doi: 10.1182/blood.V92.9.3490
- A study of efficacy and safety of ianalumab in Previously treated patients with warm autoimmune hemolytic anemia (VAYHIA). ClinicalTrials.Gov Identifier: NCT05648968. [ Updated 2023 May 25; cited 2023 Jul 5]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT05648968
- Xu ZZ, Zhao BB, Xiong H, et al. Serum BAFF and APRIL levels in patients with autoimmune hemolytic anemia and their clinical significance. Int J Hematol. 2015 Oct;102(4):394–400. doi: 10.1007/s12185-015-1851-8
- An open-label study of povetacicept in subjects with autoimmune cytopenias (RUBY-4). ClinicalTrials.Gov Identifier: NCT05757570. [ Updated 2023 Mar 7; cited 2023 Jul 5]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT05757570
- A study of obexelimab in patients with warm autoimmune hemolytic anemia (SApHiare). ClinicalTrials.Gov Identifier: NCT05786573. [ Updated 2023 May 19; cited 2023 Jul 5]. Available from: https://clinicaltrials.gov/study/NCT05786573
- Jalink M, Berentsen S, Castillo JJ, et al. Effect of ibrutinib treatment on hemolytic anemia and acrocyanosis in cold agglutinin disease/cold agglutinin syndrome. Blood. 2021 Nov 18; 138(20):2002–2005. doi: 10.1182/blood.2021012039
- Ibrutinib in steroid refractory autoimmune hemolytic anemia (ISRAEL). ClinicalTrials.Gov Identifier: NCT03827603. [ Updated 2020 Jan 10; cited 2023 Jul 5]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT03827603
- Efficacy, safety and pharmacokinetics of Rilzabrutinib in patients with warm autoimmune hemolytic anemia (wAIHA). ClinicalTrials.Gov Identifier: NCT05002777. Updated 2023 May 31; cited 2023 Jul 5]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT05002777
- Kuter DJ, Rogers KA, Boxer MA, et al. Fostamatinib for the treatment of warm antibody autoimmune hemolytic anemia: phase 2, multicenter, open-label study. Am J Hematol. 2022 Jun 1; 97(6):691–699. doi: 10.1002/ajh.26508
- Pasquale R, Giannotta JA, Barcellini W, et al. Bortezomib in autoimmune hemolytic anemia and beyond. Ther Adv Hematol. 2021 Nov 12;12:20406207211046428. doi: 10.1177/20406207211046428
- Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132(5):547–550. doi: 10.1182/blood-2018-03-835413
- Schuetz C, Hoenig M, Moshous D, et al. Daratumumab in life-threatening autoimmune hemolytic anemia following hematopoietic stem cell transplantation. Blood Adv. 2018 Oct 9; 2(19):2550–2553. doi: 10.1182/bloodadvances.2018020883
- Cooling L, Hugan S. Daratumumab in combination with standard treatment for autoimmune hemolytic anemia in a pediatric patient. Transfusion. 2019 Dec;59(12):3801–3802. doi: 10.1111/trf.15539
- Blennerhassett R, Sudini L, Gottlieb D, et al. Post-allogeneic transplant Evans syndrome successfully treated with daratumumab. Br J Haematol. 2019 Oct;187(2):e48–e51. doi: 10.1111/bjh.16171
- Even-Or E, Naser Eddin A, Shadur B, et al. Successful treatment with daratumumab for post-HSCT refractory hemolytic anemia. Pediatr Blood Cancer. 2020 Jan;67(1):e28010. doi: 10.1002/pbc.28010
- Rieger MJ, Stolz SM, Ludwig S, et al. Daratumumab in rituximab-refractory autoimmune haemolytic anaemia. Br J Haematol. 2021 Sep;194(5):931–934. doi: 10.1111/bjh.17655
- Jain A, Gupta DK. Daratumumab for refractory warm autoimmune hemolytic anemia. Ann Hematol. 2021 May;100(5):1351–1353. doi: 10.1007/s00277-020-04063-w
- Tomkins O, Berentsen S, Arulogun S, et al. Daratumumab for disabling cold agglutinin disease refractory to B-cell directed therapy. Am J Hematol. 2020 Jul 11;95(10). doi: 10.1002/ajh.25932
- Zaninoni A, Giannotta JA, Gallì A, et al. The immunomodulatory effect and clinical efficacy of daratumumab in a patient with cold agglutinin disease. Front Immunol. 2021 Mar 1;12:649441. doi: 10.3389/fimmu.2021.649441
- Driouk L, Schmitt R, Peters A, et al. Daratumumab therapy for post-HSCT immune-mediated cytopenia: experiences from two pediatric cases and review of literature. Mol Cell Pediatr. 2021 Apr 29; 8(1):5. doi: 10.1186/s40348-021-00114-y
- Safety, pharmacokinetics, and efficacy of subcutaneous isatuximab in adults with warm autoimmune hemolytic anemia (wAIHA). ClinicalTrials.Gov Identifier: NCT04661033. [ Updated 2023 Mar 13; cited 2023 Jul 5]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT04661033
- Gupta N, Wang ES. Long-term response of refractory primary cold agglutinin disease to eculizumab therapy. Ann Hematol. 2014 Feb;93(2):343–344. doi: 10.1007/s00277-013-1800-7
- Shapiro R, Chin-Yee I, Lam S. Eculizumab as a bridge to immunosuppressive therapy in severe cold agglutinin disease of anti-pr specificity. Clin Case Rep. 2015 Nov;3(11):942–944. doi: 10.1002/ccr3.399
- Makishima K, Obara N, Ishitsuka K, et al. High efficacy of eculizumab treatment for fulminant hemolytic anemia in primary cold agglutinin disease. Ann Hematol. 2019 Apr;98(4):1031–1032. doi: 10.1007/s00277-018-3521-4
- Herbreteau L, Le Calloch R, Arnaud B, et al. Eculizumab, a real-life successful treatment for refractory cold agglutinin-mediated auto-immune hemolytic anemia secondary to lymphoproliferative disorders. Ann Hematol. 2021 Aug;100(8):2105–2106. doi: 10.1007/s00277-021-04557-1
- Dawudi Y, Federici L, Debus J, et al. Cold agglutinin disease secondary to severe SARS-CoV-2 treated with eculizumab. BMJ Case Rep. 2022 Apr 29; 15(4):e242937. doi: 10.1136/bcr-2021-242937
- Chao MP, Hong J, Kunder C, et al. Refractory warm IgM-mediated autoimmune hemolytic anemia associated with Churg-Strauss syndrome responsive to eculizumab and rituximab. Am J Hematol. 2015 Jan;90(1):78–81. doi: 10.1002/ajh.23791
- Neave L, Wilson AJ, Lissack M, et al. Severe refractory idiopathic warm autoimmune haemolytic anaemia responsive to complement inhibition with eculizumab. BMJ Case Rep. 2018 Dec 13; 11(1):e226429. doi: 10.1136/bcr-2018-226429
- Gauchy AC, Hentzien M, Wynckel A, et al. Efficacy of eculizumab in refractory life-threatening warm autoimmune hemolytic anemia associated with chronic myelomonocytic leukemia. Clin Case Rep. 2020 Aug 17;8(12):2641–2644. doi: 10.1002/ccr3.3250
- Tahhan F, Huynh B, Xu P. Novel monoclonal antibody therapy in a patient with treatment-refractory warm autoimmune hemolytic anemia. Cureus. 2022 Jun 17;14(6):e26051. doi: 10.7759/cureus.26051
- Jackson EM, Harper S, Webb GJ, et al. Severe refractory warm autoimmune haemolytic anaemia after the SARS-CoV-2 pfizer-BioNTech vaccine (BNT162b2 mRNA) managed with emergency splenectomy and complement inhibition with eculizumab. BMJ Case Rep. 2022 Aug 31; 15(8):e250774. doi: 10.1136/bcr-2022-250774
- Shi J, Rose EL, Singh A, et al. TNT003, an inhibitor of the serine protease C1s, prevents complement activation induced by cold agglutinins. Blood. 2014 Jun 26; 123(26):4015–4022. doi: 10.1182/blood-2014-02-556027
- Jäger U, D’Sa S, Schörgenhofer C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. 2019 Feb 28; 133(9):893–901. doi: 10.1182/blood-2018-06-856930
- Röth A, Barcellini W, D’Sa S, et al. Sutimlimab in cold agglutinin disease. N Engl J Med. 2021 Apr 8;384(14):1323–1334. doi: 10.1056/NEJMoa2027760.
- Tvedt THA, Steien E, Øvrebø B, et al. Sutimlimab, an investigational C1s inhibitor, effectively prevents exacerbation of hemolytic anemia in a patient with cold agglutinin disease undergoing major surgery. Am J Hematol. 2022 Feb 1;97(2):E51–E54. doi: 10.1002/ajh.26409
- Röth A, Berentsen S, Barcellini W, et al. Sustained efficacy of sutimlimab, a complement c1s inhibitor, in patients with cold agglutinin disease: results from part b of the phase 3 cadenza study. Poster presented at EHA, Frankfurt, 2023
- Röth A, Barcellini W, Tvedt THA, et al. Sutimlimab improves quality of life in patients with cold agglutinin disease: results of patient-reported outcomes from the CARDINAL study. Ann Hematol. 2022 Oct;101(10):2169–2177. doi: 10.1007/s00277-022-04948-y
- Gertz M, Roman E, Fattizzo B, et al. Inhibition of C3 with APL-2 controls haemolysis and increases haemoglobin levels in subjects with autoimmune haemolytic anaemia (AIHA). EHA Lib. 2019;3:S899. doi: 10.1097/01.HS9.0000561876.96057.48
- A study to evaluate the efficacy and safety of pegcetacoplan in patients with cold agglutinin disease (CAD). ClinicalTrials.Gov Identifier: NCT05096403. [ Updated 2023 Jun 2; cited 2023 Jul 5]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT05096403
- Basket study to assess efficacy, safety and PK of iptacopan (LNP023) in autoimmune benign hematological disorders. ClinicalTrials.Gov Identifier: NCT05086744. [ Updated 2023 Jul 3; cited 2023 Jul 5]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT05086744
- Barcellini W, Zaninoni A, Giannotta JA, et al. New insights in autoimmune hemolytic anemia: from pathogenesis to therapy stage 1. J Clin Med. 2020 Nov 27;9(12):3859. doi: 10.3390/jcm9123859
- Garg M, Agarwal S, Altohami M. A single dose of eculizumab terminated life-threatening haemolysis in idiopathic IgM-mediated warm autoimmune haemolytic anaemia: a case report. Br J Haematol. 2022 Apr;197(2):e28–e31. doi: 10.1111/bjh.18011
- Study to assess the safety, tolerability, efficacy and PK of APL-2 in patients with warm type autoimmune hemolytic anemia (wAIHA) or cold agglutinin disease (CAD). ClinicalTrials.Gov Identifier: NCT03226678. [ Updated 2023 Apr 3; cited 2023 Jul 5]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT03226678
- Safety, tolerability, pharmacokinetics and pharmacodynamics of ANX005 in participants with warm autoimmune hemolytic anemia (wAIHA). ClinicalTrials.Gov Identifier: NCT04691570. [ Updated 2023 Mar 6; cited 2023 Jul 5]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT04691570
- A safety study of SYNT001 in participants with warm autoimmune hemolytic anemia (WAIHA). ClinicalTrials.Gov Identifier: NCT03075878. [ Updated 2020 May 13; cited 2023 Jul 5]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT03075878
- To assess the efficacy and safety of RVT-1401 in the treatment of warm autoimmune hemolytic anemia (ASCEND-WAIHA). ClinicalTrials.Gov Identifier: NCT04253236. [ Updated 2022 Jul 28; cited 2023 Jul 5]. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT04253236