
ABSTRACT
Background
PB006 (Polpharma Biologics S.A; marketed as Tyruko®, Sandoz) is an approved biosimilar to natalizumab (Tysabri®; Biogen [ref-NTZ]). This multicenter, double-blind, randomized, single-dose study was conducted to demonstrate pharmacokinetic/pharmacodynamic (PK/PD) similarity between PB006 and ref-NTZ.
Research design and methods
Healthy participants (N = 453) were randomized to receive 3 mg/kg infusion of PB006, US-licensed, or EU-approved ref-NTZ before an 85-day follow-up. Primary PK endpoint was total natalizumab serum concentration over time; secondary PK endpoints explored concentration changes. Primary PD endpoints compared CD19+ cell counts and percentage α4-integrin receptor saturation, per natalizumab’s mechanism of action. Secondary PD endpoints explored serum changes in sVCAM-1 and sMAdCAM-1, CD34+, and CD19+ cells. Safety, tolerability, and immunogenicity were assessed.
Results
The primary PK endpoint was met, with 90% confidence intervals (CIs) of the geometric mean for serum test/reference ratios contained within a prespecified margin (0.8–1.25). All primary PD endpoints were met, with 90% and 95% CIs within this similarity margin for baseline-adjusted CD19+ cell counts and percentage α4-integrin receptor saturation. All secondary endpoints were similarly contained, except sVCAM. No notable differences in safety, tolerability, or immunogenicity were observed.
Conclusion
Similarity was confirmed, with PB006 demonstrating PK/PD behavior consistent with that of ref-NTZ.
Clinical trial registration
EudraCT number 2019-003874-15
Plain Language Summary
PB006 (developed by Polpharma Biologics S.A; and marketed as Tyruko® by Sandoz) is an approved biosimilar to the reference medicine, natalizumab (Tysabri®, Biogen [ref-NTZ]) used to treat relapsing forms of multiple sclerosis. Approved biosimilar medicines have been shown to be as safe and effective as their reference medicines via different types of comparisons to the reference medicine, confirming that physicians and patients can expect the same clinical outcome.
This study was conducted to confirm that PB006 acts the same way in the body as ref-NTZ. Healthy participants received one dose of either PB006, ref-NTZ from the US or ref-NTZ from Europe. During the study, blood samples were tested to confirm how much of each medicine was present in participants’ blood, as well as to assess changes in immune cells or proteins related to how natalizumab works. The study also measured whether any treatment caused unwanted side effects or caused any changes in the immune system that may stop the medicine working.
The results showed that PB006 behaved in the same way as ref-NTZ in the blood. All reported side effects were similar between groups and were as expected for this medicine, and neither PB006 nor ref-NTZ caused any important or unexpected changes to the immune system. This study showed that biosimilar natalizumab, PB006, behaves in the same way as ref-NTZ, and the same treatment outcomes can be expected.
1. Background and objectives
PB006 (developed by Polpharma Biologics S.A; marketed as Tyruko®, Sandoz) has been developed as a biosimilar to natalizumab (Tysabri®; Biogen, Cambridge, MA [ref-NTZ]), a highly effective, humanized, monoclonal IgG4 antibody indicated as a monotherapy for the treatment of highly active relapsing forms of multiple sclerosis (MS) in Europe and the United States (US) [Citation1,Citation2]. In the US, natalizumab is also approved in patients with active, moderate-to-severe Crohn’s disease with inadequate response to conventional therapies [Citation2].
The clinical efficacy of natalizumab is predicated on its binding to the α4-subunit of α4β1 and α4β7 integrins, which are expressed on the surface of leukocytes, except neutrophils. Natalizumab binding inhibits the α4-integrin-mediated adhesion of leukocytes to their cognate receptors [Citation3]. Specifically, natalizumab inhibits the interaction of α4β1-integrin (also known as very late antigen 4 [VLA-4]) with vascular cell adhesion molecule-1 (VCAM-1) expressed on activated vascular endothelium and inhibits the interaction of α4β7-integrin with mucosal vascular addressin cell adhesion molecule-1 (MAdCAM-1) expressed on vascular endothelial cells of the gastrointestinal (GI) tract [Citation3–5]. Inhibition of α4-integrins prevents the adhesion and subsequent migration of leukocytes, including CD19+ B cells and CD34+ T cells from the periphery into the central nervous system or, in the case of Crohn’s disease, into the GI tract [Citation3].
Biosimilar medicines are successors to biologic medicines for which the patent has expired and market exclusivity has been lost. The reduced acquisition cost of biosimilars compared with the reference medicine may increase treatment options, expand access to biologics, and free up healthcare resources, thereby driving sustainability of care [Citation6,Citation7]. Biosimilar medicines play an increasing role in treatment in the fields of oncology, immunology, and rheumatology, where they have been integrated successfully into the respective treatment paradigms. Substantial experience with multiple biosimilars has been accrued in these fields, providing evidence for their potential in other disease areas [Citation8–10].
Approval of a biosimilar is contingent on a stepwise comparability exercise to an already approved reference medicine. This includes extensive physiochemical and functional characterization of the biosimilar molecule using state-of-the-art analytical technologies, assessment of the pharmacokinetic/pharmacodynamic (PK/PD) profile, and also a comparative clinical study evaluating efficacy and safety in an indicated patient population [Citation11,Citation12]. The results of the foundational analytical characterization, combined sequentially with the preclinical and clinical study results, are assessed together as the ‘totality of evidence,’ which is required from a regulatory perspective to establish biosimilarity between a biosimilar and its reference medicine. Once biosimilarity is established, a biosimilar is as safe and effective as its reference medicine, and physicians and patients can expect the same clinical outcome [Citation13–17].
PB006 was approved in 2023 by the US Food and Drug Administration and the European Medicines Agency [Citation18,Citation19]. Previously reported results of the Antelope study demonstrated that PB006 matches its reference medicine in terms of efficacy, safety, and immunogenicity in patients with relapsing-remitting MS (RRMS) [Citation20]. Here, we present the results of a pivotal, single-dose, randomized controlled study in 453 healthy subjects completed as part of the approval package to demonstrate PK/PD similarity between biosimilar natalizumab PB006 and its US-licensed/EU-approved reference medicine.
2. Subjects and methods
2.1. Ethical approval
This study was conducted in accordance with the principles of the Declaration of Helsinki in place at the time of study conduct, the International Council on Harmonisation (ICH) E6 Guideline for Good Clinical Practice, European Medicines Agency (EMA)/Committee for Medicinal Products for Human Use (CHMP)/ICH/135/1995, EU Clinical Trial Directive: Directive 2001/20/EC, and all applicable national and local laws and regulations. At screening, subjects were informed verbally and in writing of the objectives, procedures, and risks of study participation, including possible side effects and potential interactions. Written informed consent was obtained from all participants in the study. Independent ethics committees for sites in The Netherlands and Poland were the Evaluation of Ethics in Biomedical Research and the Bioethics Committee of the District Medical Chamber in Warsaw, respectively. For sites in the US, the independent review board was the Midlands Independent Review Board.
2.2. Design
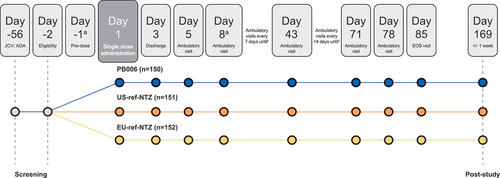
This was a randomized, double-blind, three-arm, multicenter, single-dose study (EudraCT trial number: 2019–003874–15). Healthy male and female subjects were recruited to study sites in the US, The Netherlands, and Poland, between 21 October 2019 and 11 March 2021 (first screening and last follow-up). A total of 453 subjects were randomized: they were aged 18–65 years at study Day 1, weighing 50–92 kg, and with a body mass index (BMI) of 18.5–30 kg/m2 (). Use of natalizumab in John Cunningham virus (JCV)-positive individuals is associated with increased risk of developing progressive multifocal leukoencephalopathy (PML) secondary to JCV infection [Citation21]. JCV status was determined by Stratify® JCV DxSelect™ assay (Focus Diagnostics: Cypress, CA). Subjects were required to be negative for JCV antibodies at Screening 1 (56 days to 2 days prior to Day 1), prior to Screening 2 (up to 28 days prior to Day 1) (). Subjects with a JCV-positive test result before Screening 2 were excluded from the study.
Figure 1. Study consort diagram. Definitions of protocol deviations: for the PK set, protocol deviations were defined as exiting the study before a serum natalizumab level below the lower limit of quantification was reached or two visits were missed before levels had returned to baseline. For the PD α4-integrin sensitivity set, protocol deviation was defined as three consecutive missed visits ahead of %RS returning to baseline. For the PD α4-integrin %RS set, protocol deviation was defined as three consecutive missed visits ahead of %RS returning to baseline or analysis of samples outside of the validated stability window. JCV, John Cunningham virus; PD, pharmacodynamic; PK, pharmacokinetic; ref-NTZ, reference natalizumab; %RS, percentage receptor saturation.
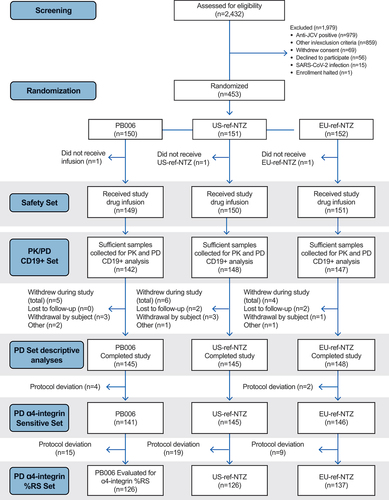
Figure 2. Study design. aSubjects were admitted to the studio center on Day −1 and discharged on Day 3, with subsequent ambulatory visits from Day 5. After the COVID-19 risk assessment, this was amended to discharge on Day 8 with ambulatory visits from Day 15. bPK/PD sampling was performed via blood sampling, once pre-infusion and then at each ambulatory visit over 85 days post-infusion. Subjects returned for ambulatory visits on days 15, 22, 29, 36, 43, 57, 71, 78, and 85. A final follow-up visit was performed to identify any new neurological symptoms potentially suggestive of PML. ADA, antidrug antibody; EOS, end of study; JCV, John Cunningham virus; PD, pharmacodynamic; PK, pharmacokinetic; ref-NTZ, reference natalizumab.
Eligible subjects were randomized 1:1:1 to receive a 3 mg/kg single intravenous infusion of either PB006, US-licensed (US-), or EU-approved (EU-) ref-NTZ administered over a 60-minute period (). Randomization was performed by S-Clinica sprl, Belgium. Randomization was stratified by body weight class (50–65 kg, >65–80 kg, and >80–92 kg) to ensure an equal distribution of subjects within each body weight class per arm, based on the known effects of body weight on the PK of natalizumab [Citation22–24]. Investigators remained blinded to each subject’s assigned study intervention throughout the course of the study. In order to maintain this blind, an otherwise uninvolved third party (e.g. site pharmacy staff) was responsible for the dilution and dispensing of all study intervention and delivery to site.
PK/PD sampling was performed as per a sampling schedule (). Safety and immunogenicity were monitored throughout the study by repeated clinical and laboratory evaluations. A final follow-up visit 6 months (24 weeks) after dosing was performed to identify any new neurological symptoms potentially suggestive of PML.
A sample size of 151 subjects per study arm was derived using the following assumptions: a test/reference ratio for the PK (total serum concentration of natalizumab) and PD endpoints (change from baseline in circulating CD19+ cells and percentage of α4-integrin receptor saturation [%RS]) of 0.95–1.00/0.95; a significance level of 5% (corresponding to two one-sided tests of 90% confidence intervals [CI] for the test/reference ratio); a similarity margin of 0.80–1.25, a 1:1:1 randomization ratio, and overall power of at least 80%. Sample size calculations assumed a 10% drop out rate for the study subjects.
Study enrollment was halted on 16 March 2020 due to the developing COVID-19 pandemic. At this time, 16 subjects had received an infusion. These subjects completed the study as planned. A participatory risk assessment with regards to the mechanism of action of natalizumab and potential SARS-CoV-2 and subsequent COVID-19 infection was then performed.
Study enrollment resumed in the US after 1 July 2020, Netherlands after 9 July 2020, and Poland after 16 September 2020. At study restart, additional COVID-19 safety and quarantine measures were implemented as a protocol amendment (). The maximum age of inclusion was lowered from 65 to 54 years. Subjects required two negative SARS-CoV-2 polymerase chain reaction tests prior to participation (n = 434 subjects), and those with a history of COVID-19 infection in the month prior to Screening 1 or contact with a COVID-19-positive subject 2 weeks ahead of dosing were excluded.
2.3. Study endpoints
2.3.1. PK analyses
The primary PK endpoint was the area under the curve from time of dosing extrapolated to infinity (AUC0-inf) of total natalizumab. PK parameters were calculated based on the PK Set, which included all subjects who received the study drug, did not experience any major protocol deviations, and provided sufficient samples to allow calculation of the AUC0-inf for total natalizumab serum concentration.
Natalizumab is a full-length IgG4 antibody that comprises two heavy and light chains connected by disulfide bonds. Its assessment in serum is complicated by the ability of human IgG4 antibodies to undergo Fab arm exchange in vivo, described below. ‘Total’ and ‘unexchanged’ natalizumab cannot be quantified independently using a single enzyme-linked immunosorbent assay (ELISA); therefore, an established quantitation strategy was employed using one ELISA measuring both total and unexchanged and another ELISA measuring only unexchanged natalizumab. The presence and amount of natalizumab in serum was calculated by the difference in values obtained from the two assays [Citation25].
Concentrations of total natalizumab in human serum were determined using a validated, quantitative ELISA sandwich format using anti-natalizumab IgG4 Fc-HRP as capture and mouse anti-human IgG4 Fc-HRP as detection. Both exchanged and unexchanged natalizumab are detected by this approach as the HRP-conjugated antibody detected the Fc fragment. Concentrations of unexchanged natalizumab in human serum were determined using a validated, quantitative ELISA sandwich format using monovalent Fab format anti-natalizumab IgG as capture and human anti-natalizumab HRP as detection. Only unexchanged natalizumab is detected by this approach as the coating antibody only binds unexchanged natalizumab. All reported results for total natalizumab and unexchanged natalizumab in serum were generated in analytical runs using appropriate calibration curves and quality control samples that met pre-established acceptance criteria. Both assays used for assessing total and unexchanged natalizumab in serum had a lower limit of quantitation of 60.0 µg/L and an upper limit of quantitation of 400.0 µg/L.
Analysis of PK parameters was performed using a three-way comparison of PB006 to both US-and EU-ref-NTZ and of US- to EU-ref-NTZ. PK similarity was established using an ANOVA model that included calculation of the least-squares means (LSM) for the treatments. The 90% CIs were calculated and back-transformed to the original scale, and PK similarity was concluded if the respective CIs for AUC0-inf were completely contained within the pre-specified margin of similarity set to 0.8–1.25. PK parameters were calculated using non-compartmental analysis via Certara Phoenix™ PK/PD platform Version 8.3.
2.3.2. Additional PK analysis: Fab-arm exchange
Natalizumab is an IgG4 antibody, and as such is able to undergo heavy – light chain recombination (Fab-arm exchange) with endogenous IgG4 antibodies, resulting in the formation of bispecific IgG4 antibodies that result in lower affinity to α4-integrins [Citation26]. Given the heterogeneity of the exchanged Fab-arm species, no direct measurement of Fab-arm exchanged natalizumab is possible [Citation25]. Therefore, the difference between ‘total’ and ‘unexchanged’ natalizumab was used as an indirect measurement of Fab-arm-exchanged natalizumab and evaluated descriptively for all treatment arms.
2.3.3. PD analyses
PD similarity was established by comparison of the area under the effect (AUEC) curve from time of dosing to 12 weeks (AUEC0-12w) for baseline-adjusted CD19+ cell counts and α4-integrin %RS using flow cytometry analysis of cells/µl and % cells for whole blood samples to within quality control limits (CD19+) or absolute percentage receptor occupancy bias between comparators of ≤20.0% (%RS).
PD endpoints were chosen based on data from a pilot study (PB006-01-01, data not shown) investigating dose-response in healthy subjects for a number of natalizumab PD biomarkers at single natalizumab doses ranging from 1 mg/kg to 6 mg/kg, which comprised sMAdCAM, sVCAM, α4-integrin %RS, cell counts for CD34+, CD19+, CD3+, CD3+CD8+, CD3+CD4+, CD3-CD16+CD56+, and CD10+CD19+ cell counts. The PD endpoints included into this study were required to comply with the essential characteristics for PD endpoints in biosimilar studies as defined by the FDA [Citation27], which include 1) relevance to the mechanism of action of natalizumab, 2) sensitivity to detect differences between PB006 and its reference medicine, 3) a sufficient dynamic range during exposure to natalizumab, 4) a rapid onset of the PD biomarker relative to dosing of natalizumab and a timely return to baseline upon discontinuation of natalizumab dosing, and 5) a validated bioanalytical assay for determination of the PD biomarker.
From the results of this pilot study, a set of PD biomarkers, covering natalizumab’s mechanism of action at different stages in concert with the translational PK/PD biomarker classification by Visser and Bueters [Citation28] was selected as follows: α4-integrin receptor saturation, to compare natalizumab target binding (α4β1-integrin, α4β7-integrin expressed on circulating leukocytes), sVCAM/sMAdCAM, to compare the interaction of α4β1-integrin and α4β7-integrin with their respective ligands and circulating CD19+ and CD34+ cells as physiologic responses to blocking the interaction of α4β1-integrin/sVCAM and α4β7-integrin/sMAdCAM. Baseline-adjusted CD19+ cell counts and α4-integrin %RS were chosen as primary PD endpoints, since they exhibited the steepest dose – response and thus the most sensitive measure of detecting potential differences in PB006 PD.
Study endpoints were analyzed using a two-stage hierarchical testing procedure. Similarity within a prespecified margin for PK endpoints was used as justification to pool the US- and EU-ref-NTZ arms for the PD endpoint analyses, in line with previous approaches reported in the literature [Citation29]. PD similarity was then evaluated via an ANCOVA model for baseline-adjusted CD19+ levels and via an ANOVA model for α4-integrin %RS, following the calculation of the LSM for each treatment arm. Geometric mean ratios and corresponding CIs (90% and 95%, reflecting the requirements of US Food and Drug Administration [FDA] and EMA, respectively) were back-transformed against the original scale. Similarity was concluded if the AUEC0-12w CIs for baseline-adjusted CD19+, and the AUEC0-12w and AUEC4-12w for α4-integrin %RS, were completely contained within the prespecified 0.8–1.25 margin for similarity.
Further PD endpoints were selected based on the known mechanism of action of natalizumab and the subsequent downstream effects of α4-integrin binding to VCAM-1 and MAdCAM-1 and analyzed descriptively [Citation3,Citation30]. Secondary PD endpoints were the levels of circulating CD34+ cells (AUEC0-t), maximum concentration of CD19+ cells (Emax), time taken to reach CD19+ Emax (tmax), minimum serum levels of soluble (s)VCAM-1 and sMAdCAM-1 (Emin), time taken to achieve minimum serum levels or sVCAM-1 and sMAdCAM-1 (tmin), duration of the decrease from baseline (AUECbase-neg) for sVCAM-1 and sMAdCAM-1, and the geometric mean ratios and corresponding 90% and 95% CIs calculated for AUECbase-neg and lowest concentration (Emin) of sVCAM and sMAdCAM.
Quantification of CD19+ and CD34+ cells was performed in whole human blood using a validated flow cytometry method. The distribution and absolute numbers of CD19+ B cells and CD34+ T cells were assessed using an immunophenotyping assay using fluorochrome-labeled antibodies that bind specifically to surface antigens. The absolute number of positive cells was determined by comparing cell with fluorescent bead events. White blood cell counts were obtained from the same whole blood samples prior to staining to ensure that WBC counts were within the linear range (CD19+, 0–863 cells/µL; CD34+, 0–1000 cells/µL). Samples were analyzed using a FACSCanto IITM flow cytometer, and BD FACSCANTOTM clinical software was used for acquisition.
Assessment of α4-integrin %RS by natalizumab was performed using flow cytometry on CD45+ leukocytes in whole human blood. Leukocytes were identified using CD45 staining. Samples were split into two aliquots, with unlabeled PB006 added to one aliquot to induce maximum receptor saturation; thereafter, mouse anti-human IgG4 Fc-PE antibody was applied to detect natalizumab using the PE detection ratio between the two aliquots. Samples were analyzed using a FACSCanto IITM flow cytometer, BD FACSDivaTM flow cytometry software and De Novo FCS Express 6 Flow Clinical EditionTM for data processing.
Quantification of VCAM-1 and MadCAM-1 was performed using the automated immunoassay platform ELLATM and Simple PlexTM ELISA assay using a validated sandwich format using capture and detection antibodies specific for each analyte (MAdCAM-1 3.36 pg/mL LLOQ 12,800 pg/mL ULOQ; VCAM-1 560 pg/mL LLOQ 83,490 pg/mL ULOQ).
2.3.4. Safety, tolerability, and immunogenicity
Descriptive safety and tolerability assessments were recorded and included local tolerability, adverse event (AE) reporting, PML monitoring, clinical laboratory evaluation, vital signs, 12-lead electrocardiogram, and physical examination.
Both PB006 and its reference medicine are humanized murine-derived monoclonal antibodies and as such may provoke an immune response [Citation1]. Thus, a comparison of immunogenicity was performed throughout the study using sensitive drug-tolerant anti-drug antibody (ADA) assays to establish ADA and neutralizing antibody (NAb) rates and titers, and the rate of seroconversion. ADA measurements were conducted using an electrochemiluminescence immunoassay (ECLIA) incorporating a sample pre-treatment step to optimize drug tolerance. A normalization factor of 1.25 determined during assay validation was used for analysis. A sample was screened positive if the electrochemiluminescence signal was equal to or greater than the run-specific screening cut-point; values below the screening cut-point were classified as ‘negative.’ The ADA assay was validated with sensitivity of 3.88 ng/mL for screening and 2.82 ng/mL for confirmation; 100 ng/mL of the positive control antibody could be detected in the presence of 62.5 µg/mL of natalizumab.
A cell-based assay format, based on measurement of inhibition of the in vitro binding of natalizumab to α4-integrin expressed on Karpas 299 CD45+ lymphocytes, was used to measure NAbs. The sensitivity of the NAb assay was 205 ng/mL; 1.4 μg/mL positive control detected in the presence of up to 16 μg natalizumab/mL. A solid-phase extraction with acid dissociation sample pre-treatment step enabled adequate sensitivity to be achieved in the presence of on-board drug levels, as reflected by the detection of NAb in the majority of ADA-positive subjects.
3. Results
3.1. Subject baseline characteristics
At the start of the study, 453 subjects were randomized to receive either PB006, US-, or EU-ref-NTZ. A total of three subjects were withdrawn from the study before administration of study drug: one subject was randomized and infused but did not receive the study treatment due to an error in the pharmacy; two subjects were withdrawn before receiving study drug due to poor venous access (n = 1), and an AE occurring between randomization and infusion (n = 1) All subjects treated with study drug were assessed as part of the Safety Set (n = 450) (). Subjects were distributed evenly across all treatment arms with respect to mean age, weight, height, BMI, sex, and ethnicity ().
Table 1. Patient baseline characteristics (Safety Set).
3.2. PK analysis
PB006 demonstrated similar serum concentration kinetics for total natalizumab compared to both US-ref-NTZ and EU-ref-NTZ throughout the post-dose evaluation period (). PK similarity between PB006 and the two reference medicines as well as between US-ref-NTZ and EU-ref-NTZ was demonstrated, with the 90% CIs for AUC0-inf (h.mg/L) within the pre-specified similarity margin of 0.8–1.25 for all comparisons ().
Figure 3. Primary PK and PD endpoints. a) Semi-logarithmic plot of mean total serum natalizumab concentration over time for PB006 versus US-ref-NTZ and EU-ref-NTZ (PK set); b) change from baseline in mean (SD) CD19+ cell counts; c) change from baseline in mean (SD) α4-integrin %RS/RO over time for PB006 versus US-ref-NTZ and EU-ref-NTZ (PK/target receptor engagement Set) ref-NTZ, reference natalizumab; RO, receptor occupancy; RS, receptor saturation; SD, standard deviation.
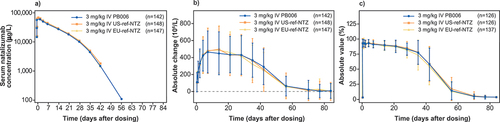
Table 2. Total natalizumab serum concentration (AUC0-inf) (PK Set).
Cmax, AUC0-inf, AUC0-t of total and unexchanged serum natalizumab were evaluated in the same fashion as the primary PK endpoints. All values were contained within the margin of 0.8–1.25 (Supplementary Material Table S1). Additional PK parameters, including the time to maximal serum concentration (tmax), the time to half maximal serum concentration (t1/2), drug clearance (CL), and volume of distribution (Vz) for total and unexchanged natalizumab were analyzed using descriptive statistics, and were found to be similar for all treatments (Supplementary Material Table S2).
3.3. %Fab-arm exchange
Subjects in all treatment arms demonstrated Fab-arm exchange occurring in >95% of all PK samples across all three treatment arms by Day 8. The mean %Fab-arm exchanged natalizumab recorded in subjects was 95.4%, 96.2%, and 95.9% for subjects treated with PB006, US-ref-NTZ, and EU-ref-NTZ, respectively. Maximum values reached 100% in individual subjects at Day 8 for all three study arms (Supplementary Material Figure S1).
3.4. PD analysis
The time-course of baseline-adjusted number of circulating CD19+ cells over time was also comparable across treatment arms (). Likewise, a similar degree of α4-integrin receptor engagement was observed across all treatment arms and time points (). Saturation of the α4-integrin receptor was calculated as AUEC0-12w α4-integrin %RS (). The α4-integrin %RS of >80% was maintained until Day 29 (), after which it declined to close to baseline levels (<5% RS) by Day 71.
Table 3. AUEC0-12w of baseline-corrected CD19+ cells and AUEC0-12w % α4-integrin RS.
Comparison of PD similarity regarding the primary endpoints, AUEC0-12w of baseline-adjusted CD19+ and AUEC0-12w of α4-integrin %RS, was the second step of the two-step hierarchical testing procedure. Both were contained within the 95% CIs (Supplementary Material Table S3), justifying the pooling of US-ref-NTZ and EU-ref-NTZ arms into a single reference. Following establishment of the pooled reference, the levels of circulating CD19+ cells and percentage α4-integrin %RS for PB006 versus US-ref-NTZ, EU-ref-NTZ, and pooled ref-NTZ groups were analyzed using ANCOVA and ANOVA models, respectively. The 90% and 95% CIs of both the back-transformed baseline-adjusted AUEC0-12w of baseline-adjusted CD19+ cell levels and α4-integrin %RS were contained within the prespecified margin of similarity 0.8–1.25 (), demonstrating PD similarity between treatment arms.
Due to α4-integrin receptor being completely saturated by a 3 mg/kg dose of natalizumab during the first 4 weeks of the study, α4-integrin %RS was also calculated over 4–12 weeks (AUEC4-12w) in addition to the AUEC0-12w (Supplementary Material Table S4). Taken together, the results support a similar PD profile for PB006 compared to the reference medicine in terms of α4-integrin %RS and the subsequent increase from baseline in circulating CD19+ cells.
A similar increase in serum circulating CD34+ cells from baseline was observed across all treatment arms and was contained within the 90% CI similarity margin of 0.8–1.25 (). Comparable decreases in mean absolute values for sVCAM and sMAdCAM in subjects across treatment arms () were also noted in all three treatment arms. All but one comparison (sVCAM levels between PB006 and EU-ref-NTZ) were contained in the similarity margin (0.8–1.25) at 90% (1.00, 1.26) or 95% CIs (0.97, 1.29). Finally, descriptive analyses of Emax for baseline-adjusted CD19+ and CD34+, Emin for sVCAM and sMAdCAM also demonstrated similar outcomes between PB006 and the reference medicines (Supplementary Material Table S5).
Table 4. AUECbase-neg baseline-corrected CD34+ cells, sVCAM, and sMadcam (PD set).
3.5. Safety analysis
No notable difference between treatments was seen in terms of the reported number and type of treatment-emergent adverse events (TEAE) (Supplementary Material Table S6). Approximately two-thirds of subjects in each treatment group reported at least one TEAE; approximately half of these were considered possibly or probably related to study treatment in each group.
The most frequently reported TEAEs (i.e. reported by >5% of subjects) were headache, injection site reactions, nausea, and nasopharyngitis.
Serious TEAEs were reported in two subjects receiving US-ref-NTZ. One subject reported acute kidney injury and ureterolithiasis on Day 51 subsequent to bilateral kidney stones reported on Day 13 (both resolved). A second subject reported injury of the left thumb. Both were assessed as likely unrelated to the study treatment. No cases of hypersensitivity were reported in the study.
The incidence of JCV seroconversion for each arm was comparable, with 9/149 (6.0%), 6/150 (4.0%), and 4/151 (2.7%) of subjects in the Safety Set showing detectable JCV antibodies at Day 85 in the PB006, US-, and EU-ref-NTZ arms, respectively.
No deaths, TEAEs leading to study discontinuation, or cases of PML were reported during the study.
3.6. Immunogenicity
No apparent differences in immunogenicity were observed following the infusion of PB006, US-ref-NTZ, or EU-ref-NTZ during the study. Incidence of ADA-positive subjects was similar across all treatment arms, with most subjects (84–92%) testing positive for ADAs at least once during the study. NAbs were detected in most ADA-positive subjects with a similar incidence across all three treatment arms (Supplementary Material Table S7).
Likewise, similar ADA and NAb titer profiles were observed across all treatment arms at the timepoints recorded as part of the immunogenicity analysis (). No treatment-related differences were observed with regards to PK or PD measures relating to ADA- or NAb-positive status (Supplementary Material Figures S2 and S3).
Table 5. ADA and NAb rates over time across treatment arms.
4. Discussion
The purpose of this study was to demonstrate PK and PD similarity of PB006 to both US-licensed ref-NTZ and EU-approved ref-NTZ following infusion of single doses of 3 mg/kg. This PK/PD similarity study employed endpoints reflecting of the mechanism of action of natalizumab, at a dose sensitive to detect differences, if any, in line with global regulatory requirements [Citation31].
Natalizumab is an IgG4 humanized monoclonal antibody that can undergo Fab-arm exchange with endogenous IgG4 molecules to form clinically inactive bi-specific antibodies of unknown specificity and lower affinity to α4-integrins [Citation25,Citation26]. Such exchange generates IgG4 molecules comprising a natalizumab heavy – light chain pair coupled to an endogenous IgG4 heavy – light chain pair. Since exchanged and unexchanged species cannot be quantified independently using a single ELISA, a quantitation strategy was developed employing two ELISAs: one measuring total natalizumab, including both unexchanged (i.e. intact) and exchanged molecules, and the second measuring only unexchanged natalizumab. The presence and amount of exchanged natalizumab in serum was calculated by the difference in values obtained in the two assays.
Demonstration of PK similarity was therefore based on the determination of total natalizumab measuring both, i.e. unexchanged as well as Fab-arm exchanged natalizumab species. The results of total natalizumab PK showed that PB006 matched EU-/US-ref-NTZ in all tested PK parameters. The 90% CIs for the geometric mean ratios of AUC0–inf were completely included in the prespecified PK similarity margin (0.80–1.25), demonstrating PK similarity for all three comparisons (PB006 vs US-ref-NTZ, PB006 vs EU-ref-NTZ, EU-ref-NTZ vs US-ref-NTZ). Additionally, the statistical comparison of the secondary PK parameter Cmax matched across treatment arms. Overall, natalizumab Fab-arm exchange was rapid and complete; serum concentration – time curves of exchanged natalizumab were similar for all three treatment groups. The 90% CIs for the geometric mean ratios of exchanged Cmax and AUC0–inf were completely included in a margin of 0.80–1.25.
The PK data generated in this study indicate that in a clinical context the administration of PB006 would be expected to result in the same exposure of natalizumab as the reference medicine. In addition, the data provide evidence that the rate and the extent of PB006 Fab-arm exchange matches its reference medicine.
There are five essential characteristics required by the FDA for demonstration of similar efficacy in the context of biosimilar development [Citation27]. To support the demonstration of similar PD of PB006, a number of PD measures were selected to compare natalizumab target binding (α4-integrin %RS), target mechanism (sVCAM; sMAdCAM), and physiologic responses of target engagement (circulating CD19+ and CD34+ cells). Determination of the increase in baseline levels of circulating CD19+ cells (AUC012w) and α4-integrin %RS (AUEC012w) have been shown to meet these essential characteristics. Neither of the PD endpoints above has, however, been statistically associated with a surrogate or clinical endpoint in MS. Therefore, the demonstration of similar efficacy was also based on a clinical endpoint, namely the cumulative number of new active lesions over 24 weeks in patients with RRMS in a comparative clinical efficacy and safety study, which is reported elsewhere [Citation20].
The ratio of geometric means for AUEC0-12w α4-integrin % receptor saturation as well as for AUEC0-12w of baseline-adjusted CD19+ for all three comparisons (PB006 vs US-ref-NTZ, PB006 vs EU-ref-NTZ, EU-ref-NTZ vs US-ref-NTZ) were close to 1.0 and the 90%/95% CIs were completely included in the prespecified PD similarity margin (0.80–1.25). For the key secondary endpoints (AUEC sVCAM, AUEC sMAdCAM, CD34+ cell counts) all 90% CIs were within the PD similarity margin (0.80–1.25). For the 95% CI, relevant for EMA, all comparisons were within the PD similarity margin, with the exception of sVCAM (ratio: 1.13; 95% CI: 0.97, 1.29). This was determined not to be clinically meaningful given that sVCAM values were otherwise similar for all treatment comparisons. The secondary PD endpoints (Emax and tmax of baseline-adjusted CD19+, Emin, tmin of sVCAM and sMAdCAM, AUEC0-t, Emax, tmax of CD34+) were overall supportive of PD similarity between PB006 and its reference medicine.
The PK and PD endpoints chosen for this PK/PD similarity study are based on the assessment of translational development risk as described earlier for reference medicine development [Citation28]. While the initial biomarker classification was developed to provide developers with a guide to designing translational and early clinical development plans, it applies equally well to the demonstration of similar efficacy for biosimilars, as follows:
Using the data on Type 1 (systemic exposure: PK of unexchanged in Fab-arm exchanged PB006), Type 2 (primary binding event: α4-integrin %RS), Type 3 (ligand binding: sVCAM; sMAdCAM), and Type 4 (physiologic response: circulating CD19+ and CD34+ cells) obtained from this PK/PD similarity study in addition to data on Type 5 (surrogate endpoint: cumulative number of new active lesions over 24 weeks) from the comparative efficacy and safety study in patients with RRMS [Citation20], it can be concluded that PB006 exhibits similar efficacy to its reference medicine. Furthermore, the data provide evidence that a clinical endpoint, although not measured in the PB006 development program, will also show similar results for PB006 as compared to its reference medicine.
The immunogenicity profile for PB006 matched that of US- and EU-ref-NTZ, as shown by the distribution of ADAs and NAbs across the three treatment arms. Of note, the increased rates of ADA and NAb reported here for PB006 as well as US- and EU-ref-NTZ are higher than historically reported rates [Citation32]. This is likely due to the increased sensitivity and drug tolerance of the ADA assays used in this study, compared to historical testing data [Citation33] as well as the definition of the threshold for ADA-positivity optimized for a sensitive detection of ADA for the purpose of a biosimilarity study. Notably, the drug tolerance of the ADA assay was close to the Cmax reported for total natalizumab in the study, leading to a more robust ADA signal. ADA titers, unlike ADA rates represent a semi-quantitative measure of the immune response to PB006 and EU-/US-ref-NTZ, and were similar across the three treatment arms. More details on the ADA assay and the immunogenicity data obtained for PB006 will be reported elsewhere (Manuscript in preparation). Matching natalizumab serum drug concentrations and α4-integrin receptor saturation over time were observed between PB006, US-, and EU-ref-NTZ in the ADA-positive and ADA-negative subgroups, confirming the absence of any difference in PD profile as a result of ADA formation.
Finally, administration of single doses of PB006 to healthy subjects showed that the overall safety profiles of PB006, US-ref-NTZ, and EU-ref-NTZ were similar. The most frequently reported AEs for all treatment groups were headache and injection site reactions, experienced by 26.0–29.5% and 18.0–21.5% of all subjects, respectively. Local tolerance was similar across all treatment arms in concert with the similar immunogenicity profile of PB006, US-, and EU-ref-NTZ. Importantly, no new safety findings were experienced by any subjects infused during the study. The incidence of JCV seroconversion for each arm was comparable, with 6.0%, 4.0%, and 2.7% of subjects showing detectable JCV antibodies at Day 85 in the PB006, US-, and EU-ref-NTZ arms, respectively. These values are in line with reported values from the literature for reference natalizumab treatment (7.3% per year) [Citation34]. No cases of PML or symptoms suggestive of PML were reported during the study or during the PML follow-up visit at Day 169, and no cases of hypersensitivity were reported.
5. Conclusion
Overall, the results reported here demonstrate PK and PD similarity of PB006 versus US-ref-NTZ and EU-ref-NTZ in healthy subjects and contribute to the ‘totality of evidence’ that PB006 matches ref-NTZ. Furthermore, no clinically meaningful differences in safety, tolerability, or immunogenicity were observed across treatment arms.
Declaration of interest
H Wessels and K Roth are employees of Polpharma Biologics S.A. O von Richter is an employee of Hexal AG (A Sandoz company). J Höfler is an employee of Staburo GmbH. P Chamberlain has received consulting fees from Polpharma Biologics S.A. for scientific and regulatory consulting related to PB006. A Kromminga has received consulting fees from Polpharma Biologics S.A. for consulting related to PB006. D Lehnick has received fees from bioeq GmbH (on behalf of Polpharma Biologics S.A.) for statistical and regulatory consulting in the context of study planning and scientific advisory meetings related to PB006. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have received an honorarium from Expert Opinion on Biological Therapy for their review work. A reviewer on this manuscript has disclosed receipt of support for attending medical conferences from Biogen, Jansen, Genzyme, Merck. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Ethics statement
This study was conducted in accordance with the principles of the Declaration of Helsinki in place at the time of study conduct, the International Council on Harmonisation (ICH) E6 Guideline for Good Clinical Practice, European Medicines Agency (EMA)/Committee for Medicinal Products for Human Use (CHMP)/ICH/135/1995, EU Clinical Trial Directive: Directive 2001/20/EC, and all applicable national and local laws and regulations. At Screening, subjects were informed verbally and in writing of the objectives, procedures, and risks of study participation, including possible side effects and potential interactions. Written informed consent was obtained from all participants in the study. Independent ethics committees for sites in The Netherlands and Poland were the Evaluation of Ethics in Biomedical Research and the Bioethics Committee of the District Medical Chamber in Warsaw, respectively. For sites in the US, the independent review board was the Midlands Independent Review Board.
Author contributions
H Wessels had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: O von Richter, K Roth, H Wessels, J Höfler, P Chamberlain, A Kromminga, D Lehnick. Acquisition, analysis, or interpretation of data: All authors. Drafting of the manuscript: All authors. Critical revision of the manuscript for important intellectual content: All authors. Statistical analysis: Josef Höfler.
EOBT PKPD of biosimilar natalizumab PB006 Supplementary Material.pdf
Download PDF (392 KB)Acknowledgments
Editorial support was provided by Syneos Health, funded by Hexal AG (a Sandoz company). Final approval of the manuscript rested solely with the scientific authors.
Data availability statement
The results of the study reported herein were disclosed within the EudraCT system on 3 March 2022 (2019–003874–15).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14712598.2023.2290530
Additional information
Funding
References
- Biogen Netherlands B.V. Tysabri® (natalizumab). Summary of Product Characteristics, 2022. [Internet]. [cited 2023 Apr 6]. Available from: https://www.ema.europa.eu/documents/product-information/tysabri-epar-product-information_en.pdf.
- Biogen Inc. Tysabri® (natalizumab). Prescribing Information. [Internet]. 2023 [cited 2023 Oct 2]. Available from: https://www.tysabri.com/content/dam/commercial/tysabri/pat/en_us/pdf/tysabri_prescribing_information.pdf
- Sheremata WA, Minagar A, Alexander JS, et al. The role of alpha-4 integrin in the aetiology of multiple sclerosis. CNS Drugs. 2005;19(11):909–922.
- Saure C, Warnke C, Zohren F, et al. Natalizumab and impedance of the homing of CD34+ hematopoietic progenitors. Arch Neurol. 2011;68:1428–1431. doi: 10.1001/archneurol.2011.238
- Warnke C, Stettner M, Lehmensiek V, et al. Natalizumab exerts a suppressive effect on surrogates of B cell function in blood and CSF. Mult Scler. 2015;21(8):1036–1044.
- McCamish M, Yoon W, McKay J. Biosimilars: biologics that meet patients’ needs and healthcare economics. Am J Manag Care. 2016;22:S439–S442. 13 Suppl
- IMS Health for Healthcare Informatics. Delivering on the potential of biosimilar medicines. The role of functioning competitive markets. [Internet]. 2016 [cited2023 Apr 6]. Available from: https://www.medicinesforeurope.com/wp-content/uploads/2016/03/IMS-Institute-Biosimilar-Report-March-2016-FINAL.pdf
- Smolen JS, Goncalves J, Quinn M, et al. Era of biosimilars in rheumatology: reshaping the healthcare environment. RMD Open. 2019;5(1):e000900.
- Gherghescu I, Delgado-Charro MB. The biosimilar landscape: an overview of regulatory approvals by the EMA and FDA. Pharmaceutics. 2020;13(1):48. doi: 10.3390/pharmaceutics13010048
- IQVIA. The impact of biosimilar competition in Europe. [Internet]. 2021 [cited 2021 Apr 6]. Available from:https://www.iqvia.com/library/white-papers/the-impact-of-biosimilar-competition-in-europe-2021
- Kurki P, Kang H-N, Ekman N, et al. Regulatory Evaluation of Biosimilars: refinement of principles based on the scientific evidence and clinical experience. BioDrugs. 2022;36(3):359–371.
- Wolff-Holz E, Tiitso K, Vleminckx C, et al. Evolution of the EU biosimilar framework: past and future. BioDrugs. 2019;33(6):621–634.
- Cohen HP, Lamanna WC, Schiestl M. Totality of evidence and the role of clinical studies in establishing biosimilarity. In: Gutka H, Yang H, and Kakar S, editors Biosimilars: regulatory, clinical, and biopharmaceutical development. Cham: Springer; 2018. p. 601–628. doi: 10.1007/978-3-319-99680-6_22
- Weise M, Bielsky M-C, De Smet K, et al. Biosimilars: what clinicians should know. Blood. 2012;120(26):5111–5117.
- FDA. Biosimilar and interchangeable Biologics: more treatment choices. [Internet]. 2023 [cited 2023 Oct 3]. Available from: https://www.fda.gov/consumers/consumer-updates/biosimilar-and-interchangeable-biologics-more-treatment-choices
- Kay J. A ‘wind of change’ to biosimilars: the nor - SWITCH trial and its extension. J Intern Med. 2019;285(6):693–695. doi: 10.1111/joim.12896
- EMA. Biosimilars in the EU. Information guide for healthcare professionals [Internet]. 2019 [cited 2023 Mar 3]. Available from https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf.
- FDA. FDA approves first biosimilar to treat multiple sclerosis [Internet]. 2023 [cited 2023 Aug 30]. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-biosimilar-treat-multiple-sclerosis.
- EMA. Tyruko® (natalizumab) EPAR Medicine Overview. [Internet]. 2023 [cited 2023 Nov 8]. Available from: https://www.ema.europa.eu/en/documents/overview/tyruko-epar-medicine-overview_en.pdf
- Hemmer B, Wiendl H, Roth K, et al. Efficacy and safety of proposed biosimilar natalizumab (PB006) in patients with relapsing-remitting multiple sclerosis. JAMA Neurol. 2023;80(3):298.
- Schwab N, Schneider-Hohendorf T, Melzer N, et al. Natalizumab-associated PML: Challenges with incidence, resulting risk, and risk stratification. Neurology. 2017;88(12):1197–1205.
- Tanaka M, Kinoshita M, Foley JF, et al. Body weight-based natalizumab treatment in adult patients with multiple sclerosis. J Neurol. 2015;262(3):781–782.
- Foley JF, Goelz S, Hoyt T, et al. Evaluation of natalizumab pharmacokinetics and pharmacodynamics with standard and extended interval dosing. Mult Scler Relat Disord. 2019;31:65–71. doi: 10.1016/j.msard.2019.03.017
- Serra López-Matencio JM, Pérez García Y, Meca-Lallana V, et al. Evaluation of natalizumab Pharmacokinetics and pharmacodynamics: toward individualized doses. Front Neurol. 2021;12:12. doi: 10.3389/fneur.2021.716548
- Shapiro RI, Plavina T, Schlain BR, et al. Development and validation of immunoassays to quantify the half-antibody exchange of an IgG4 antibody, natalizumab (tysabri®) with endogenous IgG4. J Pharm Biomed Anal. 2011;55(1):168–175.
- Labrijn AF, Buijsse AO, van den Bremer ETJ, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. 2009;27(8):767–771.
- Li J, Florian J, Campbell E, et al. Advancing biosimilar development using pharmacodynamic biomarkers in clinical pharmacology studies. Clin Pharmacol Ther. 2020;107(1):40–42.
- Visser SAG, Bueters TJH. Assessment of translational risk in drug research: role of biomarker classification and mechanism-based PKPD concepts. Eur J Pharmaceut Sci. 2017;109:S72–S77. doi: 10.1016/j.ejps.2017.08.006
- Maurer W, Jones B, Chen Y. Controlling the type I error rate in two‐stage sequential adaptive designs when testing for average bioequivalence. Stat Med. 2018;37(10):1587–1607. doi: 10.1002/sim.7614
- Khoy K, Mariotte D, Defer G, et al. Natalizumab in Multiple Sclerosis Treatment: From Biological Effects to Immune Monitoring. Front Immunol. 2020;11:11. doi: 10.3389/fimmu.2020.549842
- FDA. Clinical pharmacology data to support a demonstration of biosimilarity to a reference product. Guidance for industry [Internet]. 2016 [cited 2023 6 Apr]. Available from:https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-pharmacology-data-support-demonstration-biosimilarity-reference-product
- Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910.
- Link J, Ramanujam R, Auer M, et al. Clinical practice of analysis of anti-drug antibodies against interferon beta and natalizumab in multiple sclerosis patients in Europe: a descriptive study of test results. PLoS One. 2017;12(2):e0170395.
- Dwyer CM, Jokubaitis VG, Stankovich J, et al. High rates of JCV seroconversion in a large international cohort of natalizumab-treated patients. Ther Adv Neurol Disord. 2021;14:175628642199891. doi: 10.1177/1756286421998915