
ABSTRACT
Introduction
Major depression is a common, disabling mental health condition associated with the highest disease burden for any neuropsychiatric disorder worldwide, according to the WHO. Due to the imperfect efficacy and tolerability profiles of existing treatments, investigational compounds in novel treatment classes are needed. Opioid-receptor antagonists are a potential new class of treatments currently under investigation.
Areas covered
Major depressive disorder is first overviewed. Existing treatments, both their mechanisms of action and their place within the antidepressant space, are discussed herein. Then, the profile of Aticaprant and the wider context of kappa-opioid antagonism for depression are discussed in focus.
Expert opinion
Early evidence indicates that Aticaprant may possess desirable pharmacodynamic and pharmacokinetic properties. A lack of convincing efficacy data at the time of writing precludes any definitive statement on its potential as an antidepressant.
1. Background
Major depressive disorder (MDD) is a mental health disorder characterized by low mood, suicidal thoughts, and anhedonia, an impaired ability to experience reward-related pleasure or joy [Citation1]. Whilst definitions of the disorder have varied, they all tend to include the aforementioned core symptoms occurring nearly every day over the course of several weeks. Estimated by the World Health Organization to be the leading cause of disability worldwide [Citation2], 16% of UK adults report at least moderate depressive symptoms at any one time [Citation3], with MDD associated costs to the economy exceeding £23.8 billion annually [Citation4]. This manuscript reviews the extant literature regarding the development of the kappa-opioid receptor antagonist Aticaprant (see Appendix A for search strategy), in addition to its place in the wider antidepressant market.
1.1. Existing Treatments
Psychological interventions such as cognitive behavioral therapy are first-line MDD treatments, particularly for first-episode depression and milder episode severities. Availability of NHS psychological therapies is poor [Citation5], with a recent survey by the UK charity Mind finding that over half of patients wait 3 months for access to talking therapies, and more than 1 in 10 wait over a year [Citation6]. In addition to poor availability, success rates are modest at best [Citation7], albeit are similar to antidepressant medications. Many depressed patients are treated with psychiatric medications either instead of, or in tandem with psychological therapies depending on availability. There are a considerable range of pharmaceutical treatment options available, which can be broadly divided into six categories: selective serotonin reuptake inhibitors (SSRIs); serotonin-noradrenaline reuptake inhibitors (SNRIs); noradrenaline and specific serotonergic antidepressants (NASSAs); tricyclic antidepressants (TCAs); serotonin antagonists and reuptake inhibitors (SARIs) and Monoamine oxidase inhibitors (MAOIs). Whilst the number of licensed antidepressants varies based on locality, it is primarily SSRIs that are recommended as ‘first-line’ treatments by NICE [Citation8] for moderate-to-severe depression, i.e. citalopram, fluoxetine, paroxetine, and sertraline. In 2023, it is estimated that 15.3% of adults in England are currently prescribed an antidepressant [Citation9].
2. Medical need
Whilst the mechanism(s) of action of the above medication classes vary, meta-analytic data suggest that all have limited remission rates and frequent reporting of side effects associated with subsequent suboptimal adherence [Citation10]. Of particular interest to the present manuscript is the frequency of residual anhedonia in those who report improvements in low mood and suicidal ideation following treatment with established medications [Citation11]. Accumulating evidence suggests SSRIs in particular are fairly poor in treating the impaired reward and motivation seen in anhedonia [Citation12]. Another notable side effect is ‘emotional blunting,’ reported by 40–60% of patients treated with serotonergic antidepressants [Citation13], in which the intensity of both positive and negative emotions are chronically lessened. It is estimated that a third of those taking psychiatric medication will ‘fail,’ typically defined as a lack of an adequate response to ≥2 pharmacological treatments given for an adequate duration at an adequate dose during the same major depressive episode [Citation14]. Given the reality of likely partial-remission and notable side effects for those taking established medications, there is a clear need for novel, more tolerable antidepressant treatments that do not leave residual symptoms.
Box 1. Drug Summary Box.
Drug name: Aticaprant
Phase: III
Indication: Major depression; substance abuse
Pharmacology/mechanism of action: Kappa-opioid receptor antagonist.
Route of administration: Oral
Notable trials: NCT03559192 (completed) NCT05518149 (ongoing)
Chemical structure: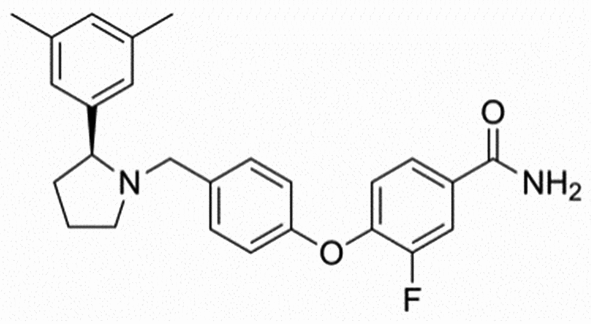
3. Market review
The global antidepressant market is large, estimated to be worth in excess of $16B in 2022, having grown from approximately $13.5B in 2020 [Citation15]. An extensive list of licensed and developmental compounds is presented in appendix B for brevity (note: additional compounds available over the counter, off-label, or as adjunct treatments for depression are not included). Of the 104 compounds presented therein, 38 (36%) have been withdrawn from the market either due to economic viability or some concern with efficacy, tolerability, or safety.
Given high rates of failure, the financial risks associated with the development of novel antidepressants are considerable. A recent systematic review reported nine compounds to be in phase II or phase III trials as of 2022 [Citation16]. Conservative estimates put the compound annual growth rate of the antidepressant market at 3% [Citation17], which would see the market value reach $21B by 2030, although some sources consider the growth rate to potentially exceed 6% [Citation15].
4. Current research goals
The process of identifying new treatments for MDD can be crudely split into two strands, with two approaches within each strand (). A recent review of promising compounds in phase 2 or 3 studies identified 19 compounds of special interest [Citation18], of which those most relevant to the present manuscript are detailed in . Increasing lines of evidence implicate the endogenous opioid system in the regulation of mood, reward, and well-being and suggest that this system may be dysregulated in depressive disorders [Citation37]. This has led to a resurgence of interest in exploring the opioid system as a target for the treatment of depression, with evidence to suggest pharmacological modulators of mu, delta, and kappa opioid receptors may have therapeutic potential [Citation38]. Here we focus on kappa-opioid receptor antagonists, drawing from both pre-clinical studies and clinical trials, to support continued exploration of this promising target for depression.
Table 1. Overview of potential antidepressant development approaches.
Table 2. K-Opioid receptor antagonists under development for MDD.
Perhaps, the most straightforward method is using existing mood disorder medications with known mechanisms of action off-label from the treatment of MDD. A clear example of this is with the established antipsychotic lurasidone, a serotonin and dopamine antagonist for the treatment of psychosis and bipolar depression. Given the overlap of bipolar depression to unipolar depression, in addition to the molecular targets of the drug, lurasidone is a clear candidate for use in MDD. Several studies of lurasidone for MDD have been conducted to date, with results indicating efficacy, albeit with a side-effect profile not dissimilar to existing treatments [Citation39,Citation40].
One might instead choose to repurpose an existing medication from a different area of medicine that exerts its effects through a mechanism of action not previously employed in MDD treatments. A recent, notable example of this is the diabetes drug metformin. Metformin acts via the enzyme AMP-activated protein kinase, albeit its purported causal relationship with antidepressant effects is still not well understood. Unsurprisingly, the potential antidepressant effects of metformin were first evidenced in diabetes cohorts, for example by Guo and colleagues [Citation41], in which metformin outperformed placebo in reducing depressive symptoms over 24 weeks as per both the MADRS and HAM-17.
For those willing to invest in research and development, the alternative approach involves identifying new compounds for MDD treatment and manufacturing a novel compound. Whilst certainly more time- and cost-intensive, this avenue allows for the potential creation of more tolerable and efficacious treatments for patients, in addition to potentially lucrative new revenue streams for the developers. There is considerable appetite in the antidepressant pharmaceutical space for the development of novel treatments with either an established or a novel mechanism of action.
Some developers opt to create novel compounds that exert their effects via mechanisms of action that are already employed in current MDD treatments. For example, several recent phase 2 and 3 trials of the NMDA receptor antagonist Rapastinel (developmental code GLYX-13; Allergan plc) have been conducted, albeit reports on efficacy have been mixed with only one of six trials finding superiority to placebo [Citation42]. NMDA receptor antagonists for depression are a relatively recent concept, with the only two FDA-approved MDD treatments in this class with nasal esketamine and Auvelity (bupropion + dextromethorphan) approved in 2019 and 2022, respectively. Nonetheless, those involved in the development of compounds acting via NMDA antagonism have at least some successful, licensed treatments as a justification for their investment.
Alternatively, developers may choose to develop novel compounds that work in a different way to existing antidepressant treatments. Of the many candidates in this category, for example vasopressin receptor antagonists [Citation43] and orexin receptor antagonists [Citation44], kappa-opioid receptor antagonists are pertinent to the present manuscript. Several k-opioid receptor antagonists are currently in development for the treatment of MDD, with several having reached phase III.
5. Rationale regarding the kappa-opioid receptor in depression
Kappa-opioid receptors (KORs) are g-protein-coupled receptors expressed throughout the brain and spinal cord. They consist of seven transmembrane alpha helix structures, each paired with an internal guanine nucleotide-binding protein (g-protein). In the case of KOR, the g-protein in question is Gi/G0, which is formed of heterotrimeric g-protein alpha subunits. The binding of an appropriate ligand, for example ketazocine, to the external binding site of the cell bestows guanine nucleotide exchange factor (GEF) ability to the receptor, i.e. the ability to swap guanine diphosphate (GDP) for guanine triphosphate (GTP) via hydrolysis. The GDP on the alpha subunit of the g-protein (in our case Gi/G0) exchanges GDP for GTP, causing a conformational change in the receptor itself that results in a structural dissociation from the rest of the g-protein, causing intracellular signaling. The g-protein therefore effectively acts as a ‘switch’ inside the cell that turns signaling ‘on’ by hydrolyzing guanosine triphosphate to guanosine diphosphate following the binding of a ligand to the outside of the cell. The signaling cascade of external ligand binding to intracellular signaling via the g-coupled protein is detailed in . In the case of KOR antagonists, the relevant ligand blocks binding to the extracellular binding site by endogenous ligands and therefore the resulting signaling cascade.
Figure 1. Kappa opioid receptor function.1. The receptor is inactive as no ligand is bound to the extracellular binding site. 2. A ligand binds to the extracellular binding site. 3. There is a resulting conformational change in the receptor. GDP is exchanged for GTP on the alpha subunit. Secondary messaging within the cell begins. Key: α, β, γ = alpha, beta, gamma subunit, respectively. GDP= guanine diphosphate. GTP= guanine triphosphate.
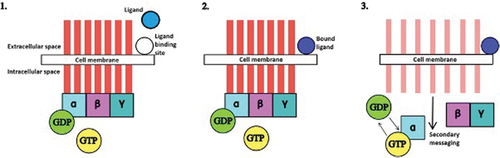
Dynorphins are endogenous peptides that primarily act on KORs, and the dynorphin-KOR system has been implicated as an endogenous anti-reward system [Citation45]. In contrast to the effects of MOR and DOR activation, KOR agonists have been demonstrated to elicit, pro-depressive [Citation46], anhedonia [Citation47], dysphoric [Citation48,Citation49] behavioral effects in pre-clinical models, in addition to being anti-manic in humans [Citation50].
KOR antagonists have been tested in preclinical depression models, consistently demonstrating an antidepressant effect on stress-induced behaviors (see Knoll [Citation51] for review). There is also compelling preclinical evidence to support the utility of KOR antagonists in reversing behavioral deficits in stress-exposed rodents [Citation52]. For example, Aticaprant restored a decrease in open arm time on the elevated plus maze [Citation53]. Furthermore, stress-naïve rodents tested on the Forced Swim Test (FST) also demonstrate reduced depressiogenic behaviors [Citation54,Citation55]. The limited human trial evidence concerning KOR antagonism for the treatment of depression is discussed below.
6. Competitive environment
6.1. Navacaprant
Developed under several codes, Navicaprant was reported to possess a potent, selective binding affinity for the KOR, in addition to a duration of action of less than 24 h [Citation56]. A phase II trial [Citation24], completed in 2022, has not yet posted results. The developer, Neumora Therapeutics, Inc., has recently begun both a 6-week RCT and a 52-week open-label continuation phase III trials [Citation25,Citation26]. As few human data regarding Navicaprant have been published, it is not possible to evaluate its potential when compared to rival compounds.
6.2. Buprenorphine/Samidorphan
This combination formulation has been under investigation since at least 2011 [Citation27,Citation28], with an early long-term safety study reporting a discontinuation rate of 50.1% over 56 weeks (n = 1485) [Citation30]. Although of a modest size (n = 66), a titration study found negligible differences between a 1-week and 2-week titration schedule [Citation31]. In a series of RCTs from 2011 to 2016 [Citation32–34], samidorphan/buprenorphine was unable to consistently demonstrate superiority to placebo on clinician rated measures of depression treatment response, albeit the most recent trial demonstrated superiority increase in a dose-dependent manner. A recent trial in a treatment resistant cohort failed to demonstrate a superiority to placebo [Citation35], with the open-label long-term continuation study only reporting safety data thus far [Citation36]. The outlook for ALKS-5461 appears fraught, given the above unconvincing data and the refusal of the U.S. Food and Drug Administration’s 2019 rejection of the developers' bid for approval.
6.3. Aticaprant
6.3.1. Pharmacodynamic profile
In the important pre-clinical pharmacodynamic investigations by Rorick-Kehn and colleagues [Citation57], the aminobenzyloxyarylamide Aticaprant has displayed a good bioavailability (F = 25%) and a rapid rate of absorption (t(max): 1–2 h). Oral administration of Aticaprant resulted in a selective and potent binding affinity for kappa opioid receptors in vivo (ED₅₀ = 0.33 mg/kg), demonstrating an approximately 30-fold preference for kappa subtype opioid receptors over mu subtype. It is also notable that Aticaprant administration did not block kappa-agonist-associated analgesia a week after administration, indicating it may be as effective as a medicine that does not possess lengthy pharmacodynamic effects. This is of particular importance given previous KOR antagonists have been shown to exert their effects for up to 2 months, a characteristic that is troublesome given the long window for drug–drug interactions [Citation58]. Further to this point, in a separate study, Aticaprant was shown to have the shortest duration of action when compared to other KOR antagonists in a pain paradigm [Citation59].
A whole-cell electrophysiology study demonstrated that Aticaprant was reasonably effective in KOR signaling blockade, with an IC50 of 3.0 ± 4.6 nM [Citation60]. Aticaprant therefore demonstrated an inhibitory effect on a specific biological process or target, with a required concentration of approximately 3.0 nanomolar (nM). It is important to note, however, that Aticaprant was significantly outperformed by BTRX-335140 in this regard (IC50 = 1.2 ± 0.9), indicating that Aticaprant may not be the most pharmacologically potent KOR antagonist in development.
6.3.2. Pre-clinical work
Aticaprant has been shown in several rodent models to ameliorate a variety of disease-like behaviors, providing a rationale to justify its exploration in humans. For example, Aticaprant has decreased the effects of both nicotine [Citation61] and alcohol withdrawal on anxiety-like behaviors [Citation62] and reduced the rates of ‘relapse’ drinking behaviors in a dose-dependent manner [Citation63]. Additionally, Aticaprant has been shown to significantly reverse stress-induced deficits produced by an unpredictable chronic mild stress paradigm on several measures [Citation64,Citation65]. Aticaprant was also effective in reducing anhedonia-like behaviors as measured by an intracranial self-stimulation test [Citation59].
Whilst mu and delta opioid receptor agonism lead to antidepressant-like behavioral effects in mouse models [Citation66–68], it is kappa-opioid antagonism that has been most strongly associated with reducing depressive-like behaviors, for example Aticaprant on the forced swim test [Citation69]. Notably in this study, Aticaprant was not superior to two other candidate KORs in this regard. The most notable rival candidate, denoted as ‘1c,’ possesses a not dissimilar binding affinity for kappa subtype receptors to that of Aticaprant (Ki (nM) 15.7 vs 11.9), respectively, albeit with a much stronger selectivity for kappa opioid receptor subtypes over both mu and delta (mu/kappa 14.0 delta/kappa 12.2 versus 7.6 and 7.6 for ‘1c’ and Aticaprant, respectively). When tested for in vitro antagonist activity in human opioid receptors, this binding/selectivity profile changes somewhat. Aticaprant’s preference for kappa over mu was much lower, albeit still present, in human opioid receptors, whilst its preference for kappa over delta was strengthened further. It is pertinent that attempts to replicate in humans the animal-model findings of delta opioid receptor agonism being associated with antidepressant effects did not succeed [Citation70]. When taken alongside the far more balanced selectivity profile of Aticaprant between mu and kappa opioid receptor subtypes observed in humans, it remains plausible that some of the antidepressant effect of Aticaprant in humans may be carried by both mu and kappa opioid receptors.
Data concerning the differences in specific opioid receptor subtype activity between humans and rodents in response to opioid modulator compounds are scant. MOR gene expression varies between humans and mice due to variations in the polypyrimidine/polypurine DNA region [Citation71]. Specifically, in mice, this region has a unique structure that allows for α-CP1 binding, thereby enhancing MOR gene expression significantly compared to humans. Schattaeur et al. [Citation72] also returned inconsistent p-38 Mitogen-Activated Protein Kinase activity between human and rodent KORs following administration of partial KOR agonists ex-vivo. Further trials into the role of subtype opioid receptor activity in depressive behaviors would help demystify the open question of which opioid receptors drive antidepressant activity and whether this differs between human and rodent models, possibly pushing compound development toward or away from joint kappa-mu opioid receptor antagonism.
6.3.3. Clinical studies
Published studies of Aticaprant in humans are currently limited, with the vast majority of published pharmacodynamic and pharmacokinetic data coming from animal studies. A pupillometry study identified a dose range of 25–60 mg as optimal for achieving maximum kappa opioid receptor occupancy [Citation73]. This finding is important as this occurred without significant mu opioid receptor occupancy, meaning this dosage profile should avoid the potential mu-associated side effects.
A 2014 study of healthy volunteers returned no clinically significant findings with respect to Aticaprant’s safety and tolerability profile, showing it to be well tolerated over single and multiple doses [Citation74]. Crucially, they reported no interactions on cognitive or motor assays, a finding which persisted when administered with ethanol. This suggests those taking Aticaprant may not have to cease alcohol consumption, which would bolster its real-world acceptability. A minority of participants in a substance-dependence sample did report pruritus [Citation75].
A randomized, double-blind, placebo-controlled proof-of-mechanism trial investigated Aticaprant in treating anhedonia in patients with mood or anxiety disorders [Citation76]. The study found that patients treated with Aticaprant (10 mg/day for 8 weeks, n = 45) showed a significant increase in ventral striatal activation during reward anticipation on the Monetary Incentive Delay Task, as measured by functional magnetic resonance imaging (fMRI), compared to the placebo group (n = 44). The observed impact of Aticaprant on brain areas associated with pleasure and the reduction in clinical anhedonia, assessed by the Snaith-Hamilton Pleasure Scale (SHAPS), indicate its potential as a therapy for anhedonia, with possible broader therapeutic effects beyond major depressive disorder (MDD) treatment. Furthermore, the authors found Aticaprant to possess good tolerability and a low side-effect profile. This study provided a rationale for carrying out larger trials to investigate the clinical impact of Aticaprant.
Whilst an early Phase II trial of the drug faltered with regard to recruitment [Citation77], a recently completed trial by Janssen, the developers of Aticaprant (JNJ-67953964), has provided valuable insight into its potential as an antidepressant [Citation20]. The trial recruited 181 depressed individuals and randomized them to either placebo or 10 mg Aticaprant, adjunct to their existing serotonergic antidepressant (to which they were only partially responsive). The primary outcome in the study assessed the improvement in MADRS score after 6 weeks in those who failed to respond to placebo during the 3-week run-in phase. Whilst the overall difference in MADRS score was only 2.1 points lower for the Aticaprant group, this was statistically significant (p = 0.04). The Aticaprant group did, however, fail to improve beyond placebo in measures of anhedonia (SHAPS), global illness severity (CGI-S), anxiety (HAM-A), and a patient-reported outcome depression scale (SMDDS). Whilst the reliability of the MADRS results benefit from the double-blind nature of the trial, it is difficult to reconcile with the lack of efficacy seen elsewhere, in particular according to global illness severity and patient-reported depression scale. Ongoing phase III trials must hope to return more robust findings in favor of the efficacy of Aticaprant’s as an antidepressant in humans.
With only one withdrawal due to adverse events in each group, and two withdrawals from the Aticaprant group due to perceived lack of efficacy, the trial demonstrated Aticaprant has good acceptability. Furthermore, 47% of participants (n = 40/85) reported at least one treatment-emergent adverse event, with zero serious adverse events being reported, meaning Aticaprant also appears to be reasonably well tolerated when compared with existing antidepressants. For reference, a 2004 study of SSRI-associated side effects reported that 86% of patients reported at least one side effect [Citation78].
6.3.4. The development path
Consistent with the challenges commonly encountered in the development of pharmaceutical agents, the principal hurdle for Aticaprant’s development is the definitive demonstration of its efficacy in alleviating symptoms of depression, which, as of the current date, has not yet been realized. A single human trial to date to post results with regard to antidepressant efficacy returned a modest MADRS reduction of 2.1 points, which whilst statistically significant, is perhaps not clinically significant. The failure of Aticaprant to outperform placebo on several other measures of core disease symptomology, including a patient reported measure of depressive symptoms, leaves pertinent outstanding questions with respect to the compound’s efficacy. Ongoing phase III studies will be expected to demonstrate good efficacy if Aticaprant is to be proven a viable antidepressant.
An additional concern regarding our appraisal of trial data concerning Aticaprant is the heavy focus toward broad depression-based outcomes, for example the MADRS, which is an issue with experimental medicine research more broadly. It remains plausible that Aticaprant may be more effective in treatment domain-specific behaviors within the context of depression, for example anhedonia. Indeed, evidence regarding anhedonia discussed previously in this manuscript makes a compelling case for the development of Aticaprant, and perhaps KOR antagonists more widely, as anti-anhedonic agents. Janssen, the developer of Aticaprant, is currently recruiting to a trial of the compound in treating a sample with a minimum threshold of anhedonia (NCT05550532). The results of this trial may shed light on the efficacy of Aticaprant in that specific domain, rather than on depression more broadly.
Another foreseeable hurdle in Aticaprant’s development is the potential for side effects associated with KOR antagonism specifically, and opioid receptor antagonism more widely.
Inhibition of opioid-mediated processes, for example hypotension, pulmonary edema, atrial and ventricular dysrhythmias [Citation79], meaning these are potential adverse events of clinical significance to be monitored in human trials. Due to their association with KOR agonism, dysphoria, hallucinations, and dissociative effects are highlighted as adverse events of special interest with respect to KOR antagonists specifically. KOR agonists are also effective in suppressing itching and treating IBS, meaning KOR antagonists should be monitored for pruritus and bowel irritation as AEs of special interest. Notably, the development of KOR antagonist candidates PF-4455242 and JDTic was ceased due to toxicity concerns [Citation80]. Although currently published data has not highlighted toxicity concerns with Aticaprant, it remains to be seen if this remains true at higher doses. Ultimately, the currently published data have reported low levels of side effects, albeit more data from phase III trials is urgently needed.
7. Conclusion
Based on early evidence, Aticaprant appears to be a safe, well-tolerated compound developed in a novel treatment class. The pharmacodynamic and pharmacokinetic profile of the compound suggests it may be available in pragmatic dosages (25–60 mg) and avoids long-term opioid receptor blockage seen in previously developed opioid-receptor antagonists. There remains limited evidence demonstrating clear, clinically significant reductions in depressive symptomology, albeit no results from phase III trials have been posted. Further, we are unable to rule out that a competitor opioids antagonist with similar specificity for KORs may prove a more suitable compound, either with respect to efficacy, tolerability, or both.
8. Expert opinion
The need for novel antidepressant compounds is clear, given the suboptimal efficacy and tolerability profiles of existing first-line treatments. The concept of using opioid-receptor antagonism is not entirely novel, with Buprenorphine emerging as a potential antidepressant candidate, both alone [Citation81] and in combination with samidorphan [Citation82,Citation83]. Although not particularly well tolerated, the efficacy of buprenorphine has undoubtedly encouraged the development of rival KOR antagonists. The reported pharmacodynamic, pharmacokinetic, and tolerability profile of Aticaprant as an adjunctive treatment have certainly encouraged further development of this compound in particular, albeit phase III data will prove decisive in this regard. Of particular interest is the anti-anhedonic potential of KOR antagonists, including Aticaprant, given that depression-related anhedonia is typically resistant to first-line treatments. Whether this profile scales to the higher doses likely required for its use as a monotherapy remains to be seen.
Preclinical work suggests Aticaprant is effective as an antidepressant in animal models of human behavior, in addition to alleviating addiction-like and anxiety-like behaviors. However, the lack of convincing human trial efficacy data published so far hampers its potential as an antidepressant. In particular, the failure of Aticaprant to achieve significant improvements in depressive symptomology as measured by patient reported outcomes in the only published phase II study to date is of concern. It is pertinent to note that the key phase II study from which extant efficacy data can be gleaned involved the use of 10 mg Aticaprant as an adjunctive treatment to those showing inadequate response to an existing treatment. When considering the place of Aticaprant’s in the antidepressant sphere, we are therefore comparing its use at a fairly low dose in a somewhat treatment-resistant sample to existing treatments delivered in monotherapy. It remains plausible that Aticaprant may prove effective when delivered as a monotherapy at a dose providing maximum KOR occupancy (determined to be 25–60 mg [Citation71]), albeit ongoing phase III studies are trialing it as an adjunct treatment.
Considering the published research concerning Aticaprant, it is difficult to label the compound as a leading investigational antidepressant. Aticaprant does appear to be further down the development path than rival KOR antagonists, albeit whether this translates into an effective treatment will be borne out by the in-progress phase III studies. Particular interest will be directed toward the outcome of the developers' trial in anhedonic participants, as this may prove a viable niche for the drug should it underwhelm with regard to broader antidepressive efficacy.
Article highlights
Treatments for major depression exist across many categories and mechanisms of action, but all are limited in their efficacy and acceptability.
Opioid-receptor antagonism has been shown to have antidepressive-like effects in pre-clinical models.
Several opioid-receptor antagonists, particularly of the kappa-opioid subtype, are in development for depression and other indications.
Aticaprant is a candidate kappa-opioid receptor antagonist under development by Janssen, of which there is published data in humans indicating acceptability.
Clear data regarding efficacy, particularly domain-specific efficacy (i.e. anhedonia) is currently being collected, precluding a definitive statement on Aticaprant’s fate.
Aticaprant: Abbreviations list
| α-CP1 | = | alpha-complex protein 1 |
| AE | = | Adverse event |
| AMP | = | Adenosine monophosphate-activated protein kinase |
| CGI-S | = | Clinical global impression severity scale |
| DNA | = | Deoxyribonucleic acid |
| FDA | = | Food and Drug Administration (of the United States of America) |
| FMRI | = | Functional magnetic resonance imaging |
| GDP | = | Guanine diphosphate |
| GEF | = | Guanine nucleotide exchange factor |
| GTP | = | Guanine triphosphate |
| KOR | = | Kappa opioid receptor |
| HAM-A | = | Hamilton Anxiety Scale |
| HAM-17 | = | Hamilton Depression Scale - 17 item |
| IBS | = | Irritable bowel syndrome |
| MADRS | = | Montgomery-Asberg Depression Rating Scale |
| MAOIs | = | Monoamine oxidase inhibitors |
| MDD | = | Major depressive disorder |
| MOR | = | Mu opioid receptor |
| NICE | = | National Institute for Care Excellence |
| NHS | = | National Health Service (of the UK) |
| NASSAs | = | Noradrenaline and specific serotonergic antidepressants |
| NMDA Receptor | = | N-methyl-D-aspartate receptor |
| RCT | = | Randomised control trial |
| SARIs | = | Serotonin antagonists and reuptake inhibitors |
| SHAPS | = | Snaith-Hamilton Pleasure Scale |
| SMDDS | = | Symptoms of Major Depressive Disorder Scale |
| SNRIs | = | Serotonin-noradrenaline reuptake inhibitors |
| SSRIs | = | Selective serotonin reuptake inhibitors |
| TCAs | = | Tricyclic antidepressants |
| UK | = | United Kingdom |
| WHO | = | World Health Organization |
Declaration of interest
At the time of article submission, E Hampsey was involved in two ongoing phase III studies of Aticaprant as a research assistant at King’s College London. He receives no financial contributions from the sponsor in any form, including consultancies, honoraria, stock ownership or options, expert testimony, grants, or patents received or pending, or royalties. E Hampsey had no relevant financial holdings with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
L Jelen is supported by a Medical Research Council (MRC) Clinical Research Training Fellowship (MR/T028084/1).
A H Young is employed by King’s College London; Honorary Consultant South London and Maudsley NHS Foundation Trust (NHS UK). He is the Editor of Journal of Psychopharmacology and Deputy Editor, BJPsych Open. Participated in paid lectures and advisory boards for the following companies with drugs used in affective and related disorders: Allegan, AstraZeneca, Bionomics Ltd, Boehringer Ingelheim, COMPASS, Eli Lilly, Janssen, LivaNova, Lundbeck, Neurocentrx, Novartis, Sage, Servier, Sumitomo Dainippon Pharma, and Sunovion. A H Young is the Principal Investigator in the Restore-Life VNS registry study funded by LivaNova; Principal Investigator on ESKETINTRD3004: ‘An Open-label, Long-term, Safety and Efficacy Study of Intranasal Esketamine in Treatment-resistant Depression’; Principal Investigator on ‘The Effects of Psilocybin on Cognitive Function in Healthy Participants’; Principal Investigator on ‘The Safety and Efficacy of Psilocybin in Participants with Treatment-Resistant Depression (p-TRD)’; UK Chief Investigator for Novartis MDD study MIJ821A12201. A H Young’s grant funding (past and present): NIMH (U.S.A.); CIHR (Canada); NARSAD (U.S.A.); Stanley Medical Research Institute (U.S.A.); MRC (UK); Wellcome Trust (UK); Royal College of Physicians (Edin); BMA (UK); UBC-VGH Foundation (Canada); WEDC (Canada); CCS Depression Research Fund (Canada); MSFHR (Canada); NIHR (UK). Janssen (UK) EU Horizon 2020. Finally, A H Young has no shareholdings in pharmaceutical companies.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Author contribution
E Hampsey: Conducted the research for the paper, drafted the manuscript, created tables and figures, prepared for journal submission, and revised based on reviewer feedback.
L Jelen & A H Young: Provided a review of the initial draft of the manuscript, made edits to the manuscript, aided in response to reviewer comments.
Reviewer disclosures
A reviewer on this manuscript has disclosed they are a Consultant to Karuna, Neurocrine, Neumarker; Advisor/Shareholder at Praxis Bioresearch, and owns stock at Merck. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- World Health Organisation. International classification of diseases, eleventh revision (ICD-11). 2019.
- World Health Organisation. Depression and other common mental disorders: global health estimates. 2017.
- Office for National Statistics. Cost of living and depression in adults, Great Britain: 29 September to 23 October 2022. 2022.
- Department for Health and Social Care. No health without mental health: a cross-government mental health outcomes strategy for people of all ages. 2011.
- Strawbridge R, McCrone P, Ulrichsen A, et al. Care pathways for people with major depressive disorder: a European brain council value of treatment study. Eur Psychiatry. 2022;65(1):e36. doi: 10.1192/j.eurpsy.2022.28
- Mind. We need to talk survey [Internet]. Available from: https://www.mind.org.uk/media-a/4248/we-still-need-to-talk_report.pdf.
- Linde K, Sigterman K, Kriston L, et al. Effectiveness of psychological treatments for depressive disorders in primary care: systematic review and meta-analysis. Ann Fam Med. 2015;13(1):56–68. doi: 10.1370/afm.1719
- National Institute for Health and Care Excellence. Depression in adults: treatment and management. 2022.
- Public Health England. Prescribed medicines review: summary. 2020.
- Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet. 2018;391(10128):1357–1366. doi: 10.1016/S0140-6736(17)32802-7
- Nierenberg AA. Residual symptoms in depression: prevalence and impact. J Clin Psychiatry. 2015;76(11):e1480–e1480. doi: 10.4088/JCP.13097TX1C
- Treadway MT, Zald DH. Reconsidering anhedonia in depression: lessons from translational neuroscience. Neurosci Biobehav Rev. 2011;35(3):537–555. doi: 10.1016/j.neubiorev.2010.06.006
- Goodwin GM, Price J, De Bodinat C, et al. Emotional blunting with antidepressant treatments: a survey among depressed patients. J Affect Disord. 2017;221:31–35. doi: 10.1016/j.jad.2017.05.048
- European Medicines Agency. Guideline on clinical investigation of medicinal products in the treatment of depression. [Internet]. 2013 [cited 2023 Nov 29]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-depression_en.pdf.
- Global Market Insights. Antidepressant drugs market size - by drug class (SSRIs, SNRIs, tricyclic antidepressants), by application (Major depressive disorder, obsessive-compulsive disorder, generalized anxiety disorder, panic disorder), competitive market share & forecast, 2021 - 2027. 2021.
- Sakurai H, Yonezawa K, Tani H, et al. Novel antidepressants in the pipeline (phase II and III): a systematic review of the US clinical trials registry. Pharmacopsychiatry. 2022;55(4):193–202. doi: 10.1055/a-1714-9097
- Newswire PR. Antidepressant drugs market global analysis by drugs class, by sales channel, by region, by country: opportunities and forecasts 2018 to 2023: inquire for antidepressant drugs market global research report 2018 with in-depth industry study, analysis and forecasting till 2023. 2018.
- Correll CU, Solmi M, Cortese S, et al. The future of psychopharmacology: a critical appraisal of ongoing phase 2/3 trials, and of some current trends aiming to de‐risk trial programmes of novel agents. World Psychiatry. 2023;22(1):48–74. doi: 10.1002/wps.21056
- Fava M, Carpenter L, Zajecka J, et al. Proof-of-concept trial of CERC-501 augmentation of antidepressant therapy i n treatment-resistant depression (RAPID KOR). 2016.
- Janssen Research & Development, LLC. A study to explore the efficacy of JNJ-67953964 in the treatment of depression. 2023.
- Janssen Research & Development, LLC. A study of aticaprant in adult and elderly participants with Major depressive disorder (MDD) (VENTURA-LT). Available from: https://clinicaltrials.gov/study/NCT05518149?term=NCT05518149&rank=1
- Janssen Research & Development, LLC. A study of aticaprant as adjunctive therapy in adult participants with Major depressive disorder (MDD) with moderate-to-severe anhedonia and inadequate response to Current antidepressant therapy (VENTURA-1). Available from: https://clinicaltrials.gov/study/NCT05455684?term=NCT05455684&rank=1
- Janssen Research & Development, LLC. A study of aticaprant 10 milligrams (mg) as adjunctive therapy in adult participants with MDD with moderate-to-severe anhedonia and inadequate response to Current antidepressant therapy (VENTURA-2). Available from: https://clinicaltrials.gov/study/NCT05550532?term=NCT05550532%20&rank=1
- BlackThorn Therapeutics, Inc. Study in Major depressive disorder with BTRX-335140 (NMRA-335140) vs placebo. Available from: https://clinicaltrials.gov/study/NCT04221230?term=NCT04221230&rank=1
- Neumora Therapeutics, Inc. Study to evaluate the effects of oral NMRA 335140 versus placebo in participants with Major depressive disorder. Available from: https://clinicaltrials.gov/study/NCT06029426?term=BTRX-335140&rank=5
- Neumora Therapeutics, Inc. Study to assess the safety and effectiveness of NMRA-335140-501. Available from: https://clinicaltrials.gov/study/NCT06029439?term=BTRX-335140&rank=3
- Alkermes, Inc. ALK33BUP-201: safety and tolerability study of ALKS 33-BUP (ALKS 5461) administration in subjects with MDD. 2011. Available from: https://clinicaltrials.gov/study/NCT01381107?term=ALKS-5461&page=2&rank=17
- Alkermes, Inc. A study of ALKS 5461 in healthy volunteers. 2014. Available from: https://clinicaltrials.gov/study/NCT02068105?term=ALKS-5461&rank=6
- Alkermes, Inc. A long-term safety study of ALKS 5461. 2014. Available from: https://clinicaltrials.gov/study/NCT02141399?term=ALKS-5461&page=2&rank=14
- Alkermes, Inc. A study of different titration schedules of ALKS 5461 in adults with Major depressive disorder (MDD). 2014. Available from: https://clinicaltrials.gov/study/NCT02085135?term=ALKS-5461&page=2&rank=15
- Alkermes, Inc. A study to evaluate ALKS 5461 in subjects with Major depressive disorder (MDD). 2011. Available from: https://clinicaltrials.gov/study/NCT01500200?term=NCT01500200&rank=1
- Alkermes, Inc. A study of ALKS 5461 for the treatment of Major depressive disorder (MDD) - the FORWARD-3 study. 2014. Available from: https://clinicaltrials.gov/study/NCT02158546?term=ALKS-5461&rank=9
- Alkermes, Inc. A study of ALKS 5461 for the treatment of Major depressive disorder (MDD) - the FORWARD-4 study. 2014. Available from: https://clinicaltrials.gov/study/NCT02158533?term=ALKS-5461&rank=7
- Alkermes, Inc. A study of ALKS 5461 for the treatment of Major depressive disorder (MDD) - FORWARD-5 study. 2014. Available from: https://clinicaltrials.gov/study/NCT02218008?term=NCT02218008&rank=1
- Alkermes, Inc. A study of ALKS 5461 for treatment refractory Major depressive disorder (MDD). 2017. Available from: https://clinicaltrials.gov/study/NCT03188185?term=ALKS-5461&rank=10
- Alkermes, Inc. A long term study of ALKS 5461 in the treatment of refractory Major depressive disorder (MDD). 2018. Available from: https://clinicaltrials.gov/study/NCT03610048?term=NCT03610048%20&rank=1
- Jelen LA, Stone JM, Young AH, et al. The opioid system in depression. Neurosci Biobehav Rev. 2022;140:104800. doi: 10.1016/j.neubiorev.2022.104800
- Jelen LA, Young AH, and Mehta MA. Current Topics in Behavioural Neurosciences. 2023. doi: 10.1007/7854_2023_448
- Suppes T, Silva R, Cucchiaro J, et al. Lurasidone for the treatment of major depressive disorder with mixed features: a randomized, double-blind, placebo-controlled study. Am J Psychiatry. 2016;173(4):400–407. doi: 10.1176/appi.ajp.2015.15060770
- Tsai J, Thase ME, Mao Y, et al. Lurasidone for major depressive disorder with mixed features and anxiety: a post-hoc analysis of a randomized, placebo-controlled study. CNS Spectr. 2017;22(2):236–245. doi: 10.1017/S1092852917000074
- Guo M, Mi J, Jiang Q, et al. Metformin may produce antidepressant effects through improvement of cognitive function among depressed patients with diabetes mellitus. Clin Exp Pharmacol Physiol. 2014;41(9):650–656. doi: 10.1111/1440-1681.12265
- Preskorn S, Macaluso M, Mehra DV, et al. Randomized proof of concept trial of GLYX-13, an N-Methyl-D-Aspartate receptor glycine site partial agonist, in major depressive disorder nonresponsive to a previous antidepressant agent. J Psychiatr Pract. 2015;21(2):140–149. doi: 10.1097/01.pra.0000462606.17725.93
- Kamiya M, Sabia HD, Marella J, et al. Efficacy and safety of TS-121, a novel vasopressin V1B receptor antagonist, as adjunctive treatment for patients with major depressive disorder: a randomized, double-blind, placebo-controlled study. J Psychiatr Res. 2020;128:43–51. doi: 10.1016/j.jpsychires.2020.05.017
- Savitz A, Wajs E, Zhang Y, et al. Efficacy and safety of seltorexant as adjunctive therapy in major depressive disorder: a phase 2b, randomized, placebo-controlled, adaptive dose-finding study. Int J Neuropsychopharmacol. 2021;24(12):965–976. doi: 10.1093/ijnp/pyab050
- Lalanne L, Ayranci G, Kieffer BL, et al. The kappa opioid receptor: from addiction to depression, and back. Front Psychiatry. 2014/12/30 ed. 2014;5:170. doi: 10.3389/fpsyt.2014.00170
- Carlezon WJ, Beguin C, DiNieri JA, et al. Depressive-like effects of the κ-opioid receptor agonist salvinorin a on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2005/10/15 ed. 2006;316(1):440–447. doi: 10.1124/jpet.105.092304
- Todtenkopf MS, Marcus JF, Portoghese PS, et al. Effects of κ-opioid receptor ligands on intracranial self-stimulation in rats. Psychopharmacol Berl. 2004/01/17 ed. 2004;172(4):463–470. doi: 10.1007/s00213-003-1680-y
- Chefer VI, Backman CM, Gigante ED, et al. Kappa opioid receptors on dopaminergic neurons are necessary for kappa-mediated place aversion. Neuropsychopharmacology. 2013/08/08 ed. 2013;38:2623–2631. doi: 10.1038/npp.2013.171
- Anderson RI, Morales M, Spear LP, et al. Pharmacological activation of kappa opioid receptors: aversive effects in adolescent and adult male rats. Psychopharmacol Berl. 2013/04/23 ed. 2014;231(8):1687–1693. doi: 10.1007/s00213-013-3095-8
- Cohen BM, Murphy B. The effects of pentazocine, a kappa agonist, in patients with mania. Int J Neuropsychopharmacol [Internet]. 2008 [cited 2023 Oct 17];11(2). Available from: https://academic.oup.com/ijnp/article-lookup/doi/10.1017/S1461145707008073
- Knoll AT, Carlezon WA. Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074
- Jacobson ML, Browne CA, Lucki I. Kappa opioid receptor antagonists as potential therapeutics for stress-related disorders. Annu Rev Pharmacol Toxicol. 2020;60(1):615–636. doi: 10.1146/annurev-pharmtox-010919-023317
- Peters MF, Zacco A, Gordon J, et al. Identification of short-acting κ-opioid receptor antagonists with anxiolytic-like activity. Eur J Pharmacol. 2011;661(1–3):27–34. doi: 10.1016/j.ejphar.2011.04.017
- Mague SD, Pliakas AM, Todtenkopf MS, et al. Antidepressant-like effects of κ-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003/03/22 ed. 2003;305(1):323–330. doi: 10.1124/jpet.102.046433
- Beardsley PM, Howard JL, Shelton KL, et al. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacol Berl. 2005/09/27 ed. 2005;183(1):118–126. doi: 10.1007/s00213-005-0167-4
- Grimwood S, Lu Y, Schmidt AW, et al. Pharmacological characterization of 2-methyl-N-((2′-(pyrrolidin-1-ylsulfonyl)biphenyl-4-yl)methyl)propan-1-amine (PF-04455242), a high-affinity antagonist selective for κ-opioid receptors. J Pharmacol Exp Ther. 2011/08/09 ed. 2011;339(2):555–566. doi: 10.1124/jpet.111.185108
- Rorick-Kehn LM, Witkin JM, Statnick MA, et al. LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology. 2014;77:131–144. doi: 10.1016/j.neuropharm.2013.09.021
- Guerrero M, Urbano M, Kim E-K, et al. Design and synthesis of a novel and selective kappa opioid receptor (KOR) antagonist (BTRX-335140). J Med Chem. 2019;62(4):1761–1780. doi: 10.1021/acs.jmedchem.8b01679
- Carlezon WA, Krystal AD. Kappa-opioid antagonists for psychiatric disorders: from bench to clinical trials: 2015 ADAA scientific research symposium: KORs in psychiatric illness. Depress Anxiety. 2016;33(10):895–906. doi: 10.1002/da.22500
- Page S, Mavrikaki MM, Lintz T, et al. Behavioral pharmacology of novel kappa opioid receptor antagonists in rats. Int J Neuropsychopharmacol. 2019:pyz054. doi: 10.1093/ijnp/pyz054
- Margolis EB, Wallace TL, Van Orden LJ, et al. Differential effects of novel kappa opioid receptor antagonists on dopamine neurons using acute brain slice electrophysiology. PLOS ONE. Taylor B, editor. 2020;15(12):e0232864. doi: 10.1371/journal.pone.0232864
- Jackson KJ, Jackson A, Carroll FI, et al. Effects of orally-bioavailable short-acting kappa opioid receptor-selective antagonist LY2456302 on nicotine withdrawal in mice. Neuropharmacology. 2015;97:270–274. doi: 10.1016/j.neuropharm.2015.05.023
- Domi E, Barbier E, Augier E, et al. Preclinical evaluation of the kappa-opioid receptor antagonist CERC-501 as a candidate therapeutic for alcohol use disorders. Neuropsychopharmacology. 2018;43(9):1805–1812. doi: 10.1038/s41386-018-0015-y
- Zhou Y. Aticaprant (clinically developed kappa-opioid receptor antagonist) combined with naltrexone prevents alcohol “relapse” drinking. J Pharm Pharmacol [Internet]. 2022 [cited 2023 Oct 17];9(1). Available from: https://www.avensonline.org/fulltextarticles/JPP-2327-204X-09-0032.html
- Jacobson ML, Wulf HA, Tsuda MC, et al. Sex differences in the modulation of mouse nest building behavior by kappa opioid receptor signaling. Neuropharmacology. 2020;177:108254. doi: 10.1016/j.neuropharm.2020.108254
- Jacobson ML, Wulf HA, Browne CA, et al. The kappa opioid receptor antagonist aticaprant reverses behavioral effects from unpredictable chronic mild stress in male mice. Psychopharmacol (Berl). 2020;237(12):3715–3728. doi: 10.1007/s00213-020-05649-y
- Filliol D, Ghozland S, Chluba J, et al. Mice deficient for δ- and μ-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25(2):195–200. doi: 10.1038/76061
- Saitoh A, Yamada M. Antidepressant-like effects of δ opioid receptor agonists in animal models. Curr Neuropharmacol. 2012;10(3):231–238. doi: 10.2174/157015912803217314
- Chen C-M, Ding H, Mabry KM, et al. Enhanced antidepressant-like effects of a delta opioid receptor agonist, SNC80, in rats under inflammatory pain. Pharmacol Biochem Behav. 2022;214:173341. doi: 10.1016/j.pbb.2022.173341
- Wang J, Song Q, Xu A, et al. Design, synthesis and biological evaluation of aminobenzyloxyarylamide derivatives as selective κ opioid receptor antagonists. Eur J Med Chem. 2017;130:15–25. doi: 10.1016/j.ejmech.2017.02.029
- AstraZeneca. Study of antidepressant efficacy of a selective, high affinity enkephalinergic agonist in anxious major depressive disorder (AMDD). 2012.
- Lee D-S, Law P-Y, Ln W, et al. Differential regulation of mouse and human mu opioid receptor gene depends on the single stranded DNA structure of its promoter and α-complex protein 1. Biomed Rep. 2017;6(5):532–538. doi: 10.3892/br.2017.877
- Schattauer SS, Miyatake M, Shankar H, et al. Ligand directed signaling differences between rodent and human κ-opioid receptors. J Biol Chem. 2012;287(50):41595–41607. doi: 10.1074/jbc.M112.381368
- Rorick-Kehn LM, Witcher JW, Lowe SL, et al. Determining pharmacological selectivity of the kappa opioid receptor antagonist LY2456302 using pupillometry as a translational biomarker in rat and human. Int J Neuropsychopharmacol [Internet]. 2015 cited 2023 Oct 17; 18(2). Available from: https://academic.oup.com/ijnp/article-lookup/doi/10.1093/ijnp/pyu036
- Lowe SL, Wong CJ, Witcher J, et al. Safety, tolerability, and pharmacokinetic evaluation of single‐ and multiple‐ascending doses of a novel kappa opioid receptor antagonist LY2456302 and drug interaction with ethanol in healthy subjects. J Clin Pharmacol. 2014;54(9):968–978. doi: 10.1002/jcph.286
- Reed B, Butelman ER, Fry RS, et al. Repeated administration of opra kappa (LY2456302), a novel, short-acting, selective KOP-r antagonist, in persons with and without cocaine dependence. Neuropsychopharmacol. 2018;43(4):739–750. doi: 10.1038/npp.2017.205
- Krystal AD, Pizzagalli DA, Smoski M, et al. A randomized proof-of-mechanism trial applying the ‘fast-fail’ approach to evaluating κ-opioid antagonism as a treatment for anhedonia. Nat Med. 2020;26(5):760–768. doi: 10.1038/s41591-020-0806-7
- Fava M, Mazzone E, Freeman M, et al. Double-blind, placebo-controlled, proof-of-concept trial of a kappa-selective opioid receptor antagonist augmentation in treatment-resistant depression. Ann Clin Psychiatry [Internet]. 2020 [cited 2023 Oct 17];33(1). Available from: https://www.aacp.com/article/abstract/double-blind-placebo-controlled-proof-of-concept-trial-of-a-kappa-selective-opioid-receptor-antagonist-augmentation-in-treatment-resistant-depression/
- Hu XH, Bull SA, Hunkeler EM, et al. Incidence and duration of side effects and those rated as bothersome with selective serotonin reuptake inhibitor treatment for depression: patient report versus physician estimate. J Clin Psychiatry. 2004;65(7):959–965. doi: 10.4088/JCP.v65n0712
- Opioid antagonists. Meylers side eff drugs int encycl adverse drug react interact [internet]. Elsevier; 2006 [cited 2023 Oct 17]. p. 2639. Available from: https://linkinghub.elsevier.com/retrieve/pii/B0444510052001972
- Urbano M, Guerrero M, Rosen H, et al. Antagonists of the kappa opioid receptor. Bioorg Med Chem Lett. 2014;24(9):2021–2032. doi: 10.1016/j.bmcl.2014.03.040
- Stanciu CN, Glass OM, Penders TM. Use of buprenorphine in treatment of refractory depression—A review of current literature. Asian J Psychiatr. 2017;26:94–98. doi: 10.1016/j.ajp.2017.01.015
- Ragguett R-M, Rong C, Rosenblat JD, et al. Pharmacodynamic and pharmacokinetic evaluation of buprenorphine + samidorphan for the treatment of major depressive disorder. Expert Opin Drug Metab Toxicol. 2018;14(4):475–482. doi: 10.1080/17425255.2018.1459564
Appendix
A
Identification of Aticaprant literature
PubMed and Embase were searched using the terms ‘Aticaprant,’ ‘JNJ-67953964,’ ‘CERC-501,’ ‘LY-2456302,’ and ‘OpraKappa,’ with no restrictions on date, region, or any other factor. This returned 25 unique articles which were synthesized in the text of the article. Identification of ongoing and unpublished trials, the same terms were searched on clinicaltrials.gov, International Clinical Trials Registry Platform, and the EU Clinical Trials Register, again with no restrictions.
Identification of supporting literature
Identification of supporting literature in this manuscript (i.e., not concerning Aticaprant directly), was identified using web of knowledge searches, reading reference lists of literature referencing Aticaprant, and our own existing knowledge base.
Identification of compounds listed in Appendix B
Appendix B lists antidepressants that either are, or were, in use across many global localities. An initial list was collated using various internet sources, including the British National Formulary, Mind.co.uk, Wikipedia, and drugs.com. Internet and PubMed searches of each category of antidepressant also returned further, more obscure compounds discussed in extant literature. Lastly, the authors existing knowledge base supplemented this list.
Appendix
B
Overview table of antidepressant medications developed internationally